

Abstract
Autophagy and apoptosis are fundamental cellular pathways that are both regulated by JNK mediated Bcl-2 phosphorylation. Several years ago, JNK-mediated Bcl-2 phosphorylation was shown to interfere with its binding to pro-apoptotic BH3 domain-containing proteins such as Bax and recently, our laboratory demonstrated that JNK1-mediated Bcl-2 phosphorylation interferes with its binding to the pro-autophagy BH3 domain-containing protein Beclin 1. Here, we examined the kinetic relationship between Bcl-2 phosphorylation, Bcl-2/Beclin 1 interactions, Bcl-2/Bax interactions and caspase 3 activation during nutrient starvation. We found that after a short period of nutrient deprivation (4 hours), a small amount of Bcl-2 phosphorylation dissociates Bcl-2 from the Bcl-2/Beclin 1 complex but not from the Bcl-2/Bax complex. After 16 hours of nutrient deprivation, Bcl-2 phosphorylation reaches maximal levels, the Bcl-2/Bax complex is disrupted, and active caspase 3 is detected, indicating the initiation of apoptosis. Based on this result, we propose a speculative model for understanding the interrelationship between autophagy and apoptosis regulated by JNK1-mediated Bcl-2 phosphorylation. According to this model, rapid Bcl-2 phosphorylation may occur initially to promote cell survival by disrupting the Bcl-2/Beclin 1 complex and activating autophagy. At a certain point when autophagy is no longer able to keep the cell alive, Bcl-2 phosphorylation might then serve to inactivate its anti-apoptotic function.
Keywords: nutrient starvation, Bcl-2, JNK1, Bax, caspase 3, autophagy, apoptosis
Given the fundamental role of autophagy in various cellular homeostatic processes1,2, it is essential for this pathway to be tightly-regulated. Previous findings suggest that mammalian autophagy can be regulated through Beclin 1, an important component of this pathway.2,3 Beclin 1 is part of a Class III PI3K complex that participates in autophagosome formation, mediating the localization of other autophagy proteins to the preautophagosomal membrane.4 The autophagy function of the Beclin 1-Class III PI3K complex is activated by UVRAG, Ambra-1, and Bif-1 and inhibited by Bcl-2 and Bcl-xL.2 Nutrient conditions have been found to regulate the interaction between endogenous Bcl-2 and Beclin 1.5 When autophagy is induced by nutrient deprivation, Bcl-2 binding to Beclin 1 is minimal; when autophagy is inhibited by nutrient excess, Bcl-2 binding to Beclin 1 is maximal. However, the mechanism(s) by which nutrient conditions regulate the interaction between Bcl-2 and Beclin 1 remained unknown until our recent discovery that starvation-induced dissociation of Beclin 1 from Bcl-2 involves phosphorylation of Bcl-2 by the stress-activated signaling molecule, c-Jun N-terminal protein kinase 1 (JNK1).6
We found that starvation induces dissociation of the cellular Bcl-2-Beclin 1 complex, while viral Bcl-2, which lacks the non-structured phosphorylation loop, escapes this starvation-induced regulation of binding to Beclin 1. This observation provided the initial insight that led to our assessment of the role of Bcl-2 phosphorylation as a gatekeeper in autophagy activation. Specifically, we asked whether and how Bcl-2 phosphorylation is induced by nutrient starvation and whether Bcl-2 phosphorylation would alter its interaction with Beclin 1. In our recent study, we found that upon nutrient withdrawal, JNK1 is activated and induces phosphorylation at multiple residues (T69, S70 and S87) in the non-structured loop of Bcl-2 that is located between BH4 and BH1 domain. JNK1 inhibition by a dominant negative JNK1 mutant or replacement of wild-type Bcl-2 by a non-phosphorylatable Bcl-2 mutant (T69A/S70A/S87A) abolishes the starvation-induced dissociation of Bcl-2 and Beclin 1 and inhibits autophagy. Expression of constitutively active JNK1 leads to Bcl-2 phosphorylation, dissociation of Bcl-2 from Beclin 1, and stimulation of autophagy during normal growth conditions; however, active JNK1 fails to stimulate autophagy when Bcl-2 is replaced by its non-phosphorylatable mutant. In jnk1-/- MEFs, starvation fails to induce Bcl-2 multi-site phosphorylation, dissociation of the Bcl-2/Beclin 1 complex, or autophagy. Furthermore, a multi-site Bcl-2 phosphomimetic mutant (T69E/S70E/S87E) fails to co-immunoprecipitate with Beclin 1 and has no autophagy-inhibitory activity. Together, these findings indicate that JNK1-mediated multi-site phosphorylation of Bcl-2 promotes its dissociation from Beclin 1, thereby inducing autophagy.
Our findings identifying a role of Bcl-2 phosphorylation in the regulation of Bcl-2/Beclin 1 binding are consistent with previous results regarding the relationship between Bcl-2 phosphorylation and binding to BH3-domain containing proteins.7 Phosphorylation of Bcl-2 inhibits its binding to both multi-domain and BH3-only pro-apoptotic family members and promotes apoptosis.7 Recently, Beclin 1 has been identified as a novel BH3-only protein that binds to Bcl-2 and Bcl-xL via its BH3 domain8,9 based on 3-D structure and mutational analyses. Thus, there are parallels between the effects of Bcl-2 phosphorylation on apoptosis regulation and on autophagy regulation. In both cases, Bcl-2 phosphorylation interferes with its binding to BH3-containing proteins, including BH3-containing pro-apoptotic proteins and the BH3-containing pro-autophagy protein, Beclin 1. Although autophagy is activated as a cell-survival mechanism during starvation,1 prolonged cellular starvation eventually results in apoptosis. Consistent with the pro-survival function of autophagy, transient JNK1 activation has also been reported to promote cell survival.10 However, prolonged JNK1 activation can mediate apoptosis .11 These observations led us to wonder why autophagy induced by the JNK1-Bcl-2 phosphorylation pathway ultimately fails to protect cells from apoptosis, and at what point nutrient-starved cells escape autophagy protection and commit to apoptosis.
To investigate this question, we starved HeLa cells in HBSS medium for different time periods, ranging from 2 hour to 30 hours, and then detected the level of Bcl-2 phosphorylation, caspase 3 activation and Bcl-2/Beclin 1 and Bcl-2/Bax interactions (Figure 1). We found that Bcl-2 multi-site phosphorylation occurs after two hours of nutrient deprivation, as detected by the appearance of a slower migration band upon immunoblot analyis with a phospho-specific anti-S70 Bcl-2 antibody. The amount of the slower migration multi-site phosphorylation band steadily increases, reaching peak levels at 16 hours after starvation. Of note, active caspase 3 is first detected after 16 hours of starvation, which indicates the onset of apoptosis in starved cells at this time point. Compared with the baseline time point (0 hour starvation), the amount of Bcl-2 that co-immunoprecipitates with Beclin 1 dramatically decreases after 2 hours of starvation, and is undetectable by 4 hours of starvation. Unlike the Bcl-2-Beclin 1 interaction, the Bcl-2-Bax interaction decreases at much slower pace in response to nutrient starvation. The dissociation of Bcl-2 from Bax begins at 16 hours of starvation when the amount of Bcl-2 multi-site phosphorylation reaches maximal levels and when caspase 3 is activated. By 24 hours of starvation, Bcl-2 completely dissociates from Bax.
Figure 1. Kinetic differences in the effects of starvation-induced multi-site phosphorylation of Bcl-2 on the disruption of Bcl-2/Beclin 1 and Bcl-2 / Bax complexes.
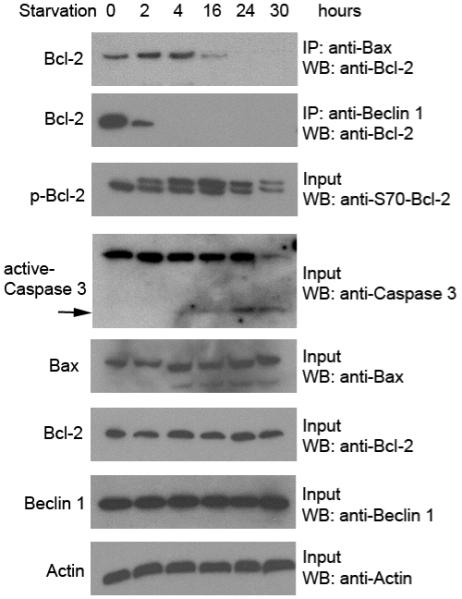
HeLa cells were starved in HBSS medium for the indicated time period. Bcl-2 co-immunoprecipitation with Bax (top panel) and Beclin 1 (second panel) was detected with an HRP-conjugated monoclonal anti-Bcl-2 (clone 100) antibody. Other panels represent the total amount of phospho-Bcl-2 (third panel), caspase 3 (fourth panel, arrow denotes active caspase 3, the lower molecular weight cleavage band), Bax (fifth panel), Bcl-2 (sixth panel), Beclin 1 (seventh panel) detected in cell lysates by Western blot analysis with the indicated antibody. Actin was used as a loading control (bottom panel).
These data suggest that, in response to starvation, Bcl-2 dissociates rapidly from Beclin 1 in the setting of low levels of Bcl-2 multi-site phosphorylation, whereas Bcl-2 dissociates more slowly from Bax when higher levels of Bcl-2 multi-site phosphorylation are observed. Further, the dissociation of Bcl-2 from Bax is temporally associated with the onset of apoptosis. Based on these findings and unpublished data from our laboratory indicating that Bcl-2 has a lower binding affinity to the BH3 domain of Beclin 1 than those of pro-apoptotic BH3 proteins including Bax, we propose a model for understanding the interrelationship between autophagy and apoptosis regulation by JNK1-mediated Bcl-2 phosphorylation (Figure 2). According to this model, at early time points during nutrient starvation, when the amount of multi-site phosphorylated Bcl-2 is low, the lower affinity binding protein Beclin 1 is released from the Bcl-2/Beclin 1 complex to promote autophagy and attempt to protect the cell from apoptosis. With prolonged starvation, there is steady accumulation of multi-site phosphorylated Bcl-2, and eventually, the higher affinity pro-apoptotic BH3 containing protein dissociates from Bcl-2. At such a point, the cells can no longer be protected by autophagy and commit to death. Thus, Bcl-2 phosphorylation may not only represent a mechanism for regulating autophagy and a mechanism for regulating apoptosis, but perhaps, a mechanism for regulating the switch between the two pathways. Further studies are needed to directly test these speculations.
Figure 2. A speculative model of the interrelationship between the roles of JNK1-mediated Bcl-2 multi-site phosphorylation in autophagy regulation and apoptosis regulation.

According to this model, a low level of Bcl-2 multi-site phosphorylation occurs initially in response to starvation to promote cell survival by disrupting the Bcl-2/Beclin 1 complex (a lower affinity interaction) and activating autophagy. With prolonged starvation, higher levels of Bcl-2 multi-site phosphorylation leads to disruption of the the complex between Bcl-2/Bax (or other pro-apoptotic BH3 domain-containing proteins) (which are higher affinity interactions), promoting apoptosis at a time point when autophagy can no longer keep cells alive.
References
- 1.Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell. 2004;6:463–77. doi: 10.1016/s1534-5807(04)00099-1. [DOI] [PubMed] [Google Scholar]
- 2.Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, Levine B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. 1999;402:672–6. doi: 10.1038/45257. [DOI] [PubMed] [Google Scholar]
- 4.Kihara A, Kabeya Y, Ohsumi Y, Yoshimori T. Beclin-phosphatidylinositol 3-kinase complex functions at the trans-Golgi network. EMBO Rep. 2001;2:330–5. doi: 10.1093/embo-reports/kve061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, Packer M, Schneider MD, Levine B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122:927–39. doi: 10.1016/j.cell.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 6.Wei Y, Pattingre S, Sinha S, Bassik M, Levine B. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol Cell. 2008;30:678–88. doi: 10.1016/j.molcel.2008.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bassik MC, Scorrano L, Oakes SA, Pozzan T, Korsmeyer SJ. Phosphorylation of BCL-2 regulates ER Ca2+ homeostasis and apoptosis. EMBO J. 2004;23:1207–16. doi: 10.1038/sj.emboj.7600104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maiuri MC, Le Toumelin G, Criollo A, Rain JC, Gautier F, Juin P, Tasdemir E, Pierron G, Troulinaki K, Tavernarakis N, Hickman JA, Geneste O, Kroemer G. Functional and physical interaction between Bcl-X(L) and a BH3-like domain in Beclin-1. EMBO J. 2007;26:2527–39. doi: 10.1038/sj.emboj.7601689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Oberstein A, Jeffrey PD, Shi Y. Crystal structure of the Bcl-XL-Beclin 1 peptide complex:Beclin 1 is a novel BH3-only protein. J Biol Chem. 2007;282:13123–32. doi: 10.1074/jbc.M700492200. [DOI] [PubMed] [Google Scholar]
- 10.Ham YM, Chun KH, Choi JS, Kim DH, Lee SK. SEK1-dependent JNK1 activation prolongs cell survival during G-Rh2-induced apoptosis. Biochem Biophys Res Commun. 2003;304:358–64. doi: 10.1016/s0006-291x(03)00591-6. [DOI] [PubMed] [Google Scholar]
- 11.Ventura JJ, Hubner A, Zhang C, Flavell RA, Shokat KM, Davis RJ. Chemical genetic analysis of the time course of signal transduction by JNK. Mol Cell. 2006;21:701–10. doi: 10.1016/j.molcel.2006.01.018. [DOI] [PubMed] [Google Scholar]