

Abstract
In an effort to probe the structure and function of a predicted mitochondrial glyoxalase 2, GLX2−1, from Arabidopsis thaliana, GLX2−1 was cloned, overexpressed, purified and characterized using metal analyses, kinetics, and UV–visible, EPR, and 1H-NMR spectroscopies. The purified enzyme was purple and contained substoichiometric amounts of iron and zinc; however, metal-binding studies reveal that GLX2−1 can bind nearly two equivalents of either iron or zinc and that the most stable analogue of GLX2−1 is the iron-containing form. UV–visible spectra of the purified enzyme suggest the presence of Fe(II) in the protein, but the Fe(II) can be oxidized over time or by the addition of metal ions to the protein. EPR spectra revealed the presence of an anti-ferromagnetically-coupled Fe(III)Fe(II) centre and the presence of a protein-bound high-spin Fe(III) centre, perhaps as part of a FeZn centre. No paramagnetically shifted peaks were observed in 1H-NMR spectra of the GLX2−1 analogues, suggesting low amounts of the paramagnetic, anti-ferromagnetically coupled centre. Steady-state kinetic studies with several thiolester substrates indicate that GLX2−1 is not a GLX2. In contrast with all of the other GLX2 proteins characterized, GLX2−1 contains an arginine in place of one of the metal-binding histidine residues at position 246. In order to evaluate further whether Arg246 binds metal, the R246L mutant was prepared. The metal binding results are very similar to those of native GLX2−1, suggesting that a different amino acid is recruited as a metal-binding ligand. These results demonstrate that Arabidopsis GLX2−1 is a novel member of the metallo-β-lactamase superfamily.
Keywords: EPR, glyoxalase 2 (GLX2), iron, metallo-β-lactamase fold, zinc
INTRODUCTION
The GLX system consists of two enzymes, lactoylglutathione lyase (glyoxalase I, GLX1) and hydroxyacylglutathione hydrolase (glyoxalase II, GLX2), that act co-ordinately to convert a variety of α-keto aldehydes into hydroxyacids in the presence of glutathione [1]. Thiohemiacetals, which are formed from the spontaneous reaction of α-oxo aldehydes with glutathione, are converted into S-(2-hydroxyacyl)glutathione derivatives by GLX1. GLX2 hydrolyses S-(2-hydroxyacyl)glutathione derivatives to regenerate glutathione and produce hydroxyacids. GLXI can utilize a number of α-oxo aldehydes; however, the primary physiological substrate of the system is thought to be MG (methylglyoxal), a cytotoxic and mutagenic compound that is formed primarily as a byproduct of carbohydrate and lipid metabolism, and from triose phosphates [2–4].
The GLX system appears to play a critical role in cellular detoxification processes and has received considerable attention as a possible antitumour and antimalarial target in animal systems [5–12]. Increased levels of GLX1 and GLX2 mRNA and protein have been detected in tumour cells, including breast carcinoma cells, while inhibitors of GLXI and II have been shown to inhibit the growth of tumour cells in vitro [4,5,8,12–19]. Plasmodium falciparum and the protozoan Leishmania donovani exhibit high rates of methylglyoxal formation and increased levels of GLX1 activity [20]. Alterations in GLX activity have also been associated with several disease states. GLX1 and 2 can inhibit the formation of hyperglycaemia-induced advanced glycation end products, suggesting that these enzymes may have a role in diabetic microangiopathy [21]. GLX enzymes may also play a role in the pathogenesis of Alzheimer's disease [22,23]. Finally, GLX2 has been identified as a target of p63 and p73, and suggested to be a pro-survival factor of the p53 family of transcription factors [24].
GLX1, which occurs as a single isoenzyme, has been studied extensively in a number of systems using biochemical computational and X-ray crystallographic approaches [25]. In contrast, GLX2 exists as multiple isoenzymes and has been considerably less well characterized. In humans, a single gene encodes for mitochondrial and cytoplasmic forms of GLX2 [26]. In contrast, plants contain distinct genes encoding cytosolic and multiple mitochondrially localized forms of GLX2 [27]. Five putative GLX2 genes were identified in the Arabidopsis thaliana genome, including three (GLX2−1, GLX2−4 and GLX2−5) that were predicted to be localized in the mitochondrion [27]. The presence of multiple forms of GLX2 in mitochondria is surprising, because GLX1 and SLG (S-D-lactoylglutathione) have been observed only in the cytosol [1,13].
GLX2 enzymes belong to the metallo-β-lactamase fold superfamily of proteins [28,29]. Metallo-β-lactamases typically bind 1−2 equivalents of Zn(II) [30], whereas the rubredoxin:oxygen oxidoreductase [31] and ZiPD (zinc phosphodiesterase) [32] families contain di-Fe and di-Zn centres, respectively. One version of Arabidopsis GLX2 (GLX2−2) was shown to bind zinc, iron and manganese [28,33,34], whereas GLX2−5 predominantly contains a Fe(III)Zn(II) centre [35]. Crystal structures of human cytoplasmic and plant mitochondrial GLX2 showed that the metal-binding and active site of GLX2 is most similar to that of metallo-β-lactamases L1 from Stenotrophomonas maltophilia [35–37]. One of the metal-binding sites (Zn1 site) consists of three conserved histidine residues, a bridging aspartic acid, and a bridging water/hydroxide. The second metal-binding site (Zn2 site) has two histidines, one bridging Asp, a non-bridging Asp, a terminally bound water and a bridging hydroxide/water (Figure 1).
Figure 1.
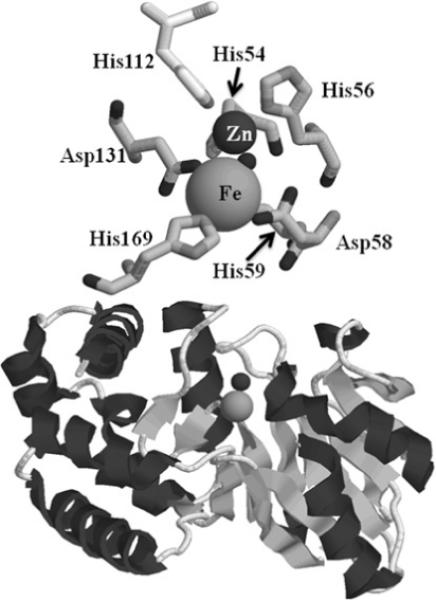
Structure of mitochondrial GLX2−5
GLX2−1, GLX2−4, and GLX2−5 share ∼80 % amino acid sequence identity and ∼88 % amino acid sequence similarity [27]; however, GLX2−1 exhibits several important differences relative to the other predicted mitochondrial GLX2 isoenzymes that led us to predict that it may in fact not be a GLX2 isoenzyme. GLX2−1 is unique among GLX2 enzymes in that it has an arginine instead of a histidine residue at position 246 of the conserved metal-binding motif (Figure 2) [35,36]. In addition, five out of eight amino acids present at the active site of human GLX2 (Lys217, Tyr219, Tyr248, Arg325 and Lys328), which are involved with substrate binding and conserved in GLX2−2, GLX2−4 and GLX2−5 [38,39], are not conserved in GLX2−1. These observations suggested that GLX2−1 may not contain a dinuclear metal centre and may not bind glutathione derivatives as substrates. We therefore overexpressed and purified GLX2−1 and characterized the protein using metal analyses and UV–visible, EPR, 1H NMR and EXAFS spectroscopies to understand how the amino acid differences affect metal binding, substrate specificity, and the structure of the enzyme. Our results show that GLX2−1 does in fact contain a dinuclear metal centre; however, it does not utlilize SLG or other GLX2 substrates, and is therefore not a GLX2 enzyme.
Figure 2. Alignment of predicted plant GLXII proteins.

The metallo-β-lactamase fold motif is shaded in grey, and substrate-binding ligands are indicated by triangles. Metal-binding ligands are indicated by #, * indicates the β-lactamase fold motif.
EXPERIMENTAL
Overexpression and purification of Arabidopsis GLX2−1
PCR was conducted on A. thaliana cDNA, with the primers CCTCCATGGTAAAAATCGAACTGGTGC and GAGTCGACTCGAGCTCTAGATCTTTTTTTTTT, which generated NcoI and XhoI sites at the 5′ and 3′ ends of the fragment. The mature N-terminus of the overexpressed GLX2−1 enzyme was chosen to be identical to that for A.thaliana GLX2−5 [35]. The GLX2−1 PCR fragment was cloned into pT7−7 using the NcoI and XhoI restriction sites, and the sequence of the resulting pGLX2−1/pT7−7 plasmid was confirmed by DNA sequencing. Plasmid pGLX2−1/pT7−7 was transformed into Escherichia coli BL21(DE3) cells and small-scale growth cultures were used to maximize the recovery of soluble protein at different temperatures (15 °C, 25 °C, 30 °C and 37 °C).
The co-expression plasmid, pGroESL, was transformed into BL21(DE3) E. coli cells containing pGLX2−1/pT7−7 to assist in proper protein folding. The large-scale overexpression of GLX2−1 was performed as follows. A 10 ml overnight culture of BL21(DE3) E. coli containing pGroESL and pGLX2−1/pT7−7 was used to inoculate 1 litre of LB (Luria–Bertani) medium containing ampicillin (150 μg/ml) and chloramphenicol (l50 μg/ml) in the presence or absence of 250 μM Fe(NH4)2(SO4)2 and 250 μM Zn(SO4)2. The cells were allowed to grow at 37 °C with shaking until they reached an attenuance at 600 nm of 0.6−0.8. Protein production was induced by making the cultures 0.3 mM in IPTG (isopropyl β-d-thiogalactoside), and the cells were shaken at 15 °C for 24 h. The cells were collected by centrifugation (15 min at 7000 g) and the cell pellets were stored at −80 °C until further use.
The cell pellet was resuspended in 10 ml of 10 mM Mops, pH 7.2, containing 0.1 mM phenylmethylsulfonyl fluoride. The cells were lysed by 3 passages through a French press at 16 000 lbf/in2 (1 lbf/in2 ≈ 6.9 kPa) and the cell debris was removed by centrifugation at 15 000 g for 30 min. The cleared supernatant was dialysed against 2 litres of 10 mM Mops, pH 7.2, overnight at 4 °C and centrifuged for 20 min at 15 000 g to remove insoluble matter. The cleared supernatant was purified using FPLC with a Q-Sepharose column (1.5 × 12 cm with a 25 ml bed volume) that was equilibrated with 10 mM Mops, pH 7.2. Bound proteins were eluted with a 0−500 mM NaCl gradient in 10 mM Mops, pH 7.2, at 2 ml/min. Protein purity was ascertained by SDS/PAGE. Fractions (8 ml) containing GLX2−1 were pooled and concentrated by an Amicon ultrafiltration cell equipped with a YM-10 membrane. Enzyme concentrations were determined by measuring the absorbance at 280 nm and using a molar absorption coefficient of 25 400 M−1 · cm−1, which was determined by using a Bio-Rad protein assay and BSA as the standard.
Metal analyses
The metal content of GLX2−1 samples was determined using a Varian-Liberty 150 ICP-AES (inductively-coupled plasma spectrometer with atomic emission spectroscopy detection) as described previously [28]. Protein samples were diluted to 10 μM with 10 mM Mops, pH 7.2. A calibration curve with four standards and a correlation coefficient of greater than 0.99 was generated using Fe, Zn and Mn reference solutions. The following emission wavelengths were chosen to ensure the lowest detection limits possible: Fe, 259.940 nm; Zn, 213.856 nm; and Mn, 257.610 nm.
To evaluate metal binding to GLX2−1, a 5-fold molar excess of Fe(NH4)2(SO4)2, Zn(SO4)2 or Fe(NH4)2(SO4)2 plus Zn(SO4)2 were added directly to 10 μM apo-GLX2−1, and the mixtures were allowed to incubate on ice for 1 h. Unbound metal ions were removed by four 1 litre dialysis steps against 10 mM Mops, pH 7.2, at 4 °C (12 h for each dialysis step). The metal content of these protein samples was determined using ICP-AES as described above.
Spectroscopic studies
UV–visible spectra were recorded using an Agilent 8453 UV-visible spectrophotometer at 25 °C. GLX2−1 concentrations were approx. 1.0 mM, and the buffer was 10 mM Mops, pH 7.2. Difference spectra were generated using Igor Pro version 4.0.5.1 (Wavemetrics).
EPR spectra were recorded using a Bruker E600 EleXsys spectrometer equipped with an Oxford Instruments ESR900 helium flow cryostat and ITC503 temperature controller, and an ER4116DM cavity operating at 9.63 GHz in perpendicular mode. Other recording parameters are given in Figure 3. Quantification of signals was carried out by double integration of spectra recorded at non-saturating power (2 mW) at 12 K. A 2 mM Cu(II)-EDTA standard in Hepes buffer, pH 7.5, recording at 60 K, 50 μW, was used. Integration limits and correction factors for S = ½ and signals where D is assumed to be small compared with temperature, were as employed elsewhere [39a] and described explicitly by Bou-Abdallah and Chasteen [39b] and references therein [39c]. Computer simulations of EPR spectra were carried out using XSophe (Bruker Biospin; [39d]).
Figure 3.
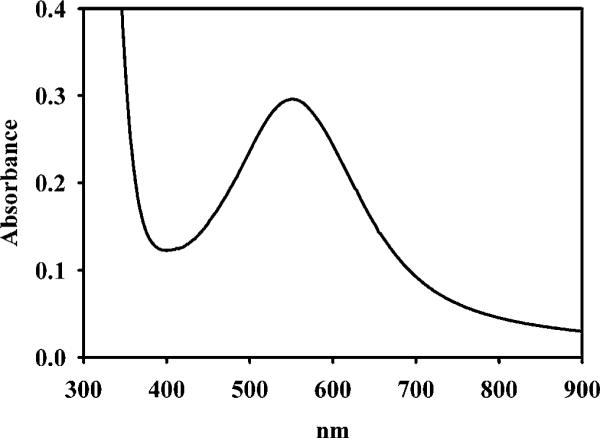
UV–visible spectrum of 1.2 mM as-isolated GLX2−1 in 10 mM Mops, pH 7.2
1H-NMR spectra were collected on a Bruker Avance 500 spectrometer operating at 500.13 MHz, 298 K, magnetic field of 11.7 T, recycle delay of 41 ms, and sweep width of 400 p.p.m. Chemical shifts were referenced by assigning the H2O signal a value of 4.70 p.p.m. A modified presaturation pulse sequence (zgpr) was used to suppress the proton signals originating from solvent and amino acids not coupled to the metal centre. The concentration of protein was approx. 1 mM, and 10 % 2H2O was included in samples for locking.
Substrate specificity studies
A series of thioesters of glutathione were used for the preliminary investigation of substrate specificity of as-isolated and Fe(II)-added GLX2−1. The substrates used were SLG (ε240 = 3100 M−1 · cm−1), S-mandeloylglutathione (ε263 = 4200 M−1 · cm−1), S-acetylglutathione (ε240 = 2980 M−1 · cm−1), S-acetoacetylglutathione (ε240 = 3400 M−1 · cm−1), S-formylglutathione (ε240 = 3300 M−1 · cm−1), S-glycolylglutathione (ε240 = 3260 M−1 · cm−1) and S-lactonylglutathione (ε240 = 3310 M−1 · cm−1). With the exception of SLG, all substrates were synthesized as described elsewhere [40]. Thioester hydrolysis was monitored at 240 nm (except S-mandeloylglutathione, 263 nm) over 60 s at 25 °C as previously reported [15]. The concentrations of as-isolated and Fe(II)-added enzyme were 10−300 μM, and substrates used were 30−600 μM.
Overexpression and purification of the R246L mutant of GLX2−1
The QuikChange® site-directed mutagenesis kit was used to prepare the R246L mutant of GLX2−1. PCR was conducted on plasmid GLX2−1/pT7−7 with the primers CAAATATATACTGCGGCCTTGAAAACACAGCAGGC and GCCTGCTGTGTTTTCAAGGCCGCAGTATATATTTG. The sequence of the resulting R246L-GLX2−1/pT7−7 plasmid was confirmed by DNA sequencing using the T7 promoter and T7 terminator primers. Plasmid R246L-GLX2−1/pT7−7 was transformed into E. coli BL21(DE3) cells. The R246L mutant was overexpressed as described above, and the resulting protein was insoluble. The co-expression plasmid, pGroESL, was transformed into BL21(DE3) E. coli cells containing R246L-GLX2−1/pT7−7 to assist in proper protein folding. The R246L mutant was then overexpressed and purified as described above.
RESULTS
Overexpression and purification of A. thaliana GLX2−1
Sequence comparisons of different GLX2 isoenzymes indicated that GLX2−1 has a N-terminal mitochondrial targeting sequence [27]. Therefore, this N-terminal leader was removed during sub-cloning to generate an N-terminus of MKIELVP (Figure 1), which is similar to that of recombinant GLX2−5, for overexpression of GLX2−1 in E. coli [35]. GLX2−1 could be overexpressed at high levels in E. coli BL21(DE3) cells; however, at temperatures > 15 °C, no soluble GLX2−1 was recovered. When GLX2−1 was overexpressed at 15 °C, some soluble protein was detected in the cleared lysates; however, the recombinant GLX2−1 did not bind to Q-Sepharose or SP-Sepharose columns at pH values ranging from 6 to 8. This result suggested that the overexpressed GLX2−1 was improperly folded. BL21(DE3) E. coli cells, containing the pGLX2−1/pT7−7 plasmid, were then transformed with pGroESL, and the previously mentioned overexpression/purification protocol was followed in an effort to produce properly folded recombinant GLX2−1. Overexpression of GLX2−1 in the presence of pGroESL at 15 °C resulted in high levels of soluble recombinant protein, which was purified with Q-Sepharose chromatography. The enzyme, which eluted from the column at 100 mM NaCl in 10 mM Mops, pH 7.2, was purple in colour; however, the colour faded in 7−10 days at 4 °C. Approx. 50 mg of purified GLX2−1 per litre of culture was obtained using this method. The resulting enzyme was shown to bind substoichiometric amounts of Fe and Zn(II), ranging from 0.1−0.5 equivalent for both metals (Table 1). The enzyme did not exhibit any SLG hydrolase activity.
Table 1.
Metal content of GLX2−1 analogues
| Fe (eq) | Zn(II) (eq) | |
|---|---|---|
| Wild-type GLX2−1 | ||
| No added metal | 0.3±0.2 | 0.3±0.1 |
| 250 μM Fe plus Zn(II) added in the medium | 0.3±0.1 | 0.3±0.2 |
| 3-fold excess of Fe added after purification | 1.9±0.1 | 0.2±0.1 |
| 3-fold excess of Zn(II) added after purification | ∼0.1 | 1.6±0.2 |
| 3-fold excess of Fe plus Zn(II) added after purification | 1.6±0.2 | 1.6±0.2 |
| GLX2−1 R246L | ||
| 250 μM Fe plus Zn(II) added in the medium | ∼0.1 | 0.6±0.1 |
| 3-fold excess of Fe added after purification | 2.4±0.5 | 0.5±0.2 |
| 3-fold excess of Zn(II) added after purification | 0.1 | 1.4±0.2 |
Metal content of recombinant GLX2−1
The substoichiometric levels of metal suggested that the enzyme may still not be folding properly or may need higher relative metal concentrations for metal binding. Therefore GLX2−1 was overexpressed in LB medium containing 250 μM Fe(NH4)2(SO4)2 and 250 μM Zn(SO4)2, conditions that had previously been shown to result in stoichiometric binding of Fe and Zn to GLX2−5 [35]. When GLX2−1 was overexpressed in the presence of 250 μM Fe(II) + 250 μM Zn(II), the purified protein was found to contain between 0.1−0.5 equivalents of Fe and between 0.1−0.5 equivalents of Zn(II) (Table 1). Therefore, the addition of metal ions to the growth medium did not greatly affect the amount of metal ions bound to purified GLX2−1. This result was surprising, since similar studies on Arabidopsis GLX2−2 showed that the amounts and identity of metal ion bound to the purified enzyme were greatly affected by metal ion present in the growth medium [33,34].
We further investigated the ability of GLX2−1 to bind metal by the addition of metal to the as isolated protein. The addition of a 5-fold molar excess of Fe(NH4)2(SO4)2, Zn(SO4)2 or both metal ions to the isolated purple protein in the presence or absence of a 10-fold molar excess of DTT (dithiothreitol) resulted in an immediate change in colour to brownish-yellow followed by protein precipitation. However, the slow addition of a 3-fold molar excess of metal ions did not result in protein precipitation. After exhaustive dialysis, the resulting protein was shown to bind 1.8−2.1 equivalent of Fe and 0.1−0.2 equivalents of Zn(II), 0.1 equivalents of Fe and 1.6 equivalents of Zn(II), and 1.6 equivalents of Fe and 1.6 equivalents of Zn(II) after addition of Fe(NH4)2(SO4)2, Zn(SO4)2, or both metals respectively (Table 1). The Zn(II)-added protein was unstable and precipitated within 1 h. None of the metal-loaded forms of GLX2−1 exhibited any SLG hydrolase activity.
Spectroscopic studies on GLX2−1
We predicted that the presence of an arginine residue at position 246 of GLX2−1 would disrupt the GLX2 binuclear metal-binding centre and interfere with the ability of the protein to bind two metal atoms. However, our observation that GLX2−1 can bind two equivalents of metal, if excess metal is added to the purified protein, suggested that either Arg246 acts as a metal-binding ligand, or more likely another amino acid is recruited as a ligand. Spectroscopic studies were conducted on GLX2−1 to probe the metal binding site of enzyme. The UV–visible spectrum of the as-isolated protein showed a broad peak with maximal absorbance at 540 nm and an absorption coefficient of 129 M−1 · cm−1 (Figure 3). The intensity of this peak suggests that it is due to a ligand field (d-d) transition of high-spin Fe(II) [41]. This assignment is consistent with the observed loss of colour of the as-isolated GLX2−1 over time and when metal ions are added to the as-isolated protein, since both of these scenarios would lead to oxidation of Fe(II) to Fe(III), which would not exhibit any spin-allowed electronic transitions [41].
We next attempted to characterize the metal-binding site of GLX2−1 by using paramagnetic 1H-NMR spectroscopy. Despite considerable effort, we were unable to obtain an NMR spectrum of the as-isolated protein, most likely due to very small levels of paramagnetic centres in the as-isolated protein. We were also unsuccessful in obtaining a spectrum for the Fe-loaded form of GLX2−1. This result is consistent with the UV–visible results that suggest that this protein probably contains only Fe(III). The presence of mononuclear Fe(III) or diamagnetic Fe(III)Fe(III) would result in no detectable paramagnetically shifted peaks [42].
We further characterized the as-isolated GLX2−1 using EPR spectroscopy. The EPR signal from as-isolated GLX2−1 (Figure 4A) contained contributions from at least two separate chemical species. The spectrum was dominated by an intense resonance at geff ∼ 4.3 due to Fe(III). Other transitions that are attributed to Fe(III) were also observed at 750 G (75 mT; geff ∼ 5.6), 900 G (90 mT; geff ∼ 7.6), 1430 G (143 mT; geff ∼ 4.8), 1730 G (173 mT; geff ∼ 4.0) and 1850 G (185 mT; geff ∼ 3.7), The 750−2000 G (75−200 mT) region of the spectrum could not be simulated using a single set of spin Hamiltonian parameters and the simple E/D-strain model in XSophe. The unsuccessful attempts at simulation suggested that there are two distinct species responsible for the multiple transitions between 750 G and 1850 G with strains in E/D that are relatively moderate compared to the large strains observed for adventitious Fe(III). The lack of resolved splitting of the geff = 4.3 line suggests that a third species may additionally contribute to the intensity at geff = 4.3. These data indicate the existence of at least one and more likely two sites in GLX2−1 that bind Fe(III) in a constrained environment, in addition to possible adventitious binding sites.
Figure 4. EPR spectra of GLX2−1.

Trace A is the EPR spectrum of as-isolated GLX2−1. Trace B is a computer simulation assuming two spin-coupled iron ions in an Fe(III)Fe(II) centre. Trace C is the spectrum of GLX2−1 after aerobic addition of Fe(II). Trace D is an expanded view of Trace A and Trace E is of GLX2−1 after aerobic addition of Fe(II). Experimental conditions were: A, 12 K, 2 mW; C, 10 K, 1 mW; D, 12 K, 2 mW; E, 10 K, 20 mW. Spectra A and C are shown normalized for temperature and microwave power; spectra D and E are shown with arbitrary intensities. 10 G (1 mT) magnetic field modulation at 100 kHz was employed and other parameters were chosen such that spectral resolution was limited by the field modulation. The parameters used for the simulation of Trace B were: for Fe(III), , gisotropic = 2.0, D = 2 cm−1 and E/D = 0; for Fe(II), S = 2, g(x,y,z) = 1.970, 2.090, 2.025, D = 15 cm−1 and E/D = 0.2; for the dinuclear centre, J = 43 cm−1 and an inter-iron distance, r, of 3.6 Å (0.36 nm) was assumed. It should be noted that (i) this is not a unique solution, and (ii) the simulation was not sensitive to all the parameters; in particular, the dependence on r and on which iron was assumed to have a rhombic zero-field splitting term was slight.
The other feature observed in the EPR spectrum of GLX2−1 was a signal that exhibited geff values of 1.946, 1.844 and 1.775. In comparison with mitochondrial GLX2−5, that exhibits this type of signal [35], the high field signal from GLX2−1 can be attributed to an Fe(III)Fe(II) dinuclear centre. The Fe(III)Fe(II) centre could be simulated to very good agreement (Figure 4B) assuming such a spin-coupled centre with reasonable spin Hamiltonian parameters (; SFe(II) = 2; gFe(III) = 2.0; gFe(II) > 2.0; DFe(III) = 2 cm−1; DFe(II) = 15 cm−1; J = 43 cm−1; the solution is not unique and detailed parameters are given in the legend to Figure 4). The presence of this feature in the spectrum supports the assignment of the lower field features to Fe(III) in two distinct iron-binding sites in GLX2−1.
Upon the aerobic addition of Fe(II) to GLX2−1, the purple colour was lost, and this loss was concomitant with the loss of the Fe(II)Fe(III) EPR signal (Figure 4C). In addition, subtle changes in the low-field region of the spectrum were observed (Figures 4C–4E). While the features at 1430 G (143 mT) and 1730 G (173 mT) were preserved, the signals at 1430 G (143 mT) and 1850 G (185 mT) were broadened considerably, and changes in the 700−900 G (70−90 mT) region were also apparent. It is noteworthy that the loss of the Fe(III)Fe(II) signal was not accompanied by an increase in the spin density due to Fe(III). The estimated spin coupling constant for the Fe(III)Fe(II) centre was much higher than the Zeeman interaction, and a Fe(III)Fe(III) centre with comparable spin coupling would not be expected to exhibit an EPR signal.
Quantitation of the EPR signals from as-isolated GLX2−1 indicated that the Fe(III)Fe(II) signal accounted for only 25 % of the spin density. As suggested by the results of the aerobic addition of Fe(II), it is likely that the fraction of the Fe(III)Fe(II) species would be highly dependent on sample history, including exposure to air and contaminating metal ions, and it is perhaps not too surprising that attempts at NMR of protons experiencing paramagnetic interactions with this centre were unsuccessful.
Efforts to identify substrates of GLX2−1
Most GLX2 enzymes exhibit a substrate preference for SLG; however, none of the recombinant GLX2−1 variants (Table 1) exhibited any detectable SLG hydrolase activity. Therefore to probe whether GLX2−1 is a GLX2 with different substrate specificity, activity assays with other thioesters of glutathione were conducted in order to determine if GLX2−1 has GLX activity, but exhibits a preference for different glutathione thio-esters. Similar to the results with SLG, the as-isolated purple protein and GLX2−1 containing 2 equivalents of Fe did not hydrolyse S-mandeloylglutathione, S-acetylglutathione, S-acetoacetylglutathione, S-formylglutathione, S-glycolylglutathione or S-lactonylglutathione. Therefore GLX2−1 is not a GLX2 enzyme.
Overexpression and purification of the R246L mutant GLX2−1
Our discovery that GLX2−1 binds two equivalents of metal ions was unexpected, given that one of the highly conserved GLX2 metal-binding ligands is replaced by an arginine (Arg246) in GLX2−1. Our observation that GLX2−1 can bind two equivalents of iron or zinc suggested either that Arg246 has the ability to act as a metal-binding ligand, or that another amino acid near the active site is recruited as a metal-binding ligand.
In order to evaluate further whether the amino acid at position 246 binds the metal, the R246L mutant was made. The rationale was that the replacement of arginine by leucine, which is not a metal-binding ligand, would result in a mutant that binds only 1 equivalent of metal. When the R246L mutant was overexpressed at 15 °C, no soluble protein was detected in cleared lysates, suggesting that the overexpressed R246L mutant was improperly folded. However, high levels of soluble protein were obtained when the R246L mutant was expressed in BL21(DE3) E. coli cells, containing pGroESL. When the mutant was overexpressed in the presence of 250 μM Fe and Zn(II), the purified protein was shown to bind 0.1 equivalent of Fe and 0.6 ± 0.1 equivalent of Zn(II) (Table 1). When excess iron was added to the purified R246L mutant, as described for wild-type GLX2−1, the resulting protein bound 2.4 ± 0.5 equivalents of Fe and 0.5 ± 0.2 equivalent of Zn(II). The Zn(II)-added analogue of R246L-GLX2−1 bound 0.1 equivalent of Fe and 1.4 ± 0.2 equivalents of Zn(II). These results are very similar to those of native GLX2−1. None of these analogues hydrolysed SLG. These results indicate that Arg246 is not acting as a metal-binding ligand and that a nearby amino acid is being recruited as part of the metal-binding centre in GLX2−1.
DISCUSSION
The metallo-β-lactamase fold consists of an αβ/βα sandwich motif, made up of a core unit of two β-sheets surrounded by solvent-exposed helices [29]. Members of this superfamily contain a conserved HXHXD motif that has been shown to bind Zn(II), Fe and Mn. There are several enzymes in the metallo-β-lactamase fold family, for example metallo-β-lactamases, GLX2, lactonase, ROO (rubredoxin:oxygen oxidoreductase), arylsulfatase, phosphodiesterase and tRNA maturase [43]. Most of the metallo-β-lactamase superfamily members (metallo-β-lactamases, tRNA maturase, phosphodiesterase, arylsulfatase and lactonase) appear to contain dinuclear Zn(II) centres. On the other hand, ROO appears to contain a dinuclear iron centre [44]. Human GLX2 was reported to contain a dinuclear Zn(II) centre [36]; however, plant mitochondrial GLX2 (GLX2−5) has been shown to contain a FeZn centre [35], and plant cytoplasmic GLX2 can exist with a number of possible metal centres, including dinuclear Fe, FeZn, MnZn and presumably dinuclear Zn(II) [33,34].
Arabidopsis GLX2−1 is highly similar to GLX2−5 and is clearly a member of the metallo-β-lactamase fold superfamily [29]. However, GLX2−1 is unique in that it contains an arginine residue at position 246, which is a metal-binding histidine in all other GLX2 enzymes [43]. Since arginine should be positively charged at neutral pH values, we hypothesized that GLX2−1 would not contain a dinuclear metal centre. Therefore, it was not surprising that as-isolated recombinant GLX2−1 contained less than 1 equivalent of total metal. This form of the protein was purple, and, based on UV–visible studies and the molar absorption coefficient of the peak at 550 nm, we predict that the colouration is due to ligand field transitions of Fe(II) bound to the protein. The Fe(II) can be oxidized to the colourless Fe(III) during short term storage or by the addition of metal ions. EPR studies were conducted to characterize further the metal-binding site in the as-isolated protein, and the spectrum clearly shows the presence of protein-bound high-spin Fe(III). In addition, the spectrum showed the presence of small amounts, up to 25 %, of an anti-ferromagnetically coupled Fe(III)Fe(II) centre that has previously been observed in GLX2−2 and GLX2−5 [33–35] and in the purple acid phosphatases [45,46]. Addition of metal ions to the sample resulted in the loss of the Fe(III)Fe(II) signal with no concomitant increase in the Fe(III) signal, which suggests that the purple colouration arises from Fe(II) in the Fe(III)Fe(II) centre. The lack of paramagnetically shifted 1H resonances in the NMR spectra strongly indicates that there are low levels of either Fe(III)Fe(II) or mononuclear Fe(II). The protons coupled to Fe(III) would probably be broadened too much for detection. Our finding that as-isolated GLX2−1 appears to contain a dinuclear Fe(III)Fe(II) centre, but overall binds less than two equivalents of metal, may be due to the fact that the metal falls out of the protein during the purification process because Arg246 does not allow the enzyme to bind tightly to metal. Taken together, these data suggest that as-isolated GLX2−1 contains small amounts of a Fe(III)Fe(II) centre and larger amounts of FeZn [Fe(III) or Fe(II)] and possibly ZnZn centres. It is also possible that there are significant amounts of mononuclear Fe(II) or a Fe(III)Fe(III) centre in the protein.
In an attempt to saturate the metal-binding site, we incubated the purified protein with excess Zn(II) or Fe, and, after dialysis to remove loosely bound or unbound metal ions, GLX2−1 was found to bind nearly 2 equivalents of metal. There does not appear to be significant discrimination in metal binding, and dinuclear Zn(II), Fe and FeZn centres can apparently be generated if the protein is incubated with Zn(II), Fe, or Fe and Zn(II). Empirically, the Zn(II)-containing analogues are not stable and precipitate relatively quickly once they are prepared, while the Fe-containing analogues are stable for long periods of time. It is not clear why the Zn(II)-containing forms of the protein are unstable. Nonetheless, the ability of GLX2−1 to bind 2 equivalents of metal was unexpected, given that the protein has an arginine residue at position 246, which is not a common metal-binding ligand due to its charge.
In order to investigate further whether Arg246 binds metal, the R246L mutant was generated and characterized. The resulting protein after being loaded with Fe and/or Zn(II) exhibited metal-binding properties similar to those of the wild-type enzyme. The ability of the R246L mutant to bind 2 moles of metal was unexpected because leucine is not a metal-binding ligand. This result strongly suggests that the amino acid at position 246 is not involved in metal binding. Since the dinuclear metal centre is present, this result suggests that another active site residue is recruited to bind metal in GLX2−1.
The results of steady-state kinetic studies with GLX2−1, using a number of different thiolester substrates, indicate that GLX2−1 is not a GLX2 enzyme. The amino acid sequences of GLX2−1 and GLX2−5 exhibit ∼80% identity and ∼88% similarity [27]. However, five of seven active-site residues involved with substrate binding (glutathione) in GLX2−2 [39] and human GLX2 [36] are not conserved in GLX2−1 (Figure 2). It is possible that the different metal-containing analogues of GLX2−1 that we generated in this study are not the physiologically relevant form(s) of the enzyme, or that the absence of GLX2 activity could be due to improperly folded protein. However, previous work on GLX2−2 has shown that this enzyme can utilize a number of different metal centres for catalysis [33,34], and this scenario certainly would offer the organism (Arabidopsis in this case) with an evolutionary advantage, because toxins could be removed regardless of the metal content. The most straightforward interpretation of the steady-state kinetic studies is that GLX2−1 is not a GLX2. At this time, the function for GLX2−1 is unknown; however, molecular studies are being conducted in an effort to identify the physiological role of this protein. In addition, we are attempting to solve the crystal structure of the protein in the hope that the active-site structure can offer some clue as to the natural substrate [47]. It is likely that GLX2−1 will represent a novel function for a metallo-β-lactamase superfamily member [43].
Our previous sequence comparison study identified five genes that were predicted to encode for GLX2 isozymes (GLX2−1, 2−2, 2−3, 2−4 and 2−5) in A. thaliana [27]. We have demonstrated that GLX2−2 and GLX2−5 are in fact GLX2 enzymes [27,28,35] and that they contain dinuclear metal-binding sites [34,35,48]. We recently showed that GLX2−3 contains a mononuclear Fe(II) site [50] and is not a GLX2 [49]. Moreover, this protein is probably a plant analogue of ethylmalonic encephalopathy protein 1 [51], which is a human protein potentially involved with β-oxidation of short-chain acyl-CoAs. The data presented herein demonstrate that GLX2−1 is also not a GLX2, but it contains a dinuclear metal binding site. To date, there is no information on the mitochondrial GLX2−4 protein, and studies are in progress to address whether this protein is a GLX2.
FUNDING
This work was supported by the National Institutes of Health [grant numbers EB001980, AI056231 to B.B., GM40052 to M.W.C.], the National Science Foundation [grant number MCB-9817083 to C.A.M.] and the Ohio Plant Biotechnology Consortium to C.A.M.
Abbreviations used
- GLX
glyoxalase
- ICP-AES
inductively coupled plasma spectrometer with atomic emission spectroscopy detection
- LB medium
Luria–Bertani medium
- ROO
rubredoxin:oxygen oxidoreductase
- SLG
S-D-lactoylglutathione
REFERENCES
- 1.Thornalley P. The glyoxalase system in health and disease. Mol. Aspects Med. 1993;14:287–371. doi: 10.1016/0098-2997(93)90002-u. [DOI] [PubMed] [Google Scholar]
- 2.Thornalley PJ. Pharmacology of methylglyoxal: formation, modification of proteins and nucleic acids, and enzymatic detoxification – a role in pathogenesis and antiproliferative chemotherapy. Gen. Pharmacol. 1996;27:565–573. doi: 10.1016/0306-3623(95)02054-3. [DOI] [PubMed] [Google Scholar]
- 3.Thornalley PJ. Methylglyoxal, glyoxalases and the development of diabetic complications. Amino Acids. 1994;6:15–23. doi: 10.1007/BF00808119. [DOI] [PubMed] [Google Scholar]
- 4.Thornalley PJ. Modification of the glyoxalase system in disease processes and prospects for therapeutic strategies. Biochem. Soc. Trans. 1993;21:531–534. doi: 10.1042/bst0210531. [DOI] [PubMed] [Google Scholar]
- 5.Tew KD. Is there a role for glyoxalase I inhibitors as antitumor drugs? Drug Resist. Updates. 2000;3:263–264. doi: 10.1054/drup.2000.0155. [DOI] [PubMed] [Google Scholar]
- 6.Kalsi A, Kavarana MJ, Lu TF, Whalen DL, Hamilton DS, Creighton DJ. Role of hydrophobic interactions in binding S-(N-aryl/alkyl-N-hydroxycarbamoyl) glutathiones to the active site of the antitumor target enzyme glyoxalase I. J. Med. Chem. 2000;43:3981–3986. doi: 10.1021/jm000160l. [DOI] [PubMed] [Google Scholar]
- 7.Vince R, Brownwell J, Akella LB. Synthesis and activity of γ-(L-γ-azaglutamyl)-S-(p-bromobenzyl)-L-cysteinylglycine: a metabolically stable inhibitor of glyoxalase I. Bioorg. Med. Chem. Lett. 1999;9:853–856. doi: 10.1016/s0960-894x(99)00097-9. [DOI] [PubMed] [Google Scholar]
- 8.Kavarana MJ, Kovaleva EG, Creighton DJ, Wollman MB, Eiseman JL. Mechanism-based competitive inhibitors of glyoxalase I: intracellular delivery, in vitro antitumor activities, and stabilities in human serum and mouse serum. J. Med. Chem. 1999;42:221–228. doi: 10.1021/jm9708036. [DOI] [PubMed] [Google Scholar]
- 9.Elia AC, Chyan MK, Principato GB, Giovannini E, Rosi G, Norton SJ. N,S-bis-fluorenylmethoxycarbonylglutathione: a new, very potent inhibitor of mammalian glyoxalase II. Biochem. Mol. Biol. Intern. 1995;35:763–771. [PubMed] [Google Scholar]
- 10.Thornalley PJ, Strath M, Wilson RJH. Antimalarial activity in vitro of the glyoxalase-I inhibitor diester, S-P-bromobenzylglutathione diethyl ester. Biochem. Pharmacol. 1994;47:418–420. doi: 10.1016/0006-2952(94)90035-3. [DOI] [PubMed] [Google Scholar]
- 11.Murthy NSRK, Bakeris T, Kavarana MJ, Hamilton DS, Lan Y, Creighton DJ. S-(N-aryl-N-hydroxycarbamoyl)glutathione derivatives are tight-binding inhibitors of glyoxalase I and slow substrates for glyoxalase II. J. Med. Chem. 1994;37:2161–2166. doi: 10.1021/jm00040a007. [DOI] [PubMed] [Google Scholar]
- 12.Norton SJ, Elia AC, Chyan MK, Gillis G, Frenzel C, Principato GB. Inhibitors and inhibition studies of mammalian glyoxalase II activity. Biochem. Soc. Trans. 1993;21:545–548. doi: 10.1042/bst0210545. [DOI] [PubMed] [Google Scholar]
- 13.Thornalley P. Advances in glyoxalase research glyoxalase expression in malignancy, anti-proliferative effects of methylglyoxal, glyoxalase I inhibitor diesters, and S-D-lactoylglutathione, and methylglyoxal-modified protein binding and endocytosis by the advanced glycation endproduct receptor. Crit. Rev. Oncol. Hematol. 1995;20:99–128. doi: 10.1016/1040-8428(94)00149-n. [DOI] [PubMed] [Google Scholar]
- 14.Edwards L, Adesida A, Thornalley P. Inhibition of human leukaemia 60 cell growth by S-D-lactoylglutathione in vitro. Mediation by metabolism to N-D-lactoylcystein and induction of apoptosis. Leukemia Res. 1996;20:17–26. doi: 10.1016/0145-2126(95)00095-x. [DOI] [PubMed] [Google Scholar]
- 15.Ratliff DM, Vander Jagt DJ, Eaton RP, Vander Jagt DL. Increased levels of methylglyoxal-metabolizing enzymes in mononuclear and polymorphonuclear cells from insulin-dependent diabetic patients with diabetic complications: aldose reductase, glyoxalase I, and glyoxalase II – a clinical research center study. J. Clin. Endocrinol. Metabol. 1996;81:488–492. doi: 10.1210/jcem.81.2.8636255. [DOI] [PubMed] [Google Scholar]
- 16.Sharkey EM, O'Neill HB, Kavarana MJ, Wang HB, Creighton DJ, Sentz DL, Eiseman JL. Pharmacokinetics and antitumor properties in tumorbearing mice of an enediol analogue inhibitor of glyoxalase I. Cancer Chemo. Pharmacol. 2000;46:156–166. doi: 10.1007/s002800000130. [DOI] [PubMed] [Google Scholar]
- 17.Rulli A, Carli L, Romani R, Baroni T, Giovannini E, Rosi G, Talesa V. Expression of glyoxalase I and II in normal and breast cancer tissues. Breast Cancer Res. Treat. 2001;66:67–72. doi: 10.1023/a:1010632919129. [DOI] [PubMed] [Google Scholar]
- 18.Antognelli C, Baldracchini F, Talesa VN, Costantini E, Zucchi A, Mearini E. Overexpression of glyoxalase system enzymes in human kidney tumor. Cancer J. 2006;12:222–228. doi: 10.1097/00130404-200605000-00011. [DOI] [PubMed] [Google Scholar]
- 19.Schober R, Buchold M, Hintersdorf A, Meixensberger J, Birkenmeier G. Differential expression of glyoxalase in human brain tumors. FASEB J. 2007;21:A27–A27. [Google Scholar]
- 20.Padmanabhan PK, Mukherjee A, Madhubala R. Characterization of the gene encoding glyoxalase II from Leishmania donovanii: a potential target for antiparasite drugs. Biochem. J. 2006;393:227–234. doi: 10.1042/BJ20050948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shinohara M, Thornalley PJ, Giardino I, Beisswenger P, Thorpe SR, Onorato J, Brownlee M. Overexpression of glyoxalase-I in bovine endothelial cells inhibits intracellular advanced glycation endproduct formation and prevents hyperglycemia-induced increases in macromolecular endocytosis. J. Clin. Invest. 1998;101:1142–1147. doi: 10.1172/JCI119885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen F, Wollmer MA, Hoerndli F, Munch G, Kuhla B, Rogaev EI, Tsolaki M, Papassotiropoulos A, Gotz J. Role for glyoxalase I in Alzheimer's disease. Proc. Natl. Acad. Sci. U.S.A. 2004;101:7687–7692. doi: 10.1073/pnas.0402338101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kuhla B, Boeck K, Schmidt A, Ogunlade V, Arendt T, Munch G, Luth HJ. Age- and stage-dependent glyoxalase I expression and its activity in normal and Alzheimer's disease brains. Neurobiol. Aging. 2007;28:29–41. doi: 10.1016/j.neurobiolaging.2005.11.007. [DOI] [PubMed] [Google Scholar]
- 24.Xu Y, Chen X. Glyoxalase II, a detoxifying enzyme of glycolysis byproduct methylglyoxal and a target of p63 and p73, is a pro-survival factor of the p53 family. J. Biol. Chem. 2006;281:26702–26713. doi: 10.1074/jbc.M604758200. [DOI] [PubMed] [Google Scholar]
- 25.Creighton DJ, Hamilton DS. Brief history of glyoxalase I and what we have learned about metal ion dependent, enzyme catalyzed isomerizations. Arch. Biochem. Biophys. 2001;387:1–10. doi: 10.1006/abbi.2000.2253. [DOI] [PubMed] [Google Scholar]
- 26.Cordell PA, Futers TS, Grant PJ, Pease RJ. The human hydroxyacylglutathione hydrolase (HAGH) gene encodes both cytosolic and mitochondrial forms of glyoxalase II. J. Biol. Chem. 2004;279:28653–28661. doi: 10.1074/jbc.M403470200. [DOI] [PubMed] [Google Scholar]
- 27.Maiti MK, Krishnasamy S, Owen HA, Makaroff CA. Molecular cloning of glyoxalase II from a higher plant: comparison of mitochondrial and cytoplasmic isozymes. Plant Mol. Biol. 1997;35:471–481. doi: 10.1023/a:1005891123344. [DOI] [PubMed] [Google Scholar]
- 28.Crowder MW, Maiti MK, Banovic L, Makaroff CA. Glyoxalase II from A. thaliana requires Zn(II) for catalytic activity. FEBS Lett. 1997;418:351–354. doi: 10.1016/s0014-5793(97)01416-6. [DOI] [PubMed] [Google Scholar]
- 29.Melino S, Capo C, Dragani B, Aceto A, Petruzzelli R. A zinc-binding motif conserved in glyoxalase II, beta-lactamase and arylsulfatases. Trends Biochem. Sci. 1998;23:381–382. doi: 10.1016/s0968-0004(98)01264-x. [DOI] [PubMed] [Google Scholar]
- 30.Crowder MW, Spencer J, Vila AJ. Metallo-β-lactamases: novel weaponry for antibiotic resistance in bacteria. Acc. Chem. Res. 2006;39:721–728. doi: 10.1021/ar0400241. [DOI] [PubMed] [Google Scholar]
- 31.Gomes CM, Silva G, Oliveira S, LeGall J, Liu MY, Xavier AV, Rodrigues-Pousada C, Teixeira M. Studies on the redox centers of the terminal oxidase from Desulfovibrio gigas and evidence for its interaction with rubredoxin. J. Biol. Chem. 1997;272:22502–22508. doi: 10.1074/jbc.272.36.22502. [DOI] [PubMed] [Google Scholar]
- 32.Vogel A, Schilling O, Niecke M, Bettmer J, Meyer-Klaucke W. ElaC encodes a novel binuclear zinc phosphodiesterase. J. Biol. Chem. 2002;277:29078–29085. doi: 10.1074/jbc.M112047200. [DOI] [PubMed] [Google Scholar]
- 33.Schilling O, Wenzel N, Naylor M, Vogel A, Crowder M, Makaroff C, Meyer-Klaucke W. Flexible metal binding of the metallo-β-lactamase domain: glyoxalase II incorporates iron, manganese, and zinc in vivo. Biochemistry. 2003;42:11777–11786. doi: 10.1021/bi034672o. [DOI] [PubMed] [Google Scholar]
- 34.Wenzel NF, Carenbauer AL, Pfiester MP, Schilling O, Meyer-Klaucke W, Makaroff CA, Crowder MW. The binding of iron and zinc to glyoxalase II occurs exclusively as di-metal centers and is unique within the metallo-β-lactamase family. J. Biol. Inorg. Chem. 2004;9:429–438. doi: 10.1007/s00775-004-0535-2. [DOI] [PubMed] [Google Scholar]
- 35.Marasinghe GPK, Sander IM, Bennett B, Periyannan G, Yang KW, Makaroff CA, Crowder MW. Structural studies on a mitochondrial glyoxalase II. J. Biol. Chem. 2005;280:40668–40675. doi: 10.1074/jbc.M509748200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cameron AD, Ridderstrom M, Olin B, Mannervik B. Crystal structure of human glyoxalase II and its complex with a glutathione thiolester substrate analogue. Structure. 1999;7:1067–1078. doi: 10.1016/s0969-2126(99)80174-9. [DOI] [PubMed] [Google Scholar]
- 37.Ullah AHJ, Dischinger HC. Identification of active site residues in Aspergillus ficuum extracellular pH 2.5 optimum acid phosphatase. Biochem. Biophys. Res. Commun. 1993;192:754–759. doi: 10.1006/bbrc.1993.1478. [DOI] [PubMed] [Google Scholar]
- 38.Cameron AD, Olin B, Ridderstrom M, Mannervik B, Jones TA. Crystal structure of human glyoxalase I – evidence for gene duplication and 3D domain swapping. EMBO J. 1997;16:3386–3395. doi: 10.1093/emboj/16.12.3386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zang TM, Hollman DA, Crawford PA, Crowder MW, Makaroff CA. Arabidopsis glyoxalase II contains a zinc/iron binuclear metal center that is essential for substrate binding and catalysis. J. Biol. Chem. 2001;276:4788–4795. doi: 10.1074/jbc.M005090200. [DOI] [PubMed] [Google Scholar]
- 39a.Purpero VM, Moran GR. Catalytic, noncatalytic, and inhibitory phenomena: kinetic analysis of (4-hydroxyphenyl)pyruvate dioxygenase from Arabidopsis thaliana. Biochemistry. 2006;45:6044–6055. doi: 10.1021/bi052409c. [DOI] [PubMed] [Google Scholar]
- 39b.Bou-Abdallah F, Chasteen ND. Spin concentration measurements of high-spin (g’ = 4.3) rhombic iron(III) ions in biological samples: theory and application. J. Biol. Inorg. Chem. 2008;13:15–24. doi: 10.1007/s00775-007-0304-0. [DOI] [PubMed] [Google Scholar]
- 39c.Aasa R, Vänngård T. EPR signal intensity and powder shapes. Reexamination. J. Magn. Reson. 1975;19:308–315. [Google Scholar]
- 39d.Hanson GR, Gates KE, Noble CJ, Mitchell A, Benson S, Griffin M, Burrage K. XSophe – Sophe – XeprView a computer simulation software suite for the analysis of continuous wave EPR spectra. In: Shiotani M, Lund A, editors. EPR of Free Radicals in Solids: Trends in Methods and Applications. Kluwer Press; Dordrect: 2003. pp. 197–237. [Google Scholar]
- 40.Uotila L. Thioesters of glutathione. Meth. Enzymol. 1981;77:424–430. [Google Scholar]
- 41.Lippard SJ, Berg JM. Principles of Bioinorganic Chemistry. University Science Books; Mill Valley, CA: 1994. [Google Scholar]
- 42.Bertini I, Banci L, Luchinat C. Proton magnetic resonance of paramagnetic metalloproteins. In: Oppenheimer NJ, James TL, editors. Methods in Enzymology. Vol. 177. Academic Press; New York: 1989. pp. 246–263. [DOI] [PubMed] [Google Scholar]
- 43.Bebrone C. Metallo-β-lactamases (classification, activity, genetic organization, structure, zinc coordination) and their superfamily. Biochem. Pharmacol. 2007;74:1686–1701. doi: 10.1016/j.bcp.2007.05.021. [DOI] [PubMed] [Google Scholar]
- 44.Frazao C, Silva G, Gomes CM, Matias P, Coelho R, Sieker L, Macedo S, Liu MY, Oliveira S, Teixeira M, et al. Structure of a dioxygen reduction enzyme from Desulfovibrio gigas. Nature Struct. Biol. 2000;7:1041–1045. doi: 10.1038/80961. [DOI] [PubMed] [Google Scholar]
- 45.Crowder MW, Vincent JB, Averill BA. Electron paramagnetic resonance studies on the high-salt form of bovine spleen purple acid phosphatase. Biochemistry. 1992;31:9603–9608. doi: 10.1021/bi00155a012. [DOI] [PubMed] [Google Scholar]
- 46.David SS, Que L. Anion binding to uteroferrin. Evidence for phosphate coordination to the iron(III) ion of the dinuclear active site and interaction with the hydroxo bridge. J. Am. Chem. Soc. 1990;112:6455–6463. [Google Scholar]
- 47.Hermann JC, Marti-Arbona R, Fedorov AA, Fedorov E, Almo SC, Shoichet BK, Raushel FM. Structure-based activity prediction for an enzyme of unknown function. Nature. 2007;448:775–781. doi: 10.1038/nature05981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schilling O, Vogel A, Meyer-Klaucke W. EXAFS studies on proteins from the metallo-beta-lactamase family reveal similar metal sites in an oxido-reductase and the hydrolase ElaC. J. Inorg. Biochem. 2001;86:422. [Google Scholar]
- 49.McCoy JG, Bingman CA, Bitto E, Holdorf MM, Makaroff CA, Phillips GNJ. Structure of an ETHE1-like protein from Arabidopsis thaliana. Acta Crystallogr. D Biol. Crystallogr. 2006;62:964–970. doi: 10.1107/S0907444906020592. [DOI] [PubMed] [Google Scholar]
- 50.Holdorf MM, Bennett B, Crowder MW, Makaroff CA. Spectroscopic studies on Arabidopsis ETHE1, a glyoxalase II-like protein. J. Inorg. Biochem. 2008;102:1825–1830. doi: 10.1016/j.jinorgbio.2008.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tiranti V, D'Adamo P, Briem E, Ferrari G, Mineri R, Lamantea E, Mandel H, Balestri P, Garcia-Silva M-T, Vollmer B, et al. Ethylmalonic encephalopathy is caused my mutations in ETHE1, a gene encoding a mitochondrial matrix protein. Am. J. Hum. Genet. 2004;74:239–252. doi: 10.1086/381653. [DOI] [PMC free article] [PubMed] [Google Scholar]