

Abstract
Hydroxylated metabolites of polychlorinated biphenyls (OH-PCBs) are inhibitors and substrates for various human sulfotransferases (SULTs). Although the rat is often used in toxicological studies on PCBs, the interactions of OH-PCBs with rat SULTs are less well understood. In the present study, 15 OH-PCBs were investigated as potential substrates or inhibitors of purified recombinant rSULT1A1 and rSULT2A3, the major family 1 and family 2 SULTs present in rat liver, respectively. None of these OH-PCBs were substrates for rSULT2A3, 11 weakly inhibited rSULT2A3-catalyzed sulfation of dehydroepiandrosterone, and 4 had no effect on the reaction. With rSULT1A1, 4-OH-PCB 8, 4′-OH-PCB 3, 9, 12, 35, and 6′-OH-PCB 35 were substrates, whereas 4′-OH-PCB 6, 4-OH-PCB 14, 4′-OH-PCB 25, 4′-OH-PCB 33, 4-OH-PCB 34, 4-OH-PCB36, 4′-OH-PCB 36, 4′-OH-PCB 68, and 4-OH-PCB 78 inhibited the sulfation of 2-naphthol catalyzed by this enzyme. OH-PCBs with a 3,5-dichloro-4-hydroxy substitution were the most potent inhibitors of rSULT1A1, and the placement of chlorine atoms in the ortho- and meta-positions on either ring of para-OH-PCBs resulted in significant differences in activity as substrates and inhibitors. The specificity of rSULT1A1 for several inhibitory OH-PCBs was altered by pretreatment of the enzyme with oxidized glutathione (GSSG). Four OH-PCBs that were inhibitors of rSULT1A1 under reducing conditions became substrates after pretreatment of the enzyme with GSSG. This alteration in specificity of rSULT1A1 for certain OH-PCBs suggests that conditions of oxidative stress may significantly alter the sulfation of some OH-PCBs in the rat.
Although the manufacture of polychlorinated biphenyls (PCBs) has been legally banned in the United States for more than 30 years, they persist in the environment in large quantities and are hazardous to public health (Agency for Toxic Substances and Disease Registry, http://www.atsdr.cdc.gov/toxprofiles/tp17.html; Robertson and Hansen, 2001). The originally released PCBs were predominantly highly chlorinated (six or more chlorines per molecule). However, PCBs in soils and marine sediments can be dechlorinated to lower chlorinated congeners (Abramowicz, 1995; Master et al., 2002). Lower chlorinated PCBs are semivolatile and are present in urban atmospheres of various areas in the United States and other countries (Wethington and Hornbuckle, 2005; Ruzickova et al., 2008), and they move in dynamic balance among the atmosphere, water, and soil.
PCBs are biotransformed by cytochrome P450 (P450) isoforms to hydroxylated PCBs (OH-PCBs) (Kaminsky et al., 1981; Safe, 1994; McLean et al., 1996; Ludewig et al., 2007). Lower chlorinated PCBs are often more susceptible than highly chlorinated congeners to biotransformation in P450-catalyzed reactions to OH-PCBs. PCBs administered to rats are initially deposited in the liver and muscles and then are translocated to the skin and adipose tissue (Matthews and Anderson, 1975). However, the OH-PCBs may be selectively concentrated in the liver compared with adipose tissue, as indicated by a study in which the concentration of OH-PCBs was approximately 20 times higher in liver than that in adipose tissues, whereas the concentrations of total PCBs were not significantly different between the two tissues (Guvenius et al., 2002). After P450-catalyzed hydroxylation of PCBs, the resulting OH-PCBs can be conjugated in reactions catalyzed by enzymes such as sulfotransferases (Liu et al., 2006; Wang et al., 2006), UDP-glucuronosyltransferases (Schnellmann et al., 1984; Tampal et al., 2002), and glutathione S-transferases (James, 2001).
The sulfotransferases (SULTs) constitute a superfamily of biotransformation enzymes catalyzing the sulfation of a spectrum of substrates ranging from endogenous hormones and neurotransmitters to xenobiotics. OH-PCBs have been observed to be inhibitors and substrates of human cytosolic sulfotransferases (hSULTs), e.g., hSULT1A1 (Wang et al., 2006), hSULT1E1 (Kester et al., 2000), and hSULT2A1 (Liu et al., 2006), major family 1 and family 2 SULTs in humans. Although hundreds of toxicological investigations on PCBs have been conducted using rats or tissues/cells derived from rats, relatively little is known about the sulfation of OH-PCBs in the rat. Moreover, the specificities of individual SULTs for OH-PCBs in that species have not been extensively studied. This gap in our knowledge could potentially cause uncertainty in the extrapolation of studies from rat to human. We hypothesized that OH-PCBs interact with rSULT1A1 (also previously known as aryl sulfotransferase IV) and rSULT2A3 (also previously known as sulfotransferase STa), two isoforms of SULT in the rat that are orthologous to human isoforms hSULT1A1 and hSULT2A1, respectively.
rSULT1A1 is also of interest because of its ability to be regulated by the thiol/disulfide status of its environment (Marshall et al., 1997, 2000; Duffel et al., 2001). There are five cysteine residues, located at positions 66, 82, 232, 283, and 289, in each of the two identical subunits (homodimers) of rSULT1A1. It has been shown that the kinetics, specificity, and pH optima of rSULT1A1 are regulated by the oxidation status of Cys66 (i.e., conversion among the free thiol, a glutathione-protein mixed disulfide, and an intramolecular disulfide between Cys66 and Cys232) (Marshall et al., 1997, 2000). These previous studies on rSULT1A1 led to our second hypothesis that interactions of OH-PCBs with the purified rSULT1A1 and rSULT2A3 would be modified by changing the oxidative environment of the enzyme with oxidized glutathione. Although the sulfation of dehydroepiandrosterone (DHEA), a known substrate of rSULT2A3, catalyzed by hepatic cytosol from male rats has been reported to be unaffected by treatment with oxidized glutathione (Maiti et al., 2004), the potential for substrate-specific modulation of a homogeneous preparation of rSULT2A3 by oxidized glutathione has not been investigated.
Thus, in the present study, 15 lower chlorinated congeners of OH-PCBs, each bearing one hydroxyl (at the para-position for 14 and the ortho-position for 1 of the congeners) and one to four chlorine atoms in different substitution patterns, were investigated for their interaction with homogeneous recombinant rSULT1A1 and rSULT2A3 as substrates and inhibitors. The potential for alteration of the specificity of rSULT1A1 and rSULT2A3 for OH-PCBs was explored by pretreatment of each enzyme with oxidized glutathione.
Materials and Methods
Chemicals. The synthesis and characterization of 12 OH-PCBs (abbreviations and chemical structures shown in Table 1) has been reported previously (Lehmler and Robertson, 2001). In addition, 3 new OH-PCBs were synthesized by Suzuki coupling of the corresponding chlorinated benzeneboronic acids and a suitable bromo chloro anisole, followed by demethylation with boron tribromide.
TABLE 1.
Inhibition of rSULT1A1 and rSULT2A3 by hydroxylated PCBs
|
OH-PCB
|
Chemical Structure
|
Solubilitya
|
IC50
|
|
|---|---|---|---|---|
| Rat SULT1A1 | Rat SULT2A3 | |||
| μM | μM | |||
| 4′-OH-PCB 3 | 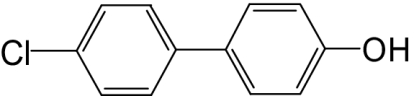 |
300 | —b | 160 |
| 4′-OH-PCB 6 | 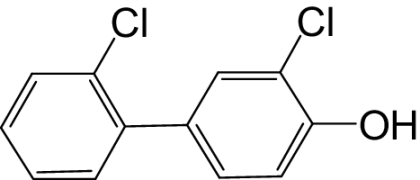 |
250 | 22.7 | —c |
| 4-OH-PCB 8 | 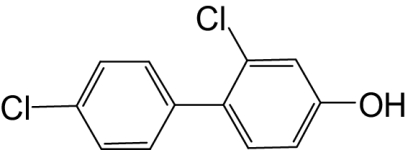 |
≥500 | —b | 400d |
| 4′-OH-PCB 9 | 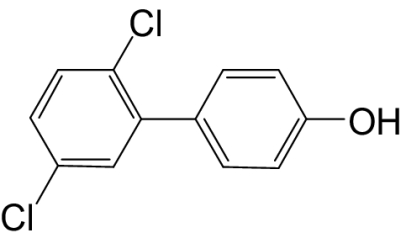 |
500 | —b | 190 |
| 4′-OH-PCB 12 | 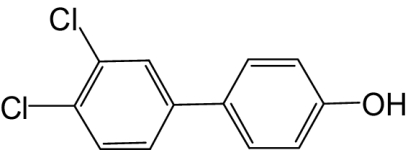 |
≥500 | —b | 160 |
| 4-OH-PCB 14 | 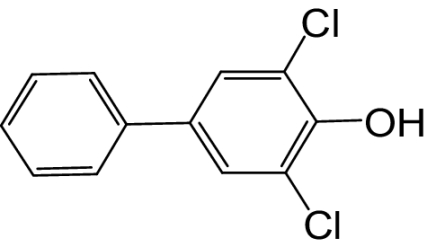 |
≥1000 | 0.27 | 64 |
| 4′-OH-PCB 25 | 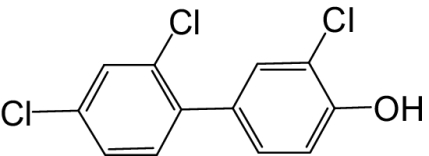 |
100 | 57.7 | —e |
| 4′-OH-PCB 33 | 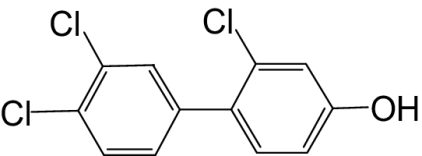 |
50 | 26.2 | —c |
| 4-OH-PCB 34 | 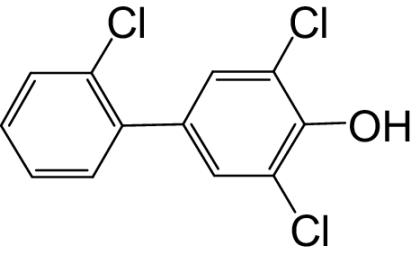 |
300 | 0.34 | 160 |
| 4′-OH-PCB 35 | 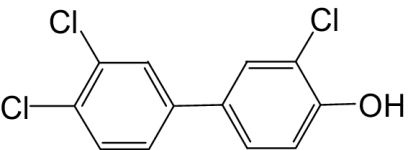 |
50 | —b | —e |
| 6′-OH-PCB 35 | 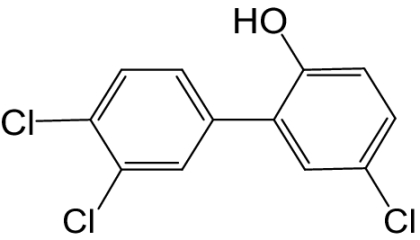 |
100 | —b | —e |
| 4-OH-PCB 36 | 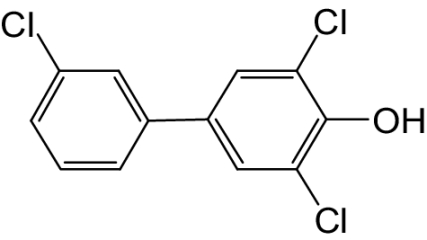 |
50 | 0.54 | —c |
| 4′-OH-PCB 36 | 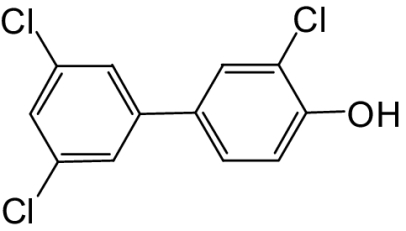 |
50 | 40 | 50d |
| 4′-OH-PCB 68 | 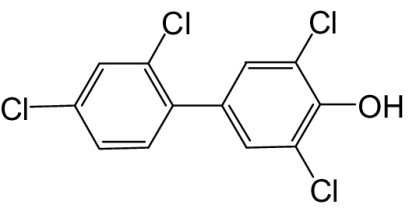 |
50 | 0.85 | 32d |
| 4-OH-PCB 78 | 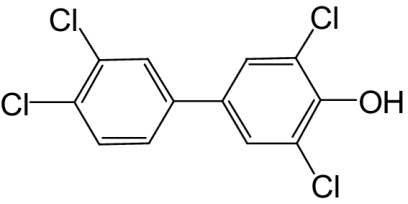 |
33 | 4.4 | —e |
Solubility of each OH-PCB was detected according to the light scattering intensity in the same solution as in the reactions.
This OH-PCB was a substrate for rSULT1A1 (Fig. 1).
Less than 50% inhibition was reached within the limit of solubility.
This is an approximate value due to 50% inhibition at the limit of solubility.
This OH-PCB was neither a substrate nor an inhibitor for rSULT2A3.
3,2′-Dichloro-biphenyl-4-ol (4′-OH-PCB 6).
White solid; mp = 46–47°C (>99% by GC); 1H NMR (400 MHz, CDCl3): δ/ppm 7.44–7.42 (m, 1H), 7.40 (d, J = 2.1 Hz, 1H), 7.28–7.23 (m, 4H), 7.05 (d, J = 8.4 Hz, 1H), 5.62 (br s, 1H, –OH); 13C NMR (100 MHz, CDCl3): δ/ppm 151.0, 139.0, 132.9, 132.7, 131.4, 130.2, 130.1, 129.9, 128.9, 127.1, 119.7, 116.0; MS m/z (relative intensity): 238 (100, M–H), 139 (80).
2,4′-Dichloro-biphenyl-4-ol (4-OH-PCB 8).
White solid; mp = 107–108°C (>99% by GC); 1H NMR (400 MHz, CDCl3): δ/ppm 7.41–7.34 (m, 4H), 7.19 (d, J = 8.4 Hz, 1H), 7.00 (d, J = 2.6 Hz, 1H), 6.82 (dd, J = 8.4, 2.6 Hz, 1H), 5.32 (br s, 1H, –OH); 13C NMR (100 MHz, CDCl3): δ/ppm 155.6, 137.6, 133.5, 133.1, 132.2, 132.1, 131.1, 128.4, 117.0, 114.5; MS m/z (relative intensity): 238 (100, M–H), 168 (36), 139 (45).
2,3′,4′-Trichloro-biphenyl-4-ol (4′-OH-PCB 33). White solid; mp = 103–104°C (>99% by GC); 1H NMR (400 MHz, CDCl3): δ/ppm 7.51–7.47 (m, 2H), 7.27–7.25 (m, 1H), 7.18 (d, J = 8.4 Hz, 1H), 6.99 (d, J = 2.6 Hz, 1H), 6.81 (dd, J = 8.4, 2.6 Hz, 1H), 5.03 (br s, 1H, –OH); 13C NMR (100 MHz, CDCl3): δ/ppm 156.0, 139.1, 133.1, 132.3, 132.1, 131.7, 131.6, 131.0, 130.2, 129.2, 117.2, 114.6; MS m/z (relative intensity): 272 (100, M–H), 202 (36), 173 (18), 139 (15).
Adenosine 3′-phosphate 5′-phosphosulfate (PAPS) was obtained from Sigma-Aldrich (St. Louis, MO) and further purified by a published procedure (Sekura, 1981) to a purity greater than 98% as determined by high-performance liquid chromatography. 2-Naphthol, DHEA, 1-octylamine, adenosine 3′,5′-diphosphate (PAP), PAP-agarose, reduced glutathione, and oxidized glutathione were used as obtained from Sigma-Aldrich. Dithiothreitol (DTT) was from Research Products International (Mt. Prospect, IL). All other chemicals and reagents were of the highest purity commercially available.
Expression and Purification of Recombinant Rat SULTs. Recombinant Escherichia coli BL 21 (DE3) cells that expressed either rSULT1A1 (Chen et al., 1992) or rSULT2A3 (Sheng and Duffel, 2001) were established using a pET-3c vector as described previously. Cells were grown, cell extract was prepared, and the enzymes were purified using minor modifications of a procedure developed for hSULT2A1 (Liu et al., 2006). In brief, each E. coli cell culture was grown in 3 ml of Luria broth medium (supplemented with 50 μg/ml ampicillin for cell selection) at 29°C. After 24 h, 100-μl aliquots of the cell suspension were transferred to each of four 20-ml portions of fresh culture medium (Luria broth medium with 50 μg/ml ampicillin) and incubated at 29°C for 24 h. Finally, each 20-ml culture was added to 400 ml of fresh culture medium and incubated with shaking (210 rpm) at 29°C for 24 h. Isopropyl-1-thio-d-galactopyranoside (1 mM) was present in the final stage of rSULT2A3-expressing cell cultures for 23 h (added 1 h after the start of final stage culture) but was not used for rSULT1A1-expressing cell cultures. The cells (weighing approximately 14 g) were disrupted by sonification in 20 ml of buffer A [10 mM Tris-HCl at pH 7.5, 0.25 M sucrose, 10% (v/v) glycerol, 1 mM phenylmethylsulfonyl fluoride, 1 μM pepstatin, 1 mM DTT, and 2 mg/l antipain]. The cell homogenate was centrifuged at 24,000g for 30 min, and the supernatant fraction was collected as cell extract. Each SULT isoform was purified using PAP-agarose affinity column chromatography (5 ml of PAP-agarose in a 1 × 10 cm column). After the loading of cell extract on the column, the column was washed with 200 ml of buffer B [10 mM Tris-HCl buffer at pH 7.5, 0.25 M sucrose, 10% (v/v) glycerol, 1 μM pepstatin, 1 mM DTT, 2 mg/l antipain, and 0.05% (v/v) Tween 20] to remove nonspecifically bound proteins. rSULT1A1 was eluted with a linear gradient formed between 20 ml of buffer B and 20 ml of buffer B containing 100 μM PAP. The linear gradient for eluting rSULT2A3 was formed between 20 ml of buffer B and 20 ml of buffer B containing 50 μM PAP. Residual PAP was removed by gel filtration chromatography on a PD-10 column (GE Healthcare, Piscataway, NJ) eluted with buffer B. The protein was concentrated by ultrafiltration with a 10-ml Amicon stirred cell and PM-10 membrane (Millipore Corporation, Billerica, MA).
Assay of OH-PCBs as Potential Substrates. The sulfation reactions were carried out in a total volume of 30 μl, with 0.25 M potassium phosphate buffer at pH 7.0, 200 μM PAPS, 7.5 mM 2-mercaptoethanol, and each OH-PCB in varied concentrations ranging from 3 μM to either its limit of solubility or 400 μM, as appropriate. Acetone was used as cosolvent for each OH-PCB and was present in each final reaction mixture at a concentration of 3.3% (v/v). Either rSULT1A1 (0.75 μg) or rSULT2A3 (0.5 μg) was added to initiate each reaction, which was carried out at 37°C for 6 and 15 min, respectively. Each reaction was terminated by the addition of 30 μl of methanol. The rate of sulfation was determined by substrate-dependent formation of PAP as analyzed by high-performance liquid chromatography (Duffel et al., 1989; Sheng et al., 2001).
Inhibition of SULTs by OH-PCBs. Those OH-PCBs that were not substrates were investigated for potential inhibition of the sulfation of 2-naphthol (for rSULT1A1) and DHEA (for rSULT2A3). Reactions were conducted under nonsaturating substrate concentrations (i.e., with 15 μM 2-naphthol or 10 μM DHEA). Each OH-PCB was studied at various concentrations up to full inhibition of the reaction or the limit of solubility, as appropriate, and either 0.75 μg of rSULT1A1 or 0.5 μg of rSULT2A3 was used in each assay. Reactions were carried out at 37°C for 6 min, and the sulfation rate was determined by substrate-dependent formation of PAP as described above.
Effect of Oxidized Glutathione on the Specificities of SULTs for OH-PCBs. For studies on the effect of disulfide on the SULTs, DTT was removed from the original enzyme preparations according to a method reported previously (Marshall et al., 1997), with minor modifications. In brief, either rSULT1A1 or rSULT2A3 in buffer B (0.5–1 ml) was added to a 5-ml PD-10 gel filtration column that had been equilibrated with buffer C (i.e., all components of buffer B except DTT). After elution with buffer C, the protein (3–5 ml) was concentrated to 0.5 to 1.0 ml by ultrafiltration using a 10-ml Amicon stirred cell with a PM-10 membrane. All manipulations were carried out at 4°C. The resulting concentration of DTT was less than 0.06 mM as determined by a standard assay for thiols using 5,5′-dithiobis(2-nitrobenzoic acid) (DTNB) (Jocelyn, 1987).
Stock solutions of reduced and oxidized glutathione (10 mM) were prepared in buffer C, and the pH was adjusted with potassium hydroxide to 7.5. Each enzyme-glutathione mixture was prepared in a 9:1 ratio (v/v), such that the final concentration of either reduced or oxidized glutathione was 1 mM, and there was a 10% decrease in protein concentration. Each enzyme in argon-saturated buffer C containing either 1 mM reduced glutathione (GSH) or 1 mM oxidized glutathione (GSSG) was incubated at 25°C for 1 h. Aliquots of these solutions of enzymes were then used in the assay of sulfation reactions as described above, but without addition of any additional reducing agents to the assay mixtures (i.e., 2-mercaptoethanol was absent from the reaction mixtures). Reaction mixtures contained 0.25 M potassium phosphate at pH 7.0, 200 μM PAPS, the OH-PCB at either 100 μM or a lower concentration if its limit of solubility was less than 100 μM, and an aliquot of each enzyme that had been pretreated with either 1 mM GSH or 1 mM GSSG (0.75 μg of rSULT1A1 or 0.5 μg of rSULT2A3). The volume of the aliquot of enzyme (1.5–2.2 μl) added to each reaction mixture did not affect the pH of the assay. Three replicates were carried out for each treatment. Reaction mixtures were incubated at 37°C for 6 min (rSULT1A1) and 15 min (rSULT2A3), respectively. An assay containing all components except OH-PCB was used to determine the OH-PCB-dependent formation of PAP with either the reduced or oxidized enzyme. These control experiments in the absence of any OH-PCB exhibited less than 18 μM PAP (rSULT1A1) and 13 μM PAP (rSULT2A3) formed in the course of the assay period, and these controls were subtracted from the PAP formed in the presence of OH-PCBs to determine the rate of substrate-dependent formation of PAP. For those OH-PCBs that were observed to be substrates for oxidized rSULT1A1, multiple concentrations of OH-PCBs were then used under the same conditions as described above. Two substrates for reduced rSULT1A1, 4′-OH-PCB 9 and 6′-OH-PCB 35 (representing kinetic profiles both with and without substrate inhibition, respectively) were also examined for differences in sulfation catalyzed by GSH- and GSSG-pretreated rSULT1A1. After each preincubation of enzyme with GSH at 25°C for 1 h, an aliquot of the mixture was analyzed with DTNB (Jocelyn, 1987) for thiol content to determine the stability of the reduced glutathione under the experimental conditions.
Reversibility of the Effect of GSSG on SULT1A1 by Reduction. After removal of reducing agents by PD-10 chromatography, rSULT1A1 was pretreated with either 5 mM DTT or 1 mM GSSG at 25°C for 1 h as described above. The resulting enzyme preparations were used to determine substrate-dependent formation of PAP with either 2-naphthol (250 μM) or 4′-OH-PCB 6 (100 μM). An aliquot of 1 mM GSSG-pretreated rSULT1A1 was further treated with 5 mM DTT (incubation at 25°C for 1 h) to reduce disulfide bonds in the enzyme, and the rate of sulfation of either 2-naphthol or 4′-OH-PCB 6 was then determined.
Solubility of OH-PCBs in the SULT Assays. The solubility of each OH-PCB was determined at 37°C in 0.25 M potassium phosphate at pH 7.0 containing 3.3% (v/v) acetone and 7.5 mM 2-mercaptoethanol. Light scattering at 400 nm was used as described previously (Blomquist et al., 1978; Liu et al., 2006) with a PerkinElmer LS-55 luminescence spectrometer (PerkinElmer Life and Analytical Sciences, Waltham, MA).
Results
Interaction of OH-PCBs with rSULT1A1 and rSULT2A3 as Substrates and Inhibitors in the Presence of Reducing Agents. Under commonly used SULT assay conditions in the presence of reducing agents (i.e., DTT during enzyme purification and 2-mercaptoethanol in reaction mixtures), six OH-PCBs were observed to be substrates for rSULT1A1. As shown in Fig. 1A, four of these six OH-PCBs demonstrated substantial substrate inhibition, whereas two, 4′-OH-PCB 35 and 6′-OH-PCB 35, did not show substrate inhibition within their limits of solubility. The maximal sulfation rates and the concentrations at which maximal sulfation rates were observed varied among the individual OH-PCBs. The concentration-velocity curves for the six OH-PCBs were complex and not amenable to description by a simple enzyme kinetic model.
Fig. 1.

OH-PCBs were either substrates or inhibitors of rSULT1A1. A, six OH-PCBs used in the present study were substrates for rSULT1A1. Data are the means ± S.E. of triplicate (4′-OH-PCB 12 and 4′-OH-PCB 35) or duplicate (4′-OH-PCB 3, 4-OH-PCB 8, 4′-OH-PCB 9, and 6′-OH-PCB 35) determinations. B, eight OH-PCBs were inhibitors of rSULT1A1 with full inhibition of sulfation of 2-naphthol. Duplicate assays were used at each OH-PCB concentration to determine IC50 values; the mean values for each concentration of OH-PCB are shown.
Although six of the OH-PCBs were substrates, the other nine OH-PCBs were found to be inhibitors of rSULT1A1 when 2-naphthol was used as the substrate. Eight of these inhibitors displayed full (100%) inhibition of the enzyme (Fig. 1B), whereas 4′-OH-PCB 36 (data not shown) reached only approximately 65% inhibition of the enzyme at its limit of solubility (i.e., 50 μM). As shown in Fig. 1B, large variations in the potency of individual OH-PCBs in inhibiting rSULT1A1 were seen. The calculated IC50 values for each OH-PCB are shown in Table 1, and these ranged from 0.27 to 57.7 μM, with 4-OH-PCB 14 being the most potent inhibitor.
Unlike the case with rSULT1A1, none of the 15 OH-PCBs showed any ability to serve as a substrate for rSULT2A3. Moreover, none of the OH-PCBs examined were potent inhibitors of the sulfation of DHEA catalyzed by rSULT2A3. Eleven OH-PCBs demonstrated weak inhibition of rSULT2A3; eight of them (4′-OH-PCB 3, 4′-OH-PCB 9, 4′-OH-PCB 12, 4-OH-PCB 14, 4-OH-PCB 34, 4′-OH-PCB 36, 4′-OH-PCB 68, and 4-OH-PCB 8) had IC50 values ranging from 32 to 400 μM, and three (4′-OH-PCB 6, 4′-OH-PCB 33, and 4-OH-PCB 36) showed less than 50% inhibition within their solubility limits. 4′-OH-PCB 9 was the only one that attained full inhibition of the enzyme. The other four OH-PCBs showed neither inhibitory nor substrate activity toward rSULT2A3 within their limits of solubility.
Structure-Activity Analysis of OH-PCBs with rSULT1A1. As shown in Fig. 2, A to C, the most potent inhibitors of rSULT1A1 have a 3,5-dichloro-4-hydroxy substitution pattern. For example, a 72-fold difference in IC50 is observed with the addition of a 5′-chlorine atom to 4′-OH-PCB 25 (i.e., 4′-OH-PCB 68). The presence or absence of a chlorine atom at the 4-position of the nonphenolic ring has a small effect on the magnitude of the IC50 value observed (Fig. 2, A and C). However, a change in chlorine atoms between the 2- and 3-positions on the aromatic ring bearing a 4-OH group (Fig. 2D) results in large changes in the interactions with rSULT1A1, as is also the case when chlorine atoms at the 2′- and 3′-positions (on the nonphenolic ring) are altered in a 3-chloro-4-OH-PCB (Fig. 2E). Thus, there appears to be a significant role for chlorine atoms in the 2 (or 2′)- and 3 (or 3′)-positions of 4′-OH-PCBs in determining their ability to serve as substrates and inhibitors of rSULT1A1.
Fig. 2.

Structure-activity relationships for OH-PCB-inhibition of rSULT1A1. A to C, relationships between IC50 values and the 3,5-dichloro-4-hydroxy substitution pattern. D, importance of chlorine atoms in the 2- and 3-positions of the aromatic ring of a PCB bearing a 4-OH group. E, effects of a chlorine atom in the 2′- or 3′-position of the nonphenolic ring of a chloro-4-OH-PCB.
Effects of Pretreatment of rSULT1A1 and rSULT2A3 with Oxidized and Reduced Glutathione on Their Abilities to Catalyze Sulfation of OH-PCBs. The nine OH-PCBs that were not substrates for either SULT under the assay conditions used for experiments in Table 1 were further examined for their potential to serve as substrates after treatment of the enzymes with oxidized and reduced glutathione. To examine the response of these enzymes to the thiol/disulfide ratio in their environment, the DTT present during purification was removed by gel filtration chromatography, and the enzymes were incubated for 1 h at 25°C under argon in the presence of either 1 mM GSH or 1 mM GSSG as described under Materials and Methods. At the end of the incubation of the enzyme with 1 mM GSH, the concentration of GSH was verified by reaction of an aliquot with DTNB. After the incubation of rSULT1A1 with either GSH or GSSG, the activity of the enzyme was determined as described under Materials and Methods. A final concentration of 100 μM in each assay was used for 4′-OH-PCB 6, 4-OH-PCB 14, 4′-OH-PCB 25, and 4-OH-PCB 34. The other OH-PCBs (i.e., 4′-OH-PCB 33, 4-OH-PCB 36, 4′-OH-PCB 36, 4′-OH-PCB 68, and 4-OH-PCB 78) were examined at concentrations equal to the limits of their solubility shown in Table 1. None of these nine OH-PCBs were substrates for the rSULT1A1 that had been pretreated with 1 mM GSH, a result similar to that obtained under standard assay conditions in which the enzyme was in a buffer containing DTT, and 2-mercaptoethanol was present in the reaction mixtures (as described above). However, with rSULT1A1 that had been pretreated with 1 mM GSSG, four of the OH-PCBs became substrates for the enzyme with the following rates of sulfation (n = 3): 4′-OH-PCB 6 (43.2 ± 9.6 nmol/min/mg protein), 4-OH-PCB 14 (23.2 ± 2.1 nmol/min/mg protein), 4′-OH-PCB 33 (12.1 ± 7.2 nmol/min/mg protein), and 4′-OH-PCB 36 (9.7 ± 1.6 nmol/min/mg protein). The rSULT2A3 that had been pretreated with either GSH or GSSG did not catalyze sulfation of any of the nine OH-PCBs.
Two of the OH-PCBs that were substrates for rSULT1A1 under reducing conditions, 4′-OH-PCB 9 and 6′-OH-PCB 35, were used to investigate the potential modulation of their sulfation by oxidation of rSULT1A1. As shown in Fig. 3A, sulfation of both OH-PCBs catalyzed by GSSG-pretreated rSULT1A1 was only slightly increased compared with that seen with GSH-pretreated enzyme. Furthermore, the absence of substrate inhibition with 6′-OH-PCB 35 and the presence of substrate inhibition with 4′-OH-PCB 9 were seen in both reduced and oxidized rSULT1A1-catalyzed reactions.
Fig. 3.

Sulfation of OH-PCBs catalyzed by oxidized and reduced rSULT1A1. A, rSULT1A1 that had been either oxidized by pretreatment with 1 mM GSSG (•) or reduced by pretreatment with 1 mM GSH (○ or □) catalyzed the sulfation of 6′-OH-PCB 35 (• or ○) and 4′-OH-PCB 9 (▪ or □). B, rSULT1A1 pretreated with 1 mM GSSG catalyzed the sulfation of 4′-OH-PCB 33 (▵), 4′-OH-PCB 6 (♦), and 4-OH-PCB 14 (▾). Data are the means ± S.E. of duplicate (A) or triplicate (B) determinations.
The sulfation of four OH-PCBs identified as substrates for oxidized rSULT1A1 was further investigated with varying concentrations of each OH-PCB after pretreatment of the enzyme with 1 mM GSSG. Three of these compounds, namely 4′-OH-PCB 6, 4-OH-PCB 14, and 4′-OH-PCB 33, showed concentration-dependent sulfation (Fig. 3B), whereas 4′-OH-PCB 36 showed sulfation only at its limit of solubility, 50 μM. The sulfation of the three OH-PCBs catalyzed by oxidized rSULT1A1 demonstrated a kinetic profile significantly different from that observed with most of the OH-PCBs that were substrates for reduced rSULT1A1.
Reversibility of Oxidized rSULT1A1 in Its Ability to Catalyze Sulfation of 4′-OH-PCB 6. As shown in Table 2, the sulfation of 2-naphthol catalyzed by rSULT1A1 was slightly enhanced by pretreatment of the enzyme with 1 mM GSSG compared with that with 1 mM GSH (p < 0.05). This effect was similar to that observed for the rates of sulfation of 4′-OH-PCB 9 and 6′-OH-PCB 35 catalyzed by oxidized versus reduced rSULT1A1. Upon reducing the oxidized rSULT1A1 by subsequent treatment with 5 mM DTT, the catalytic activity with 2-naphthol as substrate was reduced to the original level as catalyzed by the enzyme pretreated with only DTT. For 4′-OH-PCB 6, pretreatment of rSULT1A1 with 5 mM DTT or 1 mM GSSG led to a more significant difference in its catalytic activity. Under reducing conditions, 4′-OH-PCB 6 was not a substrate for rSULT1A1, but after pretreatment of the enzyme with 1 mM GSSG, the rate of sulfation was 37.3 ± 4.0 nmol product/mg/min. After reduction with 5 mM DTT, the oxidized enzyme no longer catalyzed the sulfation of 4′-OH-PCB 6. Thus, the effects of altering the thiol/disulfide environment of rSULT1A1 were fully reversible for 4′-OH-PCB 6.
TABLE 2.
Reversibility of catalytic changes in rSULT1A1 after oxidative modification of the enzyme
|
Pretreatment of
rSULT1A1a
|
|||
|---|---|---|---|
| 5 mM DTT | 1 mM GSSG | DTT after GSSGb | |
| 2-Naphthol, 250 μM | 38.3 ± 4.7 | 50.2 ± 2.1c | 37.4 ± 1.5 |
| 4′-OH PCB 6, 100 μM | 0 | 37.3 ± 4.0 | 0 |
5 mM DTT and/or 1 mM GSSG was incubated with the enzyme at 25°C for 1 h before use in an assay for sulfotransferase activity. Data are the means and S.D. of three determinations.
One aliquot of GSSG-treated enzyme was further treated with DTT at 25°C for 1 h.
This value is statistically different from that for the group treated with 5 mM DTT alone (p < 0.05 by t test).
Discussion
Metabolic hydroxylation of PCBs to OH-PCBs in humans and other mammals is catalyzed by various isoforms of cytochrome P450 and often represents an initial step in metabolism. Analyses of PCB metabolites in human populations have shown that OH-PCBs are persistent in the blood, and hydroxylation has occurred primarily at the para-position and with a lower frequency at the meta-position (Bergman et al., 1994; Sandau et al., 2000). Studies on rats exposed to PCBs have resulted in similar findings; i.e., 4(4′)-hydroxylated and 3(3′)-hydroxylated PCBs were the main metabolites (Chen et al., 1976; Schnellmann et al., 1984; Haraguchi et al., 2004). It is particularly interesting that the concentrations of these OH-PCB metabolites in blood may, in some cases, be higher than that in the parent PCBs, with additional selective concentration of OH-PCBs in liver and other tissues (Bergman et al., 1994). For example, 1 of 13 OH-PCBs detected in rat plasma after a dose of Aroclor 1254 was 4-OH-2,3,5,3′,4′-pentachlorobiphenyl, and this OH-PCB was observed at concentrations of 0.7 to 1.8 ng/mg lipid in the liver at various time points (Bergman et al., 1994). Our calculations suggest that this is approximately equivalent to a concentration range of 0.1 to 0.3 μM. Although caution is necessary in comparisons between concentrations of OH-PCBs calculated based on tissue lipid content and concentrations used with the purified enzyme, a reasonable conclusion is that the tissue concentrations likely to be seen for the OH-PCBs examined in our current study would be unlikely to have significant effects on the catalytic activity of rSULT2A3. Thus, our results indicate that the major family 2 SULT in rat liver differs significantly from the major family 2 SULT in human liver, hSULT2A1, in its interactions with OH-PCBs. For example, our recent studies indicate that 4-OH-PCB 34 and 4′-OH-PCB 68 are good substrates for hSULT2A1 and 4′-OH-PCB 9 is a potent inhibitor of hSULT2A1 (Liu et al., 2006).
In contrast with rSULT2A3, the major hepatic family 1 SULT in the rat, rSULT1A1, had significant interactions with OH-PCBs. For those OH-PCBs that were substrates for rSULT1A1, the rate of product formation would depend directly on the concentration of the OH-PCB, because they would probably be present at nonsaturating concentrations. Some inhibitory OH-PCBs, particularly those with IC50 values in the submicromolar range, may be candidates for examination of in vivo inhibition of rSULT1A1.
These results with rSULT1A1 can be compared with those in a previous report on human SULT1A1 (Wang et al., 2006), in which 18 OH-PCBs were found to inhibit the sulfation of 4-nitrophenol catalyzed by recombinant hSULT1A1 and by human liver cytosol, and at least three of these also served as substrates. Several OH-PCBs used in that study were also included in our experiments, and there are similarities in the interactions of these OH-PCBs with human and rat SULT1A1. However, there are also some notable differences. Unlike rSULT1A1, which was most potently inhibited by OH-PCBs bearing the 3,5-dichloro-4-hydroxy substitution pattern (e.g., 4-OH-PCB 14, 4-OH-PCB 34, and 4-OH-PCB 36), OH-PCBs with a 3,5-dichloro-4-hydroxy substitution pattern showed slightly weaker inhibition of hSULT1A1 than 3-chloro-4-hydroxy substituted PCBs (Wang et al., 2005). In this regard, the rSULT1A1 is more similar to hSULT1E1 (human estrogen sulfotransferase), in which the 3,5-dichloro-4-hydroxy substitution pattern in OH-PCBs provided the most potent inhibition of hSULT1E1 (Kester et al., 2000).
Taken together, the above findings with OH-PCBs indicate that the major hepatic family 2 SULTs in the rat and human (rSULT2A3 and hSULT2A1, respectively) are distinctly different in their interactions with OH-PCBs, whereas the interactions of OH-PCBs with rSULT1A1 have more similarities to the human family 1 SULTs than differences. It should be noted that this result does not necessarily mean that all family 2 SULTs in the rat do not interact strongly with OH-PCBs, as the possibility of other rat SULT2 isoforms interacting with OH-PCBs cannot be excluded in the present study.
The concentrations of OH-PCBs present in blood and tissues are important determinants of their interactions with SULTs. It is becoming increasingly apparent that concentrations of OH-PCBs vary significantly with species and tissue. Moreover, the roles of protein binding of OH-PCBs are receiving increasing attention as factors influencing these in vivo concentrations. For example, recent studies on placental transfer of PCBs and OH-PCBs in humans (Park et al., 2008) show that OH-PCBs are more efficiently transferred than the parent PCBs. This increased placental transfer to the fetus was proposed to be due to higher protein binding of OH-PCBs as opposed to the greater lipid distribution of the parent PCBs (Park et al., 2008). Further elaboration of differences in the transport and tissue concentrations of OH-PCBs will undoubtedly facilitate analysis of potential in vivo interactions with SULTs.
The kinetic characteristics of various OH-PCBs as substrates for rSULT1A1 under standard assay (reduced) conditions demonstrated significant variations, with most showing substrate inhibition. However, the kinetic data for OH-PCBs with substrate inhibition were not described well by a simple Michaelis-Menten model. 4-Nitrophenol, a prototype substrate for rSULT1A1 that has been extensively studied, displays pronounced substrate inhibition in reactions catalyzed by reduced rSULT1A1, and this has been classified as a form of uncompetitive substrate inhibition (Marshall et al., 2000). The underlying mechanism for this kinetic behavior is the formation of a dead-end ternary complex: E-PAP-ROH (where E is the rSULT1A1, PAP is the reaction product derived from PAPS, and ROH is a phenolic substrate) (Marshall et al., 2000). The binary complex of PAP bound to reduced rSULT1A1 (i.e., E-PAP) is relatively stable, and the binding of different phenolic substrates (e.g., 4-nitrophenol, OH-PCBs, and others) to E-PAP may have differential effects on the stability of the inhibitory ternary complex (Marshall et al., 2000; Duffel et al., 2001).
Compared with reduced rSULT1A1, oxidized rSULT1A1 exhibits profoundly altered kinetic interactions with its substrates. As exemplified with 4-nitrophenol and 2-naphthol, changes in pH optima (from 5.2 to 6.3 and 5.4 to 7.2, respectively) and disappearance of substrate inhibition have been observed upon oxidation of rSULT1A1 with GSSG (Marshall et al., 1997, 1998, 2000). Additional changes in substrate specificity were seen upon oxidation of the enzyme with GSSG (Marshall et al., 2000). Upon treatment of rSULT1A1 with GSSG in the present study, we observed the conversion of four of the nine inhibitory OH-PCBs to substrates. The underlying structural modification responsible for the altered kinetic behavior upon treatment of rSULT1A1 with GSSG for short time periods has been shown to be the formation of a glutathione-protein mixed disulfide at Cys66 followed by the formation of an intramolecular disulfide between Cys66 and Cys232 (Marshall et al., 1997). As rSULT1A1 is oxidized further, e.g., treatment with 1 mM GSSG at 25°C for more than 1 h, additional cysteines form disulfide bonds and the specific activity of the enzyme is decreased; after treatment for 12 to 24 h, all five cysteines are oxidized, and the enzyme is completely inactivated (Marshall et al., 1997, 2000). Homology modeling studies indicate that oxidation of Cys66 and Cys232 affects the conformation of the protein in the vicinity of the PAPS/PAP binding site, and therefore changes the dissociation of the E-PAP-ROH dead-end complex (Duffel et al., 2001). Thus, under oxidative conditions, the ternary complex E-PAP-ROH may undergo nucleotide exchange with PAPS and the catalytic cycle can proceed (Marshall et al., 2000). As noted in the previous studies on the mechanism of rSULT1A1 under reduced and oxidized conditions, the changes due to initial disulfide formation are reversible (Marshall et al., 1997, 2000). Indeed, our results also indicated that GSSG-dependent changes in the kinetic behavior of rSULT1A1 with 4′-OH-PCB 6 were reversible by reduction with DTT (Table 2).
As described in the original studies on the effect of oxidation on mechanism of the enzyme (Marshall et al., 1997, 2000), these effects of partial oxidation of cysteines in rSULT1A1 suggest an important regulatory mechanism whereby the specificity and kinetics of the enzyme can be altered by oxidative stress. This mechanism is particularly intriguing in the case of the OH-PCBs examined in the current study, because, for some OH-PCBs, cellular oxidative stress may determine whether the molecule inhibits rSULT1A1 or is sulfated in a reaction catalyzed by the enzyme. In addition to oxidative stress created by disease states or exposure to other xenobiotics, recent work on the potential role of quinone metabolites of PCBs and their involvement in creation of oxidative stress within cells (Amaro et al., 1996; Srinivasan et al., 2002) suggests that, by inducing oxidative stress, some OH-PCBs may influence sulfation of other OH-PCBs or interfere with other sulfation reactions. Finally, the recent report of disulfide-mediated regulation of hSULT1E1 (Maiti et al., 2007) and the presence of cysteine residues homologous to Cys66 in other SULTs suggest that the substrate-dependent nature of the effects seen with the OH-PCBs in our current results may also be seen in the redox regulation of other SULTs.
Acknowledgments
The contents of this article are solely the responsibility of the authors and do not necessarily represent the official views of the National Institutes of Health.
This work was supported in part by the National Institutes of Health National Institute of Environmental Health Sciences [Grants P42-ES013661, K25-ES012475, P30-ES05605]; and the National Institutes of Health National Cancer Institute [Grant R01-CA038683]. We also acknowledge programmatic support through the University of Iowa Environmental Health Sciences Research Center.
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
doi:10.1124/dmd.108.026021.
ABBREVIATIONS: PCB, polychlorinated biphenyl; OH-PCB, hydroxylated polychlorinated biphenyl; SULT, sulfotransferase; h, human; r, rat; DHEA, dehydroepiandrosterone; GC, gas chromatography; MS, mass spectrometry; PAPS, adenosine 3′-phosphate 5′-phosphosulfate; PAP, adenosine 3′,5′-diphosphate; DTT, dithiothreitol; DTNB, 5,5′-dithiobis(2-nitrobenzoic acid); GSH, reduced glutathione; GSSG, oxidized glutathione.
References
- Abramowicz DA (1995) Aerobic and anaerobic PCB biodegradation in the environment. Environ Health Perspect 103 (Suppl 5): 97–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amaro AR, Oakley GG, Bauer U, Spielmann HP, and Robertson LW (1996) Metabolic activation of PCBs to quinones: reactivity toward nitrogen and sulfur nucleophiles and influence of superoxide dismutase. Chem Res Toxicol 9 623–629. [DOI] [PubMed] [Google Scholar]
- Bergman A, Klasson-Wehler E, and Kuroki H (1994) Selective retention of hydroxylated PCB metabolites in blood. Environ Health Perspect 102 464–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blomquist CH, Kotts CE, and Hakanson EY (1978) A simple method for detecting steroid aggregation and estimating solubility in aqueous solutions. Anal Biochem 87 631–635. [DOI] [PubMed] [Google Scholar]
- Chen PR, McKinney JD, and Matthews HB (1976) Metabolism of 2,4,5,2′,5′-pentachlorobiphenyl in the rat: qualitative and quantitative aspects. Drug Metab Dispos 4 362–367. [PubMed] [Google Scholar]
- Chen X, Yang YS, Zheng Y, Martin BM, Duffel MW, and Jakoby WB (1992) Tyrosine-ester sulfotransferase from rat liver: bacterial expression and identification. Protein Expr Purif 3 421–426. [DOI] [PubMed] [Google Scholar]
- Duffel MW, Binder TP, and Rao SI (1989) Assay of purified aryl sulfotransferase suitable for reactions yielding unstable sulfuric acid esters. Anal Biochem 183 320–324. [DOI] [PubMed] [Google Scholar]
- Duffel MW, Marshal AD, McPhie P, Sharma V, and Jakoby WB (2001) Enzymatic aspects of the phenol (aryl) sulfotransferases. Drug Metab Rev 33 369–395. [DOI] [PubMed] [Google Scholar]
- Guvenius DM, Hassanzadeh P, Bergman A, and Norén K (2002) Metabolites of polychlorinated biphenyls in human liver and adipose tissue. Environ Toxicol Chem 21 2264–2269. [PubMed] [Google Scholar]
- Haraguchi K, Kato Y, Koga N, and Degawa M (2004) Metabolism of polychlorinated biphenyls by Gunn rats: identification and serum retention of catechol metabolites. Chem Res Toxicol 17 1684–1691. [DOI] [PubMed] [Google Scholar]
- James MO (2001) Polychlorinated biphenyls: Metabolism and metabolites, in PCBs: Recent Advances in Environmental Toxicology and Health Effects (Robertson LW and Hansen LG eds) pp 35–46, The University Press of Kentucky, Lexington.
- Jocelyn PC (1987) Spectrophotometric assay of thiols. Methods Enzymol 143 44–67. [DOI] [PubMed] [Google Scholar]
- Kaminsky LS, Kennedy MW, Adams SM, and Guengerich FP (1981) Metabolism of dichlorobiphenyls by highly purified isozymes of rat liver cytochrome P-450. Biochemistry 20 7379–7384. [DOI] [PubMed] [Google Scholar]
- Kester MH, Bulduk S, Tibboel D, Meinl W, Glatt H, Falany CN, Coughtrie MW, Bergman A, Safe SH, Kuiper GG, et al. (2000) Potent inhibition of estrogen sulfotransferase by hydroxylated PCB metabolites: a novel pathway explaining the estrogenic activity of PCBs. Endocrinology 141 1897–1900. [DOI] [PubMed] [Google Scholar]
- Lehmler HJ and Robertson LW (2001) Synthesis of hydroxylated PCB metabolites with the Suzuki coupling. Chemosphere 45 1119–1127. [DOI] [PubMed] [Google Scholar]
- Liu Y, Apak TI, Lehmler HJ, Robertson LW, and Duffel MW (2006) Hydroxylated polychlorinated biphenyls are substrates and inhibitors of human hydroxysteroid sulfotransferase SULT2A1. Chem Res Toxicol 19 1420–1425. [DOI] [PubMed] [Google Scholar]
- Ludewig G, Esch H, and Robertson LW (2007) Polyhalogenierte bi- und terphenyle, in Handbuch der Lebensmitteltoxicologie (Dunkelberg H, Gebel T and Hartwig A eds), pp 1031–1094, Wiley-VCH, Weinheim.
- Maiti S, Grant S, Baker SM, Karanth S, Pope CN, and Chen G (2004) Stress regulation of sulfotransferases in male rat liver. Biochem Biophys Res Commun 323 235–241. [DOI] [PubMed] [Google Scholar]
- Maiti S, Zhang J, and Chen G (2007) Redox regulation of human estrogen sulfotransferase (hSULT1E1). Biochem Pharmacol 73 1474–1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall AD, Darbyshire JF, Hunter AP, McPhie P, and Jakoby WB (1997) Control of activity through oxidative modification at the conserved residue Cys66 of aryl sulfotransferase IV. J Biol Chem 272 9153–9160. [DOI] [PubMed] [Google Scholar]
- Marshall AD, Darbyshire JF, McPhie P, and Jakoby WB (1998) A review of the effects of manipulation of the cysteine residues of rat aryl sulfotransferase IV. Chem-Biol Interact 109 107–116. [DOI] [PubMed] [Google Scholar]
- Marshall AD, McPhie P, and Jakoby WB (2000) Redox control of aryl sulfotransferase specificity. Arch Biochem Biophys 382 95–104. [DOI] [PubMed] [Google Scholar]
- Master ER, Lai VW, Kuipers B, Cullen WR, and Mohn WW (2002) Sequential anaerobic-aerobic treatment of soil contaminated with weathered Aroclor 1260. Environ Sci Technol 36 100–103. [DOI] [PubMed] [Google Scholar]
- Matthews HB and Anderson MW (1975) The distribution and excretion of 2,4,5,2′,5′-pentachlorobiphenyl in the rat. Drug Metab Dispos 3 211–219. [PubMed] [Google Scholar]
- McLean MR, Bauer U, Amaro AR, and Robertson LW (1996) Identification of catechol and hydroquinone metabolites of 4-monochlorobiphenyl. Chem Res Toxicol 9 158–164. [DOI] [PubMed] [Google Scholar]
- Park JS, Bergman A, Linderholm L, Athanasiadou M, Kocan A, Petrik J, Drobna B, Trnovec T, Charles MJ, and Hertz-Picciotto I (2008) Placental transfer of polychlorinated biphenyls, their hydroxylated metabolites and pentachlorophenol in pregnant women from eastern Slovakia. Chemosphere 70 1676–1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson LW and Hansen LG eds (2001) PCBs: Recent Advances in Environmental Toxicology and Health Effects, University of Kentucky Press, Lexington.
- Růzicková P, Klánová J, Cupr P, Lammel G, and Holoubek I (2008) An assessment of air-soil exchange of polychlorinated biphenyls and organochlorine pesticides across central and southern Europe. Environ Sci Technol 42 179–185. [DOI] [PubMed] [Google Scholar]
- Safe SH (1994) Polychlorinated biphenyls (PCBs): environmental impact, biochemical and toxic responses, and implications for risk assessment. Crit Rev Toxicol 24 87–149. [DOI] [PubMed] [Google Scholar]
- Sandau CD, Ayotte P, Dewailly E, Duffe J, and Norstrom RJ (2000) Analysis of hydroxylated metabolites of PCBs (OH-PCBs) and other chlorinated phenolic compounds in whole blood from Canadian inuit. Environ Health Perspect 108 611–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnellmann RG, Volp RF, Putnam CW, and Sipes IG (1984) The hydroxylation, dechlorination, and glucuronidation of 4,4′-dichlorobiphenyl (4-DCB) by human hepatic microsomes. Biochem Pharmacol 33 3503–3509. [DOI] [PubMed] [Google Scholar]
- Sekura RD (1981) Adenosine 3′-phosphate 5′-phosphosulfate. Methods Enzymol 77 413–415. [Google Scholar]
- Sheng J, Sharma V, and Duffel MW (2001) Measurement of aryl and alcohol sulfotransferase activity, in Current Protocols in Toxicology, pp 4.5.1–4.5.9, John Wiley & Sons, New York. [DOI] [PubMed]
- Sheng JJ and Duffel MW (2001) Bacterial expression, purification, and characterization of rat hydroxysteroid sulfotransferase STa. Protein Expr Purif 21 235–242. [DOI] [PubMed] [Google Scholar]
- Srinivasan A, Robertson LW, and Ludewig G (2002) Sulfhydryl binding and topoisomerase inhibition by PCB metabolites. Chem Res Toxicol 15 497–505. [DOI] [PubMed] [Google Scholar]
- Tampal N, Lehmler HJ, Espandiari P, Malmberg T, and Robertson LW (2002) Glucuronidation of hydroxylated polychlorinated biphenyls (PCBs). Chem Res Toxicol 15 1259–1266. [DOI] [PubMed] [Google Scholar]
- Wang LQ, Lehmler HJ, Robertson LW, Falany CN, and James MO (2005) In vitro inhibition of human hepatic and cDNA-expressed sulfotransferase activity with 3-hydroxybenzo[a]pyrene by polychlorobiphenylols. Environ Health Perspect 113 680–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang LQ, Lehmler HJ, Robertson LW, and James MO (2006) Polychlorobiphenylols are selective inhibitors of human phenol sulfotransferase 1A1 with 4-nitrophenol as a substrate. Chem-Biol Interact 159 235–246. [DOI] [PubMed] [Google Scholar]
- Wethington DM 3rd and Hornbuckle KC (2005) Milwaukee, WI, as a source of atmospheric PCBs to Lake Michigan. Environ Sci Technol 39 57–63. [DOI] [PubMed] [Google Scholar]