

Abstract
The molecular basis of general anesthetic interactions with GABAA receptors is uncertain. An accurate homology model would facilitate studies of anesthetic action. Construction of a GABAA model based on the 4 Å resolution acetylcholine receptor structure is complicated by alignment uncertainty between the acetylcholine and GABAA receptor M3 and M4 transmembrane segments. Using disulfide crosslinking we previously established the orientation of M2 and M3 within a single GABAA subunit. The resultant model predicts that the βM3 residue β2M286, implicated in anesthetic binding, faces the adjacent α1-M1 segment and not into the β2 subunit interior as some models have suggested. To assess the proximity of β2M286 to the α1-M1 segment we expressed β2M286C and γ2 with 10 consecutive α1-M1 cysteine (Cys) mutants, α1I223C to α1L232C, in and flanking the extracellular end of α1-M1. In activated states, β2M286C formed disulfide bonds with α1Y225C and α1Q229C based on electrophysiological assays and dimers on Western blots, but not with other α1-M1 mutants. β2F289, one helical turn below β2M286, formed disulfide bonds with α1I228C, α1Q229C and α1L232C in activated states. The intervening residues, β2G287C and β2C288, did not form disulfide bonds with α1-M1 Cys mutants. We conclude that the β2-M3 residues β2M286 and β2F289 face the intersubunit interface in close proximity to α1-M1 and that channel gating induces a structural rearrangement in the transmembrane subunit interface that reduces the βM3 to αM1 separation by ∼7 Å. This supports the hypothesis that some intravenous anesthetics bind in the βM3-αM1 subunit interface consistent with azi-etomidate photoaffinity labeling.
Introduction
Since the first successful use of general anesthesia in surgery in 1846 scientists have sought to understand the molecular and cellular basis of general anesthetic action. During the past decade, investigators recognized the importance of general anesthetic interactions with GABAA receptors (Franks, 2008). To elucidate the molecular basis of these interactions, research has sought to determine the GABAA receptor structure and the location of anesthetic binding sites.
GABAA receptors belong to the Cys-loop receptor neurotransmitter-gated ion channel superfamily (Akabas, 2004). Subunits have an ∼200 residue extracellular domain and four transmembrane segments (M1, M2, M3, M4). Medium to high resolution structures exist for the Torpedo nicotinic acetylcholine (ACh) receptor (4 Å resolution) (Unwin, 2005), a prokaryotic homolog (3.3 Å resolution) (Hilf and Dutzler, 2008), and the snail ACh binding proteins (Celie et al., 2004). Difficulties obtaining sufficient quantities of eukaryotic membrane proteins make a GABAA receptor crystal structure unlikely in the near future. Homology models could provide useful GABAA molecular models for the interpretation of experimental results.
Sequence alignment is a critical step in homology modeling. Absolutely conserved residues between the pre-M1 region and the extracellular end of M2 provide confidence in the GABAA-ACh sequence alignment in this region. The alignment of the M2-M3 loop and M3 segment are uncertain because of the lack of absolutely conserved residues in this region and to differences in the M2-M3 loop length of up to two amino acids. Previously, we used disulfide crosslinking between pairs of engineered cysteines (Cys) within the α1-M2 and M3 segments to demonstrate that GABAA M3 residues α1L301 and α1F304 were in close proximity to α1T267 (M2–12′) (Jansen and Akabas, 2006). In the ACh receptor structure, αI289 and αT292 in M3 are closest to M2–12′. This provides an experimental basis for constructing a GABAA receptor model to the end of the M3 segment.
Residues implicated in general anesthetic action include the M2–15′ position, aligned with β2N265, and a position near the M3 extracellular end, aligned with β2M286 (Belelli et al., 1997; Mihic et al., 1997; Hemmings et al., 2005). β2M286 mutations altered the cutoff size for long chain alcohol effects (Krasowski et al., 2001). Propofol, an intravenous general anesthetic, protected β2M286C from covalent modification by sulfhydryl-reactive reagents (Bali and Akabas, 2004). Azi-etomidate, a photo-labile anesthetic, labeled both βM286 and αM236 suggesting that the etomidate binding site is in the subunit interface (Li et al., 2006). In earlier GABAA models, βN265 and βM286 were positioned facing the interior of a single subunit (Trudell and Bertaccini, 2004; Hosie et al., 2006; Campagna-Slater and Weaver, 2007). Our GABAA receptor homology model (Jansen and Akabas, 2006) predicts that β2M286 faces the subunit interface in close proximity to the α1-M1 segment. We tested the hypothesis that β2M286 is in close proximity to residues in or flanking the extracellular end of α1-M1. The results validate our homology model and suggest that channel activation decreases the intersubunit separation between β2-M3 and α1-M1. The results are consistent with the idea that some intravenous anesthetics bind in the transmembrane subunit interface.
Materials and Methods
Constructs used, mRNA preparation, and oocyte expression.
Because the rat α1 and β2 subunits have endogenous Cys in the M1 and M3 segments we used Cys-light constructs for these subunits to ensure that the endogenous Cys did not complicate the interpretation of our results. In the Cys-light constructs the endogenous α1C234 (M1) and α1C293 (M3) were mutated to serine, and β2C288 (M3) was mutated to alanine. In this study we refer to the α1C234S-C293Sβ2C288Aγ2s as the wild-type Cys-light construct. All engineered Cys mutants were made and expressed in this background. All mutants were made using the QuikChange method (Stratagene) and sequenced completely to confirm the mutations. The α1 Cys mutants studied in this work cover the range 223-IGYFVIQTYL-232.
Preparation and injection of mRNA in oocytes was performed as described previously (Bali and Akabas, 2007). Briefly, capped mRNA was synthesized by in vitro transcription using T7 RNA polymerase mMessage mMachine kit (Ambion) from NheI-linearized (New England Biolabs) pGEMHE vectors containing rat GABAA α1, β2, γ2s or the various mutants used. Xenopus laevis oocytes were harvested and defolliculated with collagenase for 1 h at 16°C in SOS medium (in mm): 100 NaCl, 2 KCl, 1 MgCl2, 1.8 CaCl2, and 5 HEPES, pH 7.5 with 100 IU/ml penicillin, 100 μg/ml streptomycin, and 250 ng/ml amphotericin B (Invitrogen) supplemented with 5% horse serum (Sigma-Aldrich). Oocytes were injected with 50 nl of 0.2 ng/nl mRNA 24–48 h after isolation and maintained at 16°C. The ratio of mRNA α1:β2:γ2 injected was 1:1:2. We previously showed based on Zn2+ and diazepam sensitivity that this ratio gave us uniform incorporation of γ2 into the surface expressed, functional receptors (Horenstein et al., 2005).
Electrophysiology.
Two-electrode voltage-clamp experiments were conducted at room temperature 3–5 d after mRNA injection as described previously (Bali and Akabas, 2007). Oocytes, in an ∼200 μl chamber, were voltage clamped at −60 mV and perfused at 5–6 ml/min with calcium-free frog ringer (CFFR) solution (in mm) 115 NaCl, 2.5 KCl, 1.8 MgCl2, 10 HEPES, pH 7.5 with NaOH. Currents were recorded and analyzed using a TEV-200 amplifier (Dagan Instruments), Digidata 1322A analog-digital converter and pClamp 8.2 software (Molecular Devices). The ground electrode was connected to the bath via a 3 m KCl/Agar bridge. Glass microelectrodes were filled with 3 m KCl and had a resistance <2 MΩ. All experiments were performed on at least three oocytes from at least two different batches of oocytes.
Stock solutions of GABA, DTT, CuSO4 in water were prepared at concentrations of 0.1, 1.0, and 0.1 m, respectively. 1,10-phenanthroline (phen) was prepared as a 1.0 m stock solution in DMSO. Reagents were diluted into CFFR just before each experiment. DMSO concentration never exceeded 0.1%, a concentration at which no effect is observed on GABA-induced currents (data not shown). Copper phenanthroline (Cu:phen) solutions contained 100 μm CuSO4 and 200 μm phenanthroline.
Reagent applications were separated by 4–5 min wash periods to allow complete washout and recovery from desensitization. p-Chloromercuribenzenesulfonate (pCMBS−) was obtained from Toronto Research Chemicals.
Western blotting.
Four days after mRNA injection, oocytes were washed once with CFFR. Cu:phen/GABA (100:200 μm/1 μm) was then applied for 2 min on intact oocytes. Surface proteins were biotinylated with 0.5 mg/ml sulfo-NHS-LC-biotin (Pierce) for 30 min at room temperature (RT) in CFFR, pH 8. Oocytes were then washed with CFFR and Tris-NaCl buffer (100 mm NaCl, 20 mm Tris, pH 7.5), triturated at 4°C in 20 μl/oocyte lysis buffer (Tris-NaCl buffer plus 1% Triton X-100, 0.5% deoxycholate, 10 mm N-ethyl maleimide, and HALT protease inhibitor cocktail (Pierce), solubilized by rotating at 4°C for 2 h, and spun twice at 16,000 × g, 4°C, 20 min to remove debris and yolk. Biotinylated proteins were bound to streptavidin beads (Pierce) by rotating for 4 to 16 h at 4°C. Beads were washed with lysis buffer and bound proteins eluted with 4× SDS-sample buffer for 10 min, 37°C. Proteins were separated by SDS-PAGE (4–15% PAGE gel) (Bio-Rad), transferred to PVDF membranes (Bio-Rad) and processed according to standard procedures using a primary rabbit anti-GABAA α1 polyclonal (AB9820, Millipore Bioscience Research Reagents) and secondary horseradish peroxidase-conjugated goat anti-rabbit IgG (Pierce) antibodies and ECL (Pierce).
Homology model.
The construction of the homology model was described previously (Jansen and Akabas, 2006). All distance measurements are based on α carbon separation and were made using Deepview Swiss PDB Viewer version 3.7 (http://spdbv.vital-it.ch/). Figures were generated either in Deepview and rendered using PovRay version 3.6 (Persistence of Vision Pty. Ltd.) or using Pymol (DeLano Scientific LLC).
Statistical/data analysis.
Results are presented as mean ± SEM. GABA concentration-response relationships were determined by nonlinear regression fits to the Hill equation using Prism 4 software (GraphPad Software) as we have done previously (Bali and Akabas, 2004).
Results
Expression and functional characterization of the α1-M1 Cys mutants
Coinjection of mRNA encoding each of the 10 individual α1-M1 Cys mutants (223-IGYFVIQTYL-232) with wild-type Cys-light β2 and wild-type γ2 resulted in functional GABA-activated currents for all of the single α1 Cys mutants. The GABA EC50 for wild-type Cys-light was 4.0 ± 0.9 μm and for the mutants ranged from 1.5 ± 0.5 μm for α1L232C to 20 ± 4 μm for α1I223C (Table 1). The average currents induced by saturating concentrations of GABA were 4300 ± 240 nA for wild-type Cys-light and for the mutants ranged from 20 nA for α1Y231C to 5800 ± 220 nA for α1I228C (Table 1). A 90 s application of 5 mm DTT to α1Y231Cβ2γ2 receptors did not affect subsequent GABA-induced current amplitude (data not shown). The currents for α1Y231C were too small and this mutant was not studied in additional experiments. The initial leak current before application of pCMBS− was not significantly different for any of the single Cys mutants compared with wild type. We infer that the Cys mutations did not significantly increase the spontaneous open probability for the mutant receptors.
Table 1.
Characterization of the GABA-activated currents of the single Cys mutants
| EC50 (μm) | nH | Imax (nA) | n | |
|---|---|---|---|---|
| WT Cys-light | 4.0 ± 0.9 | 1.5 ± 0.1 | 4350 ± 240 | 4 |
| α1I223C | 20 ± 4 | 1.2 ± 0.2 | 560 ± 320 | 3 |
| α1G224C | 15 ± 5 | 0.8 ± 0.2 | 2100 ± 570 | 3 |
| α1Y225C | 2.0 ± 0.5 | 1.7 ± 0.3 | 4500 ± 1030 | 3 |
| α1F226C | 2.7 ± 0.4 | 1.7 ± 0.1 | 1700 ± 190 | 5 |
| α1V227C | 2.2 ± 0.4 | 1.2 ± 0.05 | 1800 ± 450 | 3 |
| α1I228C | 1.6 ± 0.3 | 0.7 ± 0.2 | 5780 ± 220 | 6 |
| α1Q229C | 2.0 ± 1.0 | 1.1 ± 0.4 | 3860 ± 290 | 5 |
| α1T230C | 16 ± 3.5 | 1.1 ± 0.1 | 2430 ± 250 | 3 |
| α1Y231C | a | a | 20 nA at 200 μm | 5 |
| α1L232C | 1.5 ± 0.5 | 0.9 ± 0.2 | 2670 ± 260 | 3 |
| β2M286C | 15 | 1.2 | ||
| β2F289C | 3.5 ± 0.75 | 1.5 ± 0.11 | 1700 ± 260 | 3 |
GABA concentration–response relationships were determined by measuring the currents induced by sequential application of increasing concentrations of GABA to oocytes expressing the single Cys mutants in α1β2γ2 receptors. α1 and β2 subunits were the Cys-light versions. Currents were normalized to the maximal current and fitted by nonlinear regression to the Hill equation. EC50 is the GABA concentration inducing half-maximal responses, nH is the Hill coefficient, and Imax is the maximal GABA-induced current. Means ± SEM are given. n, Number of oocytes tested. Data for β2M286C are from Bali and Akabas (2004).
aCurrents too small to determine EC50.
Accessibility of α1-M1 Cys mutants to pCMBS−
We tested the ability of the sulfhydryl-reactive reagent pCMBS− to react with each of the α1-M1 Cys mutants. Application of 200 μm pCMBS− for 2 min to oocytes expressing the wild-type Cys-light α1β2γ2 had no effect on the subsequent GABA-induced currents (data not shown). In contrast, a 2 min application of 200 μm pCMBS− in the absence of GABA to the α1-M1Cysβ2γ2 single Cys mutant receptors irreversibly modified the subsequent GABA-induced currents in all nine of the α1-M1 Cys mutants (Fig. 1). pCMBS− application inhibited the subsequent GABA-induced currents for four of the mutants (α1Y225C, α1F226C, α1V227C, α1Q229C) but potentiated subsequent GABA currents in the other five mutants (Fig. 1). For the β2-M3 Cys mutant, we previously showed that application of pCMBS− to α1β2M286Cγ2 receptors increased the subsequent GABA-induced currents by 75% (Bali and Akabas, 2004). For the β2F289C mutant, pCMBS− application both increased the holding current and diminished subsequent GABA currents. However, the total current, holding + GABA-activated, was similar to the initial currents before pCMBS− application. We infer that pCMBS− covalently modified the engineered Cys residue in each of these mutant receptors and that to react with pCMBS−, the engineered Cys residues were at least transiently on the water-accessible surface of the protein.
Figure 1.

Effect of pCMBS− on GABA-induced current in α1 Cys mutants expressed in Xenopus oocytes. GABAA receptors with mutated α1 subunits were treated with 200 μm pCMBS− for 2 min and the resulting GABA-induced current was normalized relative to the current before treatment. To maximize the effect of pCMBS− modification to avoid missing reactive Cys mutants the GABA test concentration was adjusted based on the effect of pCMBS− modification. For mutants in which the subsequent currents were potentiated the GABA test concentration was EC20-EC30. For mutants in which pCMBS− application inhibited subsequent GABA currents the GABA test concentration was ∼EC60–80. The bars represent the percentage change for at least 3 cells per mutant. One way ANOVA followed by Holm-Sidak post hoc test shows a statistically significant difference for all nine α1M1 Cys mutants relative to WT Cys-light receptors. Hatched bars indicate Cys mutants that after modification by pCMBS− showed an increased holding current in the absence of GABA most likely due to an increase in spontaneous opening. The initial holding currents for oocytes expressing all of the mutants were similar to those in oocytes expressing wild-type receptors indicating that there was no significant increase in spontaneous channel opening in the unmodified Cys mutants.
The extensive accessibility of the M1 Cys mutants to reaction with pCMBS− suggests that this region of the protein is not tightly packed with other protein regions because tight protein packing would preclude pCMBS− from entering the region. pCMBS− would fit into a right cylinder that is ∼6 Å in diameter and 10 Å in length. Thus, crevices or cavities at least this size must exist to allow pCMBS− to access these Cys mutants. Alternatively, this region may be relatively mobile, transiently exposing residues to the water accessible surface of the protein to allow reaction. Previous substituted cysteine accessibility method (SCAM) studies of the aligned region in the ACh receptor α subunit showed extensive accessibility to reaction with methanethiosulfonate reagents although a more limited pattern of accessibility was observed in the ACh β subunit M1 segment (Akabas and Karlin, 1995; Zhang and Karlin, 1997). The basis of these differences is uncertain, however, the ACh β subunit corresponds to the GABAA γ2 subunit, because neither forms part of the agonist binding sites.
Disulfide bond formation of α1-M1 Cys mutants with β2M286C under ambient oxidizing conditions
Coexpression of the nine individual α1-M1 Cys mutants with β2M286C and γ2 also resulted in GABA-induced currents for all nine double Cys mutant pairs. We noted that for the α1Q229Cβ2M286Cγ2 receptors, with repeated GABA applications, the amplitude of the GABA-induced current diminished with each subsequent GABA application until the currents plateaued at a level significantly smaller than the magnitude of the first GABA application (Fig. 2A). To determine whether channel activation was necessary for this reduction in current amplitude with repetitive GABA applications we examined the effect of the time interval between GABA applications on the decrease in GABA-induced current amplitude. Regardless of whether the GABA applications were separated by five or 10 min intervals, the extent of current decline was independent of the duration between GABA applications and was directly related to the cumulative duration of GABA application (Fig. 2B). We infer that channel activation was essential for this process. In the presence of GABA the channels fluctuate between open and desensitized states. We cannot distinguish whether the reaction occurred in one or both of these states, thus, we will refer to them collectively as the activated states to distinguish them from the closed, resting state that the channels occupy in the absence of GABA.
Figure 2.
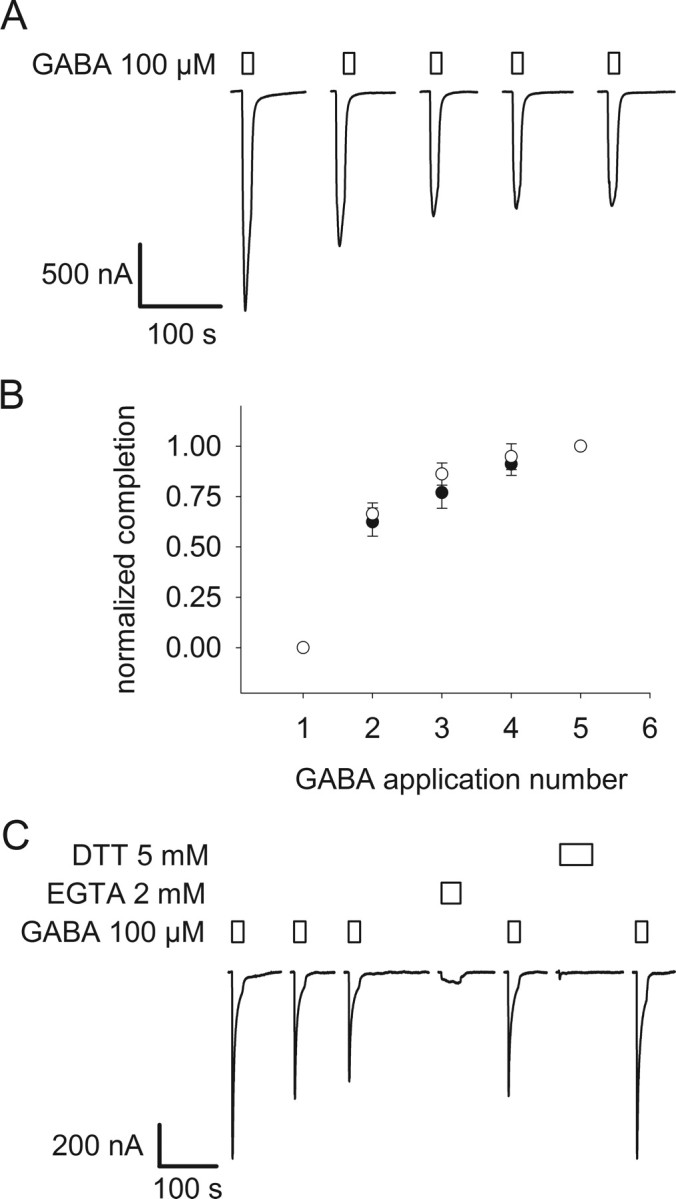
GABA-induced decrease of current in the double mutant α1Q229Cβ2M286Cγ2. A, GABA-induced currents in α1Q229Cβ2M286Cγ2 receptors decrease as a function of the application of GABA, here using a saturating concentration (100 μm). B, Mean GABA-induced current normalized for the maximum effect (normalized completion = (Ii − It)/(Ii − If), where Ii is the initial GABA current, It is the current at a time, t, and If is the final current) ± SEM (n = 3) plotted as a function of the number of 10 s applications of an EC30 GABA concentration (0.5 μm) on α1Q229Cβ2M286Cγ2. Plain circles are the mean current ± SEM for three oocytes plotted against the number of GABA applications, pulse frequency is one per 5 min; filled circles, pulse frequency is one per 10 min. C, The decrease in GABA-induced current is only reversed by 2 mm DTT in this representative experiment. Bars over traces indicate the period of application of the reagents shown to the left of each set of traces. Holding potential is −60 mV.
To determine whether the decreased current was due to disulfide bond formation or heavy metal binding we assayed the effects of EGTA and DTT application. EGTA binds polyvalent heavy metals with high affinity whereas DTT can both chelate heavy metals and reduce disulfide bonds. After reaching the plateau level, a 60 s application of 2 mm EGTA had no effect on the subsequent GABA-induced current amplitude (Fig. 2C). The lack of effect of EGTA implies that the decline in currents with GABA application was not due to heavy metal binding to the engineered Cys residues. In contrast, a 90 s application of 5 mm DTT largely reversed the decrease in GABA-induced currents and restored the currents to 85% of their initial level. Because EGTA had no effect and DTT restored the GABA-induced currents, we infer that the decrease was due to formation of a disulfide bond between α1Q229C and β2M286C. This implies that under ambient oxidizing conditions the rate of disulfide bond formation was significantly faster in the activated channel states, open and/or desensitized, compared with the resting state in which the rate was negligible.
For the other eight α1 Cys mutants coexpressed with β2M286Cγ2, there were no changes in GABA-induced currents with repetitive applications of GABA. Furthermore, a 90 s application of 5 mm DTT did not alter the subsequent GABA currents indicating that disulfide bonds had not formed before the initial GABA application (data not shown). We infer that β2M286C did not form disulfide bonds with the other eight α1-M1 Cys mutants either spontaneously at rest or during channel activation under ambient oxidizing conditions.
Copper phenanthroline induced oxidation at β2M286C
Cu:phen creates a more oxidizing environment than exists under ambient conditions by catalyzing the formation of superoxide and hydroxyl radicals (Kobashi, 1968). We assayed the ability of Cu:phen to induce disulfide bond formation between the engineered Cys in the nine double mutants. A 60–90 s application of 100:200 μm Cu:phen irreversibly diminished the subsequent GABA-induced currents for two of the Cys mutant pairs, α1Y225Cβ2M286Cγ2 and α1Q229Cβ2M286Cγ2 by 51 ± 6% (n = 8) and 62 ± 5% (n = 8), respectively (Fig. 3A,B). In the α1Y225Cβ2M286Cγ2 mutant, Cu:phen application also caused an increase in the holding current after Cu:phen washout suggesting that it increased the spontaneous open probability of the receptors (Fig. 3A). For this double mutant a subsequent 90 s application of 5 mm DTT only restored a small percentage of the initial GABA-induced current. EGTA application (2 mm, 2 min) had no effect (data not shown). For the α1Q229Cβ2M286Cγ2 mutant, after Cu:phen application, a 60–90 s application of 2 mm EGTA, whether applied in the presence or absence of GABA, had no effect on the GABA-induced currents. In contrast, a 60–90 s application of 2 mm DTT restored the GABA currents to 86 ± 5% (n = 8) of their initial value. We also tested another reducing agent, tris(2-carboxyethyl)phosphine (TCEP) and the results were similar (data not shown). Thus, 86% of the Cu:phen induced current decrease was restored by reduction with DTT. We infer that at least this fraction of the current decrease in response to Cu:phen application was due to disulfide bond formation. The inability to completely restore the GABA-induced currents suggests either that the disulfide bond was formed in a restricted space that limited access of the reducing agents DTT or TCEP to the disulfide, or some of the Cys residues may have been oxidized to higher order oxidation states (Cline et al., 2004). Neither DTT nor TCEP can reduce higher order sulfur oxidation states. Application of 100 μm CuSO4 had no irreversible effects on either of these double mutants.
Figure 3.

Oxidation alters GABA-induced currents in α1Y225Cβ2M286Cγ2 and α1Q229Cβ2M286Cγ2 receptors. A, Experiment illustrating the effect of application of 100:200 μm Cu:phen in the presence of a saturating GABA concentration (50 μm) on α1Y225Cβ2M286Cγ2 receptors. Subsequent GABA test currents are significantly diminished and the holding current in the absence of GABA after Cu:phen-induced oxidation is increased. The increased holding currents were blocked by picrotoxin (data not shown) and presumably result from an increased spontaneous open probability after disulfide bond formation. 2 mm DTT applied in the presence of GABA did not reverse the observed effect; a similar result was obtained with DTT applied in the absence of GABA (data not shown). B, Experiment showing a decrease in GABA-induced current after Cu:phen treatment of an oocyte expressing α1Q229Cβ2M286Cγ2 receptors. Application of 2 mm DTT significantly restored the currents to close to their initial level. The increase in current during Cu:phen application was reversible and was also observed in WT Cys-light receptors. This is due to an agonist effect of phenanthroline. Bars over traces indicate the period of application of the reagents shown to the left of each set of traces.
Both Cu:phen and phenanthroline by itself appeared to act as GABAA receptor activators. Application of 200 μm phenanthroline induced a current in wild type and in some of the double Cys mutants that was as large as 20% of maximal GABA currents. This was completely reversible after phenanthroline washout. Thus, during the Cu:phen application the channels were in a mixture of closed and activated (open/desensitized) states.
In each α1β2γ2 GABAA receptor there are two β2M3-α1M1 interfaces. We tested whether any of the α1Q229Cβ2M286Cγ2 engineered Cys residues were accessible to react with pCMBS− after Cu:phen oxidation. pCMBS− can only react with free thiols and not with disulfide-bonded Cys or with higher order Cys oxidation states. As a control, we assayed for the effect of pCMBS− application without previous Cu:phen oxidation. In the absence of Cu:phen oxidation, application of 200 μm pCMBS− inhibited the subsequent GABA currents and increased the holding current (Fig. 4A). Furthermore, the increase in holding current during and after pCMBS− application occurred even if the pCMBS− was applied in the presence of bicuculline, a GABA competitive antagonist (Fig. 4A). This suggests that the current was not due to an agonist effect of pCMBS− at the GABA binding site but rather to its irreversible modification of the free cysteine(s). The increased holding current induced by the reaction of pCMBS− was inhibited by picrotoxin indicating that it was mediated via spontaneously opened GABAA receptor channels (Fig. 4A, final trace). This demonstrates that pCMBS− modification causes a functional effect for this double mutant.
Figure 4.
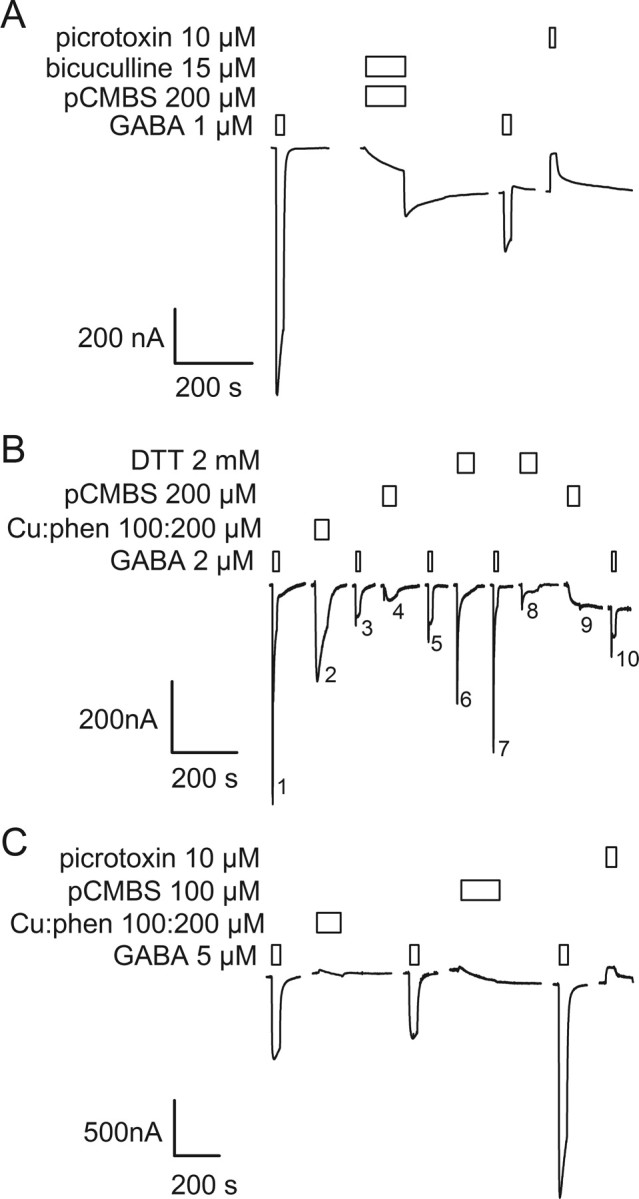
Application of the Cys-specific reagent pCMBS− probes the absence or presence of disulfide bonds in double Cys mutant receptors. A, In the absence of previous oxidation, application of 200 μm pCMBS− in the presence of 15 μm bicuculline to α1Q229Cβ2M286Cγ2 receptors induced an increased holding current in the absence of GABA. The holding current increased further after washout of pCMBS− and bicuculline. The current elicited by a subsequent GABA application was significantly decreased. Picrotoxin inhibited the increased holding current suggesting that it was due to the spontaneous opening of pCMBS− modified GABAA receptors. B, Treatment of α1Q229Cβ2M286Cγ2 receptors with the oxidizing reagent Cu:phen (100:200 μm) induced a decrease in subsequent GABA-induced current, as in Figure 3B. Subsequent treatment with 200 μm pCMBS− had no statistically significant effect on GABA-induced currents (compare traces 3 and 5). The lack of effect of pCMBS− indicates that the engineered Cys were inaccessible for reaction because they were part of disulfide bonds. Reduction with DTT restored the GABA-induced currents (compare traces 5 and 7). Application of pCMBS− after reduction (trace 9) increased the holding current and decreased the subsequent GABA-induced current (compare traces 7 and 10). C, Application of Cu:phen to α1I228Cβ2M286Cγ2 receptors either in the absence of GABA, or in the presence of GABA (data not shown) had no effect on subsequent GABA-induced currents. Subsequent application of pCMBS− caused an irreversible increase in the subsequent GABA-induced current, as well as an increase in the holding current. The increased holding current was inhibited by picrotoxin consistent with it being mediated by spontaneously opened GABAA receptors. Bars over traces indicate the period of application of the reagents shown to the left of each set of traces.
In contrast, in oocytes expressing α1Q229Cβ2M286Cγ2 receptors, after Cu:phen application, a 60 s application of 200 μm pCMBS− had little effect on the subsequent GABA-induced currents (Fig. 4B, compare traces 3 and 5). There are several possible interpretations of the result that after Cu:phen oxidation, pCMBS− application had no effect: (1) both pairs of Cys residues were involved in disulfide bonds and thus unavailable to react with pCMBS−, (2) one member of a pair was oxidized to a higher order oxidation state and either precluded pCMBS− reaction with the other Cys or it precluded a functional effect of pCMBS− reaction with the other Cys thiol, (3) both Cys in a pair could be oxidized to higher order oxidation states, or (4) if a disulfide bond formed only at one β-α subunit interface and not the other then pCMBS− modification of Cys at the nonmodified β-α interface had no effect. To determine whether Cu:phen-induced oxidation caused the formation of disulfide bonds we first oxidized α1Q229Cβ2M286Cγ2 receptors with Cu:phen, then reduced with DTT, and finally applied pCMBS− which caused an increase in the holding current and a 70% reduction in subsequent currents elicited by a submaximal (1 μm) GABA application (Fig. 4B, compare traces 7 and 10). Because DTT cannot reduce Cys oxidized to higher oxidation states, such as sulfenic acid, this result implies that a significant portion of the receptors contained at least one pair of disulfide bonded engineered Cys after Cu:phen application. Similar results were obtained with α1Y225Cβ2M286Cγ2 receptors. Thus, oxidation by Cu:phen induced reversible disulfide bond formation indicating that α1Y225C and α1Q229C are in close proximity to β2M286C in the activated state.
For the other seven Cys mutant pairs, Cu:phen application had no irreversible effects on the subsequent GABA-induced currents. When applied after Cu:phen, 200 μm pCMBS− (2 min) irreversibly altered the subsequent GABA currents for all seven Cys mutant pairs (Fig. 4C). Thus, the Cys must have been free to react with pCMBS− implying that Cu:phen oxidation did not induce disulfide bond formation or higher order oxidation states for these other pairs of Cys residues.
Disulfide crosslinking between β2F289C and α1-M1 Cys mutants
In an α helix, β2F289 would lie on the same face of the helix as β2M286, one helical turn closer to the cytoplasmic end. In the homology model it is also predicted to face into the subunit interface toward α1-M1. In the model, the α carbon separation distance between β2F289 and the closest α1-M1 residues, α1I228, α1Q229 and α1L232 is <11 Å. We tested whether disulfide bonds would form between β2F289C and Cys substituted for these residues and α1T230, predicted to lie on the side of the α1-M1 helix facing away from the subunit interface. α1Y231C was not tested due to its low expression level (Table 1).
In oocytes expressing α1L232Cβ2F289Cγ2, repeated GABA applications resulted in a progressive reduction in the peak current to a plateau level ∼31 ± 5% (n = 3) of the initial peak current (Fig. 5A). A 60 s application of 2 mm EGTA did not have a significant effect on the subsequent GABA current amplitude but a 60–90 s application of 5 mm DTT restored 75% of the initial current (Fig. 5B). A 60 s application of 100:200 μm Cu:phen caused an 80 ± 3% (n = 3) decrease in the subsequent GABA current. We conclude that channel activation induced disulfide bond formation between these two engineered Cys residues under ambient oxidizing conditions. Disulfide bonds did not form spontaneously in the resting state between this Cys pair.
Figure 5.
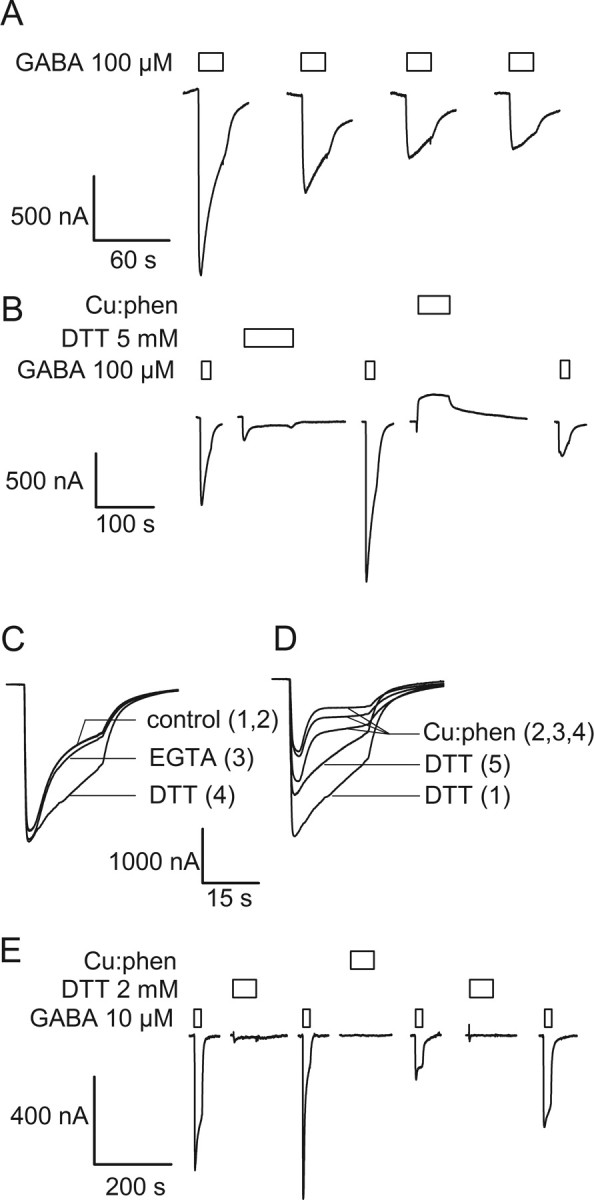
Electrophysiological effects of disulfide bond formation between β2F289C and α1L232C, α1Q229C and α1I228C. Currents recorded from oocytes expressing the double Cys mutants. A, Sequential GABA applications caused a decrease in the currents elicited from an oocyte expressing the α1L232Cβ2F289Cγ2 mutant. B, Same cell as in A. After the initial GABA-induced decrease observed in A, application of 2 mm DTT for 90 s induced a recovery of the initial current. A subsequent application of 100:200 μm Cu:phen inhibited the subsequent GABA currents to a greater extent than sequential GABA applications in ambient oxygen. C, Superposition of four consecutive GABA pulses on α1Q229Cβ2F289Cγ2. Each trace is numbered sequentially. A reagent was applied to the oocyte (data not shown) and the subsequent GABA-induced current is shown and labeled with the reagent that was applied before the GABA pulse. The two control traces (1,2) are indistinguishable. A 60 s application of 2 mm EGTA did not significantly alter the desensitization rate (3), whereas a 60 s application of 5 mm DTT induced a significant decrease in the desensitization rate (4). D, Same cell as in C; trace 4 of C is trace 1 of D. Three successive 60 s Cu:phen pulses (data not shown) induced a gradual increase in the desensitization rate and a decrease in the peak amplitude of the GABA-induced current (traces 2, 3, 4). A 60 s application of 5 mm DTT completely reversed the increase in desensitization rate and partially reversed the decrease in current amplitude. E, Illustrative experiment showing that a 100:200 μm Cu:phen application caused a 75% inhibition of a near-saturating GABA-induced current in α1I228Cβ2F289Cγ2 receptors. Prior treatment with 2 mm DTT caused a small increase in the GABA-induced currents. Application of 2 mm DTT after Cu:phen application caused a partial recovery of the initial current amplitude. Boxes over traces indicate the period of application of the reagents shown to the left of each set of traces.
For the mutant α1I228Cβ2F289Cγ2 repeated GABA applications caused a 50% reduction in the GABA-induced currents. Once a plateau was reached, a 90 s application of 5 mm DTT restored the GABA-induced currents to ∼80% of their initial value. A 60 s application of 100:200 μm Cu:phen caused a 72 ± 2% (n = 3) decrease in the subsequent GABA-induced test currents. The effect of Cu:phen was partially reversed by 2 mm DTT (2 min) but 2 mm EGTA had no effect (data not shown). We infer that a disulfide bond formed in the activated state between these two engineered Cys residues.
In contrast to the two double mutants above, oocytes expressing α1Q229Cβ2F289Cγ2 showed no progressive reduction or change in desensitization rate for the GABA currents with repeated GABA applications (Fig. 5C, control traces 1 and 2). EGTA application had little effect on the desensitization rate of a subsequent GABA test pulse (Fig. 5C, trace 3). However, the rate of desensitization of a GABA test pulse 5 min after a 60 s application of 5 mm DTT was significantly slower than the initial rate of desensitization (Fig. 5C, trace 4). This suggests that a disulfide bond had formed spontaneously under ambient oxidizing conditions in the resting state. DTT application had no effects on the desensitization rate in the single mutant α1Q229Cβ2γ2. A 60 s application of 100:200 μm Cu:phen accelerated the desensitization rate and reduced the peak current induced by subsequent GABA applications (Fig. 5D). These changes were reversed by a 60 s DTT application (Fig. 5D, trace 5) but not by EGTA (data not shown). We conclude that at least one disulfide bond had formed spontaneously in the resting state of the receptor. The major functional effect of this bond was to increase the desensitization rate after GABA application. Application of Cu:phen in the presence of GABA seemed to accelerate the decrease in subsequent GABA currents suggesting that the disulfide bond formation rate was faster in the activated than in the resting state. We conclude that a disulfide bond formed spontaneously in the resting state of the α1Q229Cβ2F289Cγ2 receptors. Disulfide bonds also formed in the more oxidizing environment created by Cu:phen in which the rate of disulfide bond formation was faster in the activated state of the receptor.
There was no effect of Cu:phen or DTT application either in the absence or presence of GABA on the double Cys mutant α1T230Cβ2F289Cγ2. We infer that no disulfide bonds were formed by this pair of engineered Cys consistent with α1T230 being on the opposite side of the α1-M1 helix from the subunit interface.
β2G287C and β2C288 do not form disulfide bonds with α1-M1 Cys mutants
We tested the ability of β2G287C and β2C288, the two amino acids between β2M286 and β2F289, to form disulfide bonds with α1-M1 Cys mutants from α1I228 to α1L232 (excluding α1Y231C). A 1 min application of 100:200 μm Cu:phen had no effect on β2G287C or β2C288 when coexpressed with the α1-Cys mutants and wild-type γ2. In these experiments, Cu:phen was applied in the absence and then in the presence of saturating GABA concentration. Furthermore, the oxidizing treatment of the receptors did not prevent the effect of a subsequent pCMBS− application (Fig. 6), strongly suggesting that the cysteines were not engaged in a disulfide bond. Figure 6A shows an example of absence of cross link for α1T230Cβ2G287Cγ2, and Figure 6B an example for α1Q229Cβ2C288(WT)γ2. Consistent with our model, β2G287 and β2C288, which are predicted to face into the β2 subunit interior, do not form disulfide bonds with α1-M1 Cys mutants.
Figure 6.

Absence of disulfide bond formation for α1T230Cβ2G287Cγ2 and α1Q229Cβ2C288(WT)γ2 receptors. A, α1T230Cβ2G287Cγ2 receptors were insensitive to a 2 mm DTT application, and to 100:200 μm Cu:phen applied in the absence (first pulse) or in the presence (second pulse) of saturating GABA. Furthermore, these oxidizing treatments did not prevent a large 200 μm pCMBS−-induced decrease in the current measured at saturating GABA concentration (80 ± 12%, n = 3). Similar results were observed using submaximal GABA concentration (data not shown). B, The intensity of a submaximal GABA-induced current in α1Q229Cβ2C288Cγ2 receptors was not modified after treatment with 100:200 μm Cu:phen in the presence of a saturating GABA concentration. Furthermore, subsequent pCMBS− reaction still induced a 50 ± 10% (n = 3) reduction of the current intensity, indicating that the cysteines were not involved in a disulfide bond. Similar results were obtained when the receptors were treated by Cu:phen in the absence of GABA (data not shown).
Detection of disulfide crosslinked α1-β2 subunits by immunoassay
To confirm the presence of disulfide bonds after Cu:phen induced oxidation, we assayed for the presence of disulfide-linked dimers using Western blots. We tested the α1Y225Cβ2M286Cγ2 and α1Q229Cβ2M286Cγ2 double Cys mutants and the corresponding single Cys mutants. Intact oocytes expressing the single and double Cys mutants were incubated with 1 μm GABA + 100:200 μm Cu:phen for 2 min, washed and surface biotinylated with sulfo-NHS-LC-biotin. The oocytes were lysed and the membranes solubilized. Biotinylated proteins were purified using streptavidin-coated beads, separated by SDS-PAGE electrophoresis, and transferred to PVDF for Western blotting. Using an anti-GABAA α1 subunit primary antibody, bands consistent with dimer formation were present in the lanes from oocytes expressing the double Cys mutants α1Y225Cβ2M286Cγ2 and α1Q229Cβ2M286Cγ2 but not the corresponding single mutants (Fig. 7A). Treatment of the samples with 40 mm DTT before PAGE analysis diminished the dimer band intensity, consistent with dimer formation being due to the presence of disulfide bonds (Fig. 7B).
Figure 7.

Western blots demonstrate disulfide linked dimers in double Cys mutant receptors inferred to form disulfide bonds in the electrophysiological assays. A, Representative Western-blot of the double Cys mutants containing β2M286 (α1Q229Cβ2M286Cγ2 and α1Y225Cβ2M286Cγ2), the single Cys mutants (α1β2M286Cγ2, α1Q229Cβ2γ2, and α1Y225Cβ2γ2), and the Cys-light WT after a 2 min treatment in intact oocytes with 100:200 μm Cu:phen in the presence of 1 μm GABA. Cys-light receptors (right) and the single mutants show a minimal level of dimers after the 2 min Cu:phen application. The two double mutants showed a significant amount of dimer on similar treatment. The positions of molecular size markers are indicated on the left. B, Effect of reduction with DTT after Cu:phen oxidation in the α1Q229Cβ2M286Cγ2 and α1Y225Cβ2M286Cγ2 double Cys mutants. Intact oocytes were treated with a 2 min application of Cu:phen/GABA 100 μm:200 μm/1 μm. Left two lanes were treated only with Cu:phen + GABA, oocytes for the right two lanes were treated with Cu:phen + GABA and then with 40 mm DTT for 2 min. Note the decrease in dimers after reduction with DTT. C, Representative Western-blot of the double Cys mutants containing β2F289C (α1L232Cβ2F289Cγ2, α1Q229Cβ2F289Cγ2 and α1I228Cβ2F289Cγ2), and the individual single mutants (α1L232Cβ2γ2, α1Q229Cβ2γ2, α1I228Cβ2γ2 and α1β2F289Cγ2). The experimental conditions were the same as in A. Bands consistent with a dimer were observed in lanes containing the double Cys mutants (right panel), but at significantly lower levels or not at all, in the lanes loaded with membrane preparations from oocytes expressing the single mutant constructs (left panel).
For the β2F289C double Cys mutants, α1L232Cβ2F289Cγ2, α1Q229Cβ2F289Cγ2 and α1I228Cβ2F289Cγ2, where electrophysiological experiments were consistent with disulfide bond formation, dimers were observed on Western blots from oocytes expressing the double Cys mutants after treatment with Cu:phen + GABA but not with the corresponding single Cys mutants (Fig. 7C). Controls with the corresponding single Cys mutants showed little or no evidence of dimer formation (Fig. 7C).
Discussion
In the absence of a high resolution GABAA receptor x-ray crystal structure, a homology model based on the 4 Å resolution model of the Torpedo ACh receptor closed state would be the best alternative (Unwin, 2005). A significant barrier to building an accurate homology model of the GABAA transmembrane domain is the uncertainty in the GABAA and ACh sequence alignment for the M2-M3 loop and M3 segment due to the lack of absolutely conserved residues in this region. We have determined experimentally validated proximity constraints using disulfide crosslinking that provide the foundation for constructing a realistic model of the transmembrane domain. Here we tested the ability of four consecutive β2-M3 Cys-substitution mutants (β2M286C, β2G287C, β2C288, β2F289C) to form disulfide bonds with α1-M1 Cys-substitution mutants. In activated states β2M286C formed disulfide bonds with α1Y225C and α1Q229C but not with neighboring α1-M1 Cys-substitution mutants. β2G287C and β2C288 did not form disulfide bonds with any α1-M1 Cys mutants. However, β2F289C, predicted to be on the same helical face as β2M286, formed disulfide bonds with α1I228C, α1Q229C and α1L232C but not with α1T230C that is predicted to face into the α subunit interior (Fig. 8). Thus, we conclude that β2M286 and β2F289 face into the subunit interface in close proximity to the α1-M1 residues α1Y225, α1I228, α1Q229 and α1L232. These results are consistent with these regions being α helical and provide strong experimental support for our GABAA model, which is based on the ACh receptor structure with the sequence alignment constrained by our previous M2-M3 crosslinking results (Unwin, 2005; Jansen and Akabas, 2006).
Figure 8.
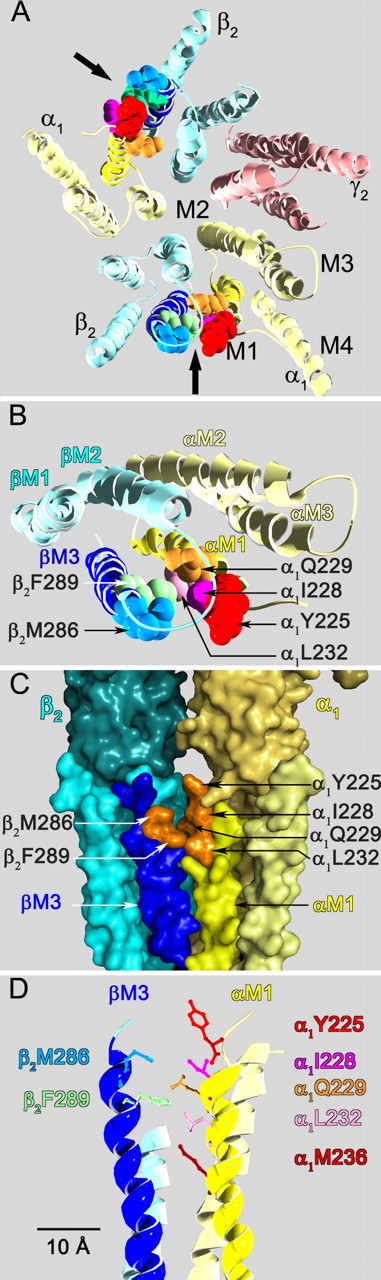
Views based on our GABAA receptor homology model of the interface between the β2-M3 and α1-M1 transmembrane segments illustrating the positions of the crosslinked residues. A, View from the extracellular space of the entire transmembrane domain. The extracellular domain has been removed. The subunits are color coded α1 (pale yellow), β2 (light blue), γ2 (pink). The transmembrane segments (M1, M2, M3, M4) are indicated on the α1 subunit in the lower right. The black arrows indicate the β2M3-α1M1 subunit interfaces. The crosslinked residues in the β2M3-α1M1 subunit interface are shown in space filling format. The residues are colored as follows in this and all other panels in this figure: α1Y225 (red), α1I228 (dark pink), α1Q229 (orange), α1L232 (light pink), α1M236 (maroon), β2M286 (gray blue), β2F289 (green). B, Closer top view of the β2M3-α1M1 subunit interface. Only two subunits are shown, the extracellular domain and the M4 segments in each of these subunits have been removed for clarity. C, Side view of the β2M3-α1M1 subunit interface from the lipid bilayer looking toward the channel lumen in the background, rendered as a molecular surface. The β2M3 segment is in dark blue, the α1M1 segment is in bright yellow. Part of the extracellular domain is visible above the transmembrane segments. The molecular surfaces of the disulfide crosslinked residues identified in this study are represented in orange. D, Side view of the residues in the β2M3-α1M1 subunit interface. Residues are shown in wireframe representation. Separation distances between α carbons are as follows β2M286 to: α1Y225 (12 Å), α1Q229 (11 Å), α1L232 (13 Å), and α1M236 (17 Å). From β2F289 to: α1Q229 (9 Å), α1L232 (11 Å), α1M236 (13 Å).
Other GABAA receptor models have positioned β2M286 facing into the interior of the β2 subunit four-helix bundle (Trudell and Bertaccini, 2004; Ernst et al., 2005; Hosie et al., 2007; Lobo et al., 2008). These models are incompatible with our current and previous crosslinking data (Jansen and Akabas, 2006) and therefore we conclude that the orientations of the M3 segments in those models are incorrect. This has important implications for interpreting experiments aimed at identifying the residues forming the binding sites for general anesthetics on the GABAA receptor.
As noted above, work by several investigators suggests a role for residues aligned with β2M286 in anesthetic action (Mihic et al., 1997; Krasowski et al., 2001; Bali and Akabas, 2004; Li et al., 2006). The fact that this residue points into the subunit interface rather than into the subunit interior suggests that a general anesthetic binding site may lie in the transmembrane domain subunit-subunit interfaces rather than in the interior of individual subunits. This would be consistent with azi-etomidate photoaffinity labeling of both αM236 and βM286, because both of these residues face a common subunit interface in our model and thus could be labeled as parts of a single binding site (Li et al., 2006). α1M236 is ∼17 Å from β2M286 in our model (Fig. 8D). Mutation of α1L232 was previously reported to alter anesthetic potentiation (Jenkins et al., 2001). In our model, this residue faces into the β-α subunit interface, one helical turn above α1M236. It is ∼13 Å from β2M286 and Cys substituted at this position formed a disulfide bond with β2F289C. Other studies have implicated the M2–15′ position, aligned with β2N265, in the actions of etomidate and other anesthetics (Belelli et al., 1997; Mihic et al., 1997; Jurd et al., 2003). These studies, including the in vivo knock-in mouse data, suggest that the amino acid at this position has important effects on the efficacy of various anesthetics. In our model β2N265 is ∼15 Å from β2M286 and although it largely faces toward the β2-M3 segment, it may have some exposure to the subunit interface, particularly if channel opening involves a coupled movement of βM2 and βM3 away from the channel axis toward αM1 in the adjacent subunit, which would be consistent with our current results.
Disulfide trapping has been used to study the proximity and mobility of pairs of residues in membrane proteins (Careaga and Falke, 1992; Yu et al., 1995; Wu and Kaback, 1996; Hastrup et al., 2001; Horenstein et al., 2001). In proteins of known crystal structure, the maximum α carbon separation for disulfide bonded Cys is 5.6 Å (Careaga and Falke, 1992). To form a disulfide bond two Cys must approach to this separation distance with sufficient frequency to allow covalent bond formation. This is not necessarily their average separation. The formation rate depends on the collision frequency, the orientation of the sulfur atoms and the presence of an oxidizing environment (Kobashi, 1968; Careaga and Falke, 1992). If the collision frequency and energetics are adequate, then ambient oxygen is sufficient to promote a measurable disulfide bond formation rate. In less favorable situations, a more oxidizing environment may be necessary to induce a measurable rate of disulfide bond formation. In the resting state, β2M286C did not form disulfide bonds with any of the α1-Cys mutants. We cannot distinguish whether in the resting state this is due to insufficient proximity, constrained mobility or an unfavorable orientation of β2M286C relative to α1-M1. In our model based on the ACh receptor closed state structure, the α carbon separation from β2M286 to α1Y225 and α1Q229 is ∼12 Å and the separation of β2F289 to α1Q229 and α1L232 is ∼11 Å (Fig. 8). These distances are too large to form a disulfide bond and imply that a conformational change during GABA-induced activation must bring these residues into closer proximity.
In the GABA-activated state a disulfide bond formed between β2M286C and α1Q229C under ambient oxidizing conditions and with α1Y225C in the presence of Cu:phen, which creates a more oxidizing environment. Because phenanthroline acts as an agonist on some of the receptors studied, we could not test whether a more oxidizing environment would promote disulfide bond formation with either of these α1-M1 Cys mutants in the absence of activation. Because the α1Q229C-β2M286C disulfide bond formed under ambient conditions, whereas the α1Y225C-β2M286C disulfide only formed with Cu:phen, we infer that in the activated state the collision frequency and/or orientation are more favorable between β2M286C and α1Q229C than with α1Y225C. For β2F289C, disulfide bonds formed with both α1Q229C and α1L232C under ambient oxidizing conditions during channel activation.
This suggests that if the closed state model is correct, these residues must move at least 6–7 Å during channel activation to reduce the α carbon separation sufficiently to allow disulfide bond formation. Whether channel activation causes these residues to move into closer proximity or causes an increase in the mobility of one or both of the regions containing these Cys to allow increased collision frequency is uncertain. In a previous SCAM study of α1-M3, we found that GABA activation significantly increased the number of pCMBS−-reactive α1-M3 substituted Cys (Williams and Akabas, 1999). We inferred that in the activated state there was looser protein packing around M3 allowing water-filled crevices to extend, at least transiently, from the extracellular solution into the transmembrane protein interior. Our previous disulfide crosslinking between α1-M2 and α1-M3 indicates that M3 does not undergo significant rotational motion or translation perpendicular to the membrane plane. Thus, the motion is more likely to be a rigid translation of β2-M3 toward α1-M1. Consistent with this, we previously suggested that during activation M2 translates away from the central channel axis toward M1 in the adjacent subunit (Jansen and Akabas, 2006). We conclude that channel activation may involve a conformational movement in the β2M3-α1M1 subunit interface in which the βM2-M3 segments move as a relatively rigid body toward αM1 thereby narrowing the separation between the two subunits. The 6–7 Å movement needed for disulfide bond formation in our experiments would be sufficient to separate the M2 segments thereby opening the channel gate and allowing ion translocation through the pore. Similar conformational changes probably occur in other Cys-loop receptors during the process of channel opening.
Thus, our GABAA receptor model, based on both intrasubunit and intersubunit constraints, shows that residues implicated in intravenous anesthetic action lie along the transmembrane domain β-α subunit interface. These transmembrane interfaces may form the binding sites for many volatile and intravenous general anesthetics in a manner similar to the agonist binding sites that are formed at the β-α subunit interface in the extracellular domain. The presence of two β-α interfaces suggests that anesthetic binding to one site may be responsible for the potentiation of GABA-induced currents observed at clinically relevant anesthetic concentrations. Anesthetic binding at both sites may be responsible for the direct activation observed at higher anesthetic concentrations (Rüsch et al., 2004). The length of the interface may in part account for differences in the efficacy of different anesthetics which may bind at slightly different positions along the interface. The fact that disulfide bond formation occurred in the GABA-activated but not in the closed state suggests that channel activation involves a rigid body movement of βM3 toward αM1. Thus, just as GABA binding promotes contraction of the agonist site in the extracellular domain β-α interface (Wagner and Czajkowski, 2001), anesthetic binding in the transmembrane β-α interface may promote the concerted movement of βM3 toward αM1.
Footnotes
This work was supported in part by National Institutes of Health Grants NS030808 and GM077660 to M.H.A. and K99NS059841 to M.J. We thank I. J. Frame and Jarrett Linder for technical assistance. We thank Nicole McKinnon, David Reeves, and Paul Riegelhaupt for helpful discussions and comments on this manuscript.
References
- Akabas, 2004.Akabas MH. GABAA receptor structure-function studies: a reexamination in light of new acetylcholine receptor structures. Int Rev Neurobiol. 2004;62:1–43. doi: 10.1016/S0074-7742(04)62001-0. [DOI] [PubMed] [Google Scholar]
- Akabas and Karlin, 1995.Akabas MH, Karlin A. Identification of acetylcholine receptor channel-lining residues in the M1 segment of the alpha-subunit. Biochemistry. 1995;34:12496–12500. doi: 10.1021/bi00039a002. [DOI] [PubMed] [Google Scholar]
- Bali and Akabas, 2004.Bali M, Akabas MH. Defining the propofol binding site location on the GABAA receptor. Mol Pharmacol. 2004;65:68–76. doi: 10.1124/mol.65.1.68. [DOI] [PubMed] [Google Scholar]
- Bali and Akabas, 2007.Bali M, Akabas MH. The location of a closed channel gate in the GABAA receptor channel. J Gen Physiol. 2007;129:145–159. doi: 10.1085/jgp.200609639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belelli et al., 1997.Belelli D, Lambert JJ, Peters JA, Wafford K, Whiting PJ. The interaction of the general anesthetic etomidate with the gamma-aminobutyric acid type A receptor is influenced by a single amino acid. Proc Natl Acad Sci U S A. 1997;94:11031–11036. doi: 10.1073/pnas.94.20.11031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campagna-Slater and Weaver, 2007.Campagna-Slater V, Weaver DF. Molecular modelling of the GABAA ion channel protein. J Mol Graph Model. 2007;25:721–730. doi: 10.1016/j.jmgm.2006.06.001. [DOI] [PubMed] [Google Scholar]
- Careaga and Falke, 1992.Careaga CL, Falke JJ. Thermal motions of surface alpha-helices in the D-galactose chemosensory receptor. Detection by disulfide trapping. J Mol Biol. 1992;226:1219–1235. doi: 10.1016/0022-2836(92)91063-u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celie et al., 2004.Celie PH, van Rossum-Fikkert SE, van Dijk WJ, Brejc K, Smit AB, Sixma TK. Nicotine and carbamylcholine binding to nicotinic acetylcholine receptors as studied in AChBP crystal structures. Neuron. 2004;41:907–914. doi: 10.1016/s0896-6273(04)00115-1. [DOI] [PubMed] [Google Scholar]
- Cline et al., 2004.Cline DJ, Redding SE, Brohawn SG, Psathas JN, Schneider JP, Thorpe C. New water-soluble phosphines as reductants of peptide and protein disulfide bonds: reactivity and membrane permeability. Biochemistry. 2004;43:15195–15203. doi: 10.1021/bi048329a. [DOI] [PubMed] [Google Scholar]
- Ernst et al., 2005.Ernst M, Bruckner S, Boresch S, Sieghart W. Comparative models of GABAA receptor extracellular and transmembrane domains: important insights in pharmacology and function. Mol Pharmacol. 2005;68:1291–1300. doi: 10.1124/mol.105.015982. [DOI] [PubMed] [Google Scholar]
- Franks, 2008.Franks NP. General anaesthesia: from molecular targets to neuronal pathways of sleep and arousal. Nat Rev Neurosci. 2008;9:370–386. doi: 10.1038/nrn2372. [DOI] [PubMed] [Google Scholar]
- Hastrup et al., 2001.Hastrup H, Karlin A, Javitch JA. Symmetrical dimer of the human dopamine transporter revealed by cross-linking Cys-306 at the extracellular end of the sixth transmembrane segment. Proc Natl Acad Sci U S A. 2001;98:10055–10060. doi: 10.1073/pnas.181344298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemmings et al., 2005.Hemmings HC, Jr, Akabas MH, Goldstein PA, Trudell JR, Orser BA, Harrison NL. Emerging molecular mechanisms of general anesthetic action. Trends Pharmacol Sci. 2005;26:503–510. doi: 10.1016/j.tips.2005.08.006. [DOI] [PubMed] [Google Scholar]
- Hilf and Dutzler, 2008.Hilf RJ, Dutzler R. X-ray structure of a prokaryotic pentameric ligand-gated ion channel. Nature. 2008;452:375–379. doi: 10.1038/nature06717. [DOI] [PubMed] [Google Scholar]
- Horenstein et al., 2001.Horenstein J, Wagner DA, Czajkowski C, Akabas MH. Protein mobility and GABA-induced conformational changes in GABA(A) receptor pore-lining M2 segment. Nat Neurosci. 2001;4:477–485. doi: 10.1038/87425. [DOI] [PubMed] [Google Scholar]
- Horenstein et al., 2005.Horenstein J, Riegelhaupt P, Akabas MH. Differential protein mobility of the gamma-aminobutyric acid, type A, receptor alpha and beta subunit channel-lining segments. J Biol Chem. 2005;280:1573–1581. doi: 10.1074/jbc.M410881200. [DOI] [PubMed] [Google Scholar]
- Hosie et al., 2006.Hosie AM, Wilkins ME, da Silva HM, Smart TG. Endogenous neurosteroids regulate GABAA receptors through two discrete transmembrane sites. Nature. 2006;444:486–489. doi: 10.1038/nature05324. [DOI] [PubMed] [Google Scholar]
- Hosie et al., 2007.Hosie AM, Wilkins ME, Smart TG. Neurosteroid binding sites on GABA(A) receptors. Pharmacol Ther. 2007;116:7–19. doi: 10.1016/j.pharmthera.2007.03.011. [DOI] [PubMed] [Google Scholar]
- Jansen and Akabas, 2006.Jansen M, Akabas MH. State-dependent cross-linking of the M2 and M3 segments: Functional basis for the alignment of GABAA and acetylcholine receptor M3 segments. J Neurosci. 2006;26:4492–4499. doi: 10.1523/JNEUROSCI.0224-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins et al., 2001.Jenkins A, Greenblatt EP, Faulkner HJ, Bertaccini E, Light A, Lin A, Andreasen A, Viner A, Trudell JR, Harrison NL. Evidence for a common binding cavity for three general anesthetics within the GABAA receptor. J Neurosci. 2001;21(RC136):1–4. doi: 10.1523/JNEUROSCI.21-06-j0002.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurd et al., 2003.Jurd R, Arras M, Lambert S, Drexler B, Siegwart R, Crestani F, Zaugg M, Vogt KE, Ledermann B, Antkowiak B, Rudolph U. General anesthetic actions in vivo strongly attenuated by a point mutation in the GABA(A) receptor beta3 subunit. FASEB J. 2003;17:250–252. doi: 10.1096/fj.02-0611fje. [DOI] [PubMed] [Google Scholar]
- Kobashi, 1968.Kobashi K. Catalytic oxidation of sulfhydryl groups by o-phenanthroline copper complex. Biochim Biophys Acta. 1968;158:239–245. doi: 10.1016/0304-4165(68)90136-0. [DOI] [PubMed] [Google Scholar]
- Krasowski et al., 2001.Krasowski MD, Nishikawa K, Nikolaeva N, Lin A, Harrison NL. Methionine 286 in transmembrane domain 3 of the GABAA receptor beta subunit controls a binding cavity for propofol and other alkylphenol general anesthetics. Neuropharmacology. 2001;41:952–964. doi: 10.1016/s0028-3908(01)00141-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li et al., 2006.Li GD, Chiara DC, Sawyer GW, Husain SS, Olsen RW, Cohen JB. Identification of a GABAA receptor anesthetic binding site at subunit interfaces by photolabeling with an etomidate analog. J Neurosci. 2006;26:11599–11605. doi: 10.1523/JNEUROSCI.3467-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobo et al., 2008.Lobo IA, Harris RA, Trudell JR. Cross-linking of sites involved with alcohol action between transmembrane segments 1 and 3 of the glycine receptor following activation. J Neurochem. 2008;104:1649–1662. doi: 10.1111/j.1471-4159.2007.05090.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihic et al., 1997.Mihic SJ, Ye Q, Wick MJ, Koltchine VV, Krasowski MD, Finn SE, Mascia MP, Valenzuela CF, Hanson KK, Greenblatt EP, Harris RA, Harrison NL. Sites of alcohol and volatile anaesthetic action on GABA(A) and glycine receptors. Nature. 1997;389:385–389. doi: 10.1038/38738. [DOI] [PubMed] [Google Scholar]
- Rüsch et al., 2004.Rüsch D, Zhong H, Forman SA. Gating allosterism at a single class of etomidate sites on alpha1beta2gamma2L GABAA receptors accounts for both direct activation and agonist modulation. J Biol Chem. 2004;279:20982–20992. doi: 10.1074/jbc.M400472200. [DOI] [PubMed] [Google Scholar]
- Trudell and Bertaccini, 2004.Trudell JR, Bertaccini E. Comparative modeling of a GABAA alpha1 receptor using three crystal structures as templates. J Mol Graph Model. 2004;23:39–49. doi: 10.1016/j.jmgm.2004.03.004. [DOI] [PubMed] [Google Scholar]
- Unwin, 2005.Unwin N. Refined structure of the nicotinic acetylcholine receptor at 4A resolution. J Mol Biol. 2005;346:967–989. doi: 10.1016/j.jmb.2004.12.031. [DOI] [PubMed] [Google Scholar]
- Wagner and Czajkowski, 2001.Wagner DA, Czajkowski C. Structure and dynamics of the GABA binding pocket: a narrowing cleft that constricts during activation. J Neurosci. 2001;21:67–74. doi: 10.1523/JNEUROSCI.21-01-00067.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams and Akabas, 1999.Williams DB, Akabas MH. gamma-aminobutyric acid increases the water accessibility of M3 membrane-spanning segment residues in gamma-aminobutyric acid type A receptors. Biophys J. 1999;77:2563–2574. doi: 10.1016/s0006-3495(99)77091-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu and Kaback, 1996.Wu J, Kaback HR. A general method for determining helix packing in membrane proteins in situ: helices I and II are close to helix VII in the lactose permease of Escherichia coli. Proc Natl Acad Sci U S A. 1996;93:14498–14502. doi: 10.1073/pnas.93.25.14498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu et al., 1995.Yu H, Kono M, McKee TD, Oprian DD. A general method for mapping tertiary contacts between amino acid residues in membrane-embedded proteins. Biochemistry. 1995;34:14963–14969. doi: 10.1021/bi00046a002. [DOI] [PubMed] [Google Scholar]
- Zhang and Karlin, 1997.Zhang H, Karlin A. Identification of acetylcholine receptor channel-lining residues in the M1 segment of the beta-subunit. Biochemistry. 1997;36:15856–15864. doi: 10.1021/bi972357u. [DOI] [PubMed] [Google Scholar]