

Abstract
Objective
Our objective was to delineate the potential role of adipogenesis in insulin resistance and type 2 diabetes. Obesity is characterized by an increase in adipose tissue mass resulting from enlargement of existing fat cells (hypertrophy) and/or from increased number of adipocytes (hyperplasia). The inability of the adipose tissue to recruit new fat cells may cause ectopic fat deposition and insulin resistance.
Research Methods and Procedures
We examined the expression of candidate genes involved in adipocyte proliferation and/or differentiation [CCAAT/enhancer-binding protein (C/EBP) α, C/EBPδ, GATA domain-binding protein 3 (GATA3), C/EBPβ, peroxisome proliferator-activated receptor (PPAR) γ2, signal transducer and activator of transcription 5A (STAT5A), Wnt-10b, tumor necrosis factor α, sterol regulatory element-binding protein 1c (SREBP1c), 11 beta-hydroxysteroid dehydrogenase, PPARG angiopoietin-related protein (PGAR), insulin-like growth factor 1, PPARγ coactivator 1α, PPARγ coactivator 1β, and PPARδ] in subcutaneous adipose tissue from 42 obese individuals with type 2 diabetes and 25 non-diabetic subjects matched for age and obesity.
Results
Insulin sensitivity was measured by a 3-hour 80 mU/m2 per minute hyperinsulinemic glucose clamp (100 mg/dL). As expected, subjects with type 2 diabetes had lower glucose disposal (4.9 ± 1.9 vs. 7.5 ± 2.8 mg/min per kilogram fat-free mass; p < 0.001) and larger fat cells (0.90 ± 0.26 vs. 0.78 ± 0.17 μm; p = 0.04) as compared with obese control subjects. Three genes (SREBP1c, p < 0.01; STAT5A, p = 0.02; and PPARγ2, p = 0.02) had significantly lower expression in obese type 2 diabetics, whereas C/EBPβ only tended to be lower (p = 0.07).
Discussion
This cross-sectional study supports the hypothesis that impaired expression of adipogenic genes may result in impaired adipogenesis, potentially leading to larger fat cells in subcutaneous adipose tissue and insulin resistance.
Keywords: insulin resistance, gene expression, adipogenesis, fat cell size, type 2 diabetes
Introduction
The World Health Organization estimates that by the year 2025, there will be ∼300 million individuals affected by type 2 diabetes worldwide (1). Although the exact molecular mechanisms are not fully understood, it is clear that obesity constitutes a major risk for the development of insulin resistance and type 2 diabetes (2). There is, however, considerable evidence that individuals with similar degrees of obesity can have strikingly different risks of developing diabetes (3). Therefore, understanding the underlying mechanisms of insulin resistance and the transition from normal glucose tolerance to type 2 diabetes in individuals at risk will help to develop new therapeutic strategies for the treatment and eventually the prevention of diabetes.
A major function of the adipose organ is to store excess energy as triglycerides under conditions of nutrient excess (4). However, in response to prolonged periods of calorie excess, the adipose organ may become overloaded and unable to recruit new fat cells, resulting in adipose tissue hypertrophy of existing fat cells and increased ectopic fat deposition in tissues such as skeletal muscle, liver, myocardium, and pancreas. For example, independent of total body fat, Pima Indians with larger abdominal fat cells are more insulin resistant and more likely to develop diabetes than those with smaller fat cells (5,6). In a recent review, Shulman (7) hypothesized that insulin resistance develops because of alterations in the partitioning of fat between adipose tissue and muscle or liver. It is also now recognized that a lack of adipose tissue is similarly associated with insulin resistance and increased risk for development of type 2 diabetes (8–10). Similarly, in humans, lipodystrophy is associated with insulin resistance and often type 2 diabetes (11). Recently, Danforth (12) proposed that impaired adipocyte proliferation and differentiation may cause the progressive filling of existing adipocytes, leading to overflow of excess calories as fat into other tissues and insulin resistance. Thus, it is hypothesized that insulin resistance and eventually full-blown type 2 diabetes can be triggered by a failure of new adipocytes to differentiate.
Adipose differentiation is a complex process accompanied by coordinated changes in cell morphology, hormone sensitivity, and gene expression. This process of adipogenesis is controlled by interplay of transcription factors including but not limited to peroxisome proliferator-activated receptor (PPAR)1 γ, CCAAT/enhancer-binding proteins (C/EBPs), and adipocyte determination and differentiation factor-1(ADD1)/sterol regulatory element-binding protein 1c (SREBP1). Adipocyte differentiation is characterized by a shift in gene expression between transcripts that determine early stages of the adipocyte proliferation/differentiation process to those transcripts causing the final maturation of adipocytes (13,14).
In the present study, we investigated the potential role of adipose tissue proliferation/differentiation in insulin resistance and type 2 diabetes by measuring the expression of genes involved in adipogenesis in obese type 2 diabetic and obese non-diabetic subjects. More specifically, we analyzed genes involved in early adipocyte differentiation stages such as ADD1/SREBP1c, signal transducer and activator of transcription (STAT) 5A, C/EBPδ, PPARδ, GATA domain-binding protein 3 (GATA3), and C/EBPβ and in terminal differentiation such as C/EBPα and PPARγ2. In addition, we studied genes related to the regulation of PPARγ such as the PPARγ coactivator 1 (α and β), the expression of genes involved in the inhibition of adipocyte differentiation, including the signaling molecules Wnt-10b and tumor necrosis factor-α, and genes known to have a positive role in in vitro adipogenesis, insulin-like growth factor-1 and 11 beta-hydroxysteroid dehydrogenase.
Research Methods and Procedures
Subjects
Forty-two obese individuals with type 2 diabetes and 25 obese non-diabetic subjects matched for sex, age, BMI, and race were enrolled in this study (Table 1). All diabetic subjects were participants in Look AHEAD (Baton Rouge, LA, Pittsburgh, PA, and New York, NY), a randomized multicenter, controlled trial of a lifestyle intervention for weight loss in overweight or obese adults (45 to 75 years old) with type 2 diabetes (15). Subjects were studied before a lifestyle intervention for weight loss. Obese type 2 diabetic subjects were required to have fasting blood glucose < 180 mg/dL, a BMI between 27 and 35 kg/m2, and untreated with insulin or thiazolidinedione. The non-diabetic subjects were recruited separately. The Institutional Review Boards of the three clinical sites (see Appendix) approved the study. Participants were informed of the nature, purpose, and possible risks of the study, and provided written consent to participate.
Table 1.
Clinical and metabolic characteristics of the subjects
| Type 2 diabetics | Non-diabetics | |
|---|---|---|
| N (men/women) | 42 (19/23) | 25 (11/14) |
| Race (white/black) | 34/8 | 20/5 |
| Age (years) | 58 ± 8 | 54 ± 8 |
| Weight (kg) | 96 ± 10 | 95.4 ± 14 |
| BMI (kg/m2) | 33.8 ± 2.4 | 32.2 ± 2.1 |
| Body fat (%)* | 37 ± 6 | 37 ± 8 |
| Ratio VAT/SAT | 0.87 ± 0.54 | 0.72 ± 0.47 |
| Fat cell size (μL) | 0.90 ± 0.26 | 0.78 ± 0.17† |
| Fat cell number/mg tissue | 3578 ± 1103 | 3565 ± 1009 |
| Fasting glucose (mg/dL) (mM) (SI units) |
145 ± 27 8 ± 1.5 |
100 ± 7‡ 6 ± 0.4‡ |
| Fasting Insulin (μU/mL) (pM) (SI units) |
12 ± 4 83 ± 28 |
10 ± 7 69 ± 49 |
| HbA1c (%) | 6.9 ± 1.0 | 5.3 ± 0.7‡ |
| Clamp study (last 30 minutes) | ||
| Glucose concentration (mg/dl) (mM) (SI units) |
103 ± 6 6 ± 0.3 |
107 ± 5† 5.9 ± 0.3† |
| Insulin concentration (μU/mL) (pM) (SI units) |
158 ± 40 1097 ± 278 |
182 ± 47† 1264 ± 326† |
| Glucose disposal (mg/min per kilogram FFM) | 4.9 ± 1.9 | 7.5 ± 2.8‡ |
VAT, visceral adipose tissue; SAT, subcutaneous adipose tissue; SI, systéme internationale; HbA1c, hemoglobin A1c; FFM, fat-free mass.
DXA was used to assess percentage body fat. Data are mean ± standard deviation.
p < 0.05.
p < 0.001.
Experimental Protocol
Subjects were admitted to the respective inpatient clinics at 4 PM on the day before the fat biopsy and clamp procedures. Body composition was measured in the late afternoon by DXA using the Hologic QDR 4500A whole-body scanner (Hologic, Bedford, MA), and participants were fed a standard dinner between 6 and 7 PM. After an overnight fast (6:30 to 7:30 AM), a 250- to 350-mg sample of adipose tissue was obtained by needle biopsy after local anesthesia (5 mL of 50%–50% mixture of lidocaine and bupivacaine). Samples of superficial subcutaneous adipose tissue (SAT) were obtained lateral to the umbilicus, cleaned of visible connective tissues and vascular elements, and washed in sterile phosphate-buffered saline before snap freezing in liquid nitrogen. Another 50-mg sample was placed in osmium tetraoxide. Fat cell size and number, RNA extraction, gene expression, and clinical chemistry were all conducted at the Pennington Biomedical Research Center. Insulin sensitivity was assessed by a 3-hour hyperinsulinemic euglycemic clamp with a primed insulin infusion at 80 mU/m2 per minute, resulting in a plasma insulin concentration of ∼170 μU/mL. Briefly, an intravenous catheter was placed in an antecubital vein for infusion of insulin and glucose, and a second catheter was placed retrograde in the dorsal vein of the contralateral hand for blood withdrawal at -25, -15, and -5 minutes (baseline) and 145, 160, and 175 minutes (clamp). Glucose was clamped at 100 mg/dL by a variable glucose infusion (20% dextrose; Baxter Healthcare, Deerford, IL).
Fat Cell Size
Fat cell size and number was measured as previously described (16). Briefly, adipose tissue (≈50 mg) was fixed in a solution containing collidine HCL (0.2 M) and osmium tetraoxide (31 mg/mL collidine HCL buffer). After fixation, the samples were diluted in 154 mM NaCl, filtered over a 10-micron nylon screen, recollected in 10 mL of 154 mM NaCl, and dissociated over 1 week by the addition of 10 mL of a solution containing 8 M urea and 154 mM NaCl. The samples were then filtered through a 250-micron nylon filter into a weighed beaker and counted on a Multisizer-3 (Beckman Coulter, Fullerton, CA) using a 400-μm aperture (dynamic linear range, 12 to 320 μm). Average cell size for each sample was calculated for cells > 22 μm and expressed in microliters. Adipocyte cell number (cells per milligram wet weight of tissue) is then determined from the amount of sample (milliliters), the quantity of cells (per milliliter), and the tissue weight (milligrams).
Real-Time Quantitative Reverse Transcription (RT)-Polymerase Chain Reaction (PCR)
Total RNA was isolated following the method of Chomczynski and Sacchi (17) and using the total RNA extraction kit from Qiagen (Valencia, CA). The quality and quantification of RNA were determined by spectrophotometry at 260/280 nm. The average yield of total RNA was 1.37 ± 0.17 μg/100 mg adipose tissue (wet weight), and the average optical density ratio 260/280 was 1.87 ± 0.02. The Taqman real-time RT-PCR technique was used to quantify the mRNA level of each gene (Applied Biosystems, Foster City, CA). RT-PCR was carried out using 30 ng of total RNA on a Bio-Rad I Cycler (Bio-Rad, Hercules, CA). PCR conditions were 48 °C for 30 minutes and 95 °C for 10 minutes, followed by 40 cycles of 95 °C for 15 seconds and 60 °C for 1 minute. Each gene expression was normalized for cyclophilin A expression. No significant difference was observed in cyclophilin A expression between obese type 2 diabetics and obese non-diabetic subjects. A standard curve for each primer probe set was generated by serial dilution of adipose tissue RNA. Each sample was run in duplicate, and the mean value was normalized for the cyclophilin transcript level. Primers and probes sequences of the selected genes are listed in Table 2.
Table 2.
Sequences of primers and probes used for RT-PCR
| Genes | Primers | Probes |
|---|---|---|
| C/EBPα F | 5′-agggtctctagttccacgcc-3′ | C/EBPα 5′tcccacctccctccgcacacacc-3′ |
| C/EBPα R | 5′-caaggggaagcccagcctata-3′ | |
| C/EBPδ F | 5′-cggcagttcttcaagcagct-3′ | C/EBPδ 5′cccagcccgcccttcctgcc-3′ |
| C/EBPδ R | 5′-gggtctgaggtatgggtcgtt-3′ | |
| C/EBPβ F | 5′-ggcagcaccacgacttcct-3′ | C/EBPβ 5′cgacctcttctccgacgactacggg-3′ |
| C/EBPβ R | 5′-cgccccaggctcacgtag-3′ | |
| PPARδ F | 5′-agcatcctcaccggcaaa-3′ | PPARδ 5′-ccagccacacggcgccct-3′ |
| PPARδ R | 5′-ttctctgcctgccacaatg-3′ | |
| PPARγ2 F | 5′-tagatgacagcgacttggcaatat-3′ | PPARγ2 5′-cagtggagaccgcccaggtttgct-3′ |
| PPARγ2 R | 5′-gaatgtcttcaatgggcttcaca-3′ | |
| ANGPTL4F | 5′-ccacttgggaccaggatcac-3′ | ANGPTL4 5′-aactgcgccaagagcctctctggag-3′ |
| ANGPTL4 R | 5′-atggctgcaggtgccaaa-3′ | |
| PGC1-α F | 5′-tcctcttcaagatcctgctattac-3′ | PGC1-α 5′-aagccactacagacaccgcacgca-3′ |
| PGC1-α R | 5′-gacggctgtagggcgatc-3′ | |
| PGC1-β F | 5′-tgcaagagccctgagtatgac-3′ | PGC1-β 5′-cgccgctgctgctgctgctg-3′ |
| PGC1-β R | 5′-ctctgggaggaagctgctc-3′ | |
| SREBP-1c F | 5′-ggtgagtggcggaaccatc-3′ | SREBP-1c 5′-caacagtcccactggtcgtagatgcg-3′ |
| SREBP-1c R | 5′-gccggttgataggcagctt-3′ | |
| STAT5A F | 5′-gcatcaccatcgcctggaag-3′ | STAT5A 5′-tgactccccggaacgcaacctgt-3′ |
| STAT5A R | 5′-gcggtcaggaaacacatagatgag-3′ | |
| Wnt-10b F | 5′-tgatacccacaaccgcaattct-3′ | Wnt-10b 5′-agccccgtctgcgtccccgt-3′ |
| Wnt-10b R | 5′-tctcgctcacagaagtcagga-3′ | |
| TNFα F | 5′-gtgctcctcacccacaccat-3′ | TNFα 5′-agccgcatcgccgtctcctacc-3′ |
| TNFα R | 5′-gggctcataccagggcttgg-3′ | |
| IGF1 F | 5′-cctcctcgcatctcttctacct-3′ | IGF1 5′-cgctgtgcctgctcaccttcacca-3′ |
| IGF1 R | 5′-gttgaaataaaagcccctgtctcc-3′ | |
| 11βHSD-1 F | 5′-ggcttatcatctggcgaagatg-3′ | 11βHSD-1 5′-acctcgctgtcaccaccacatggg-3′ |
| 11βHSD-1 R | 5′-ggataccaccttctgtagagtttctttt-3′ | |
| GATA3 F | 5′-ggaggtggatgtgctttttaaca-3′ | GATA3 5′-acggtcaaggcaaccacgtccc-3′ |
| GATA3 R | 5′-ccctgaccgagtttccgtagt-3′ |
RT-PCR, reverse transcription-polymerase chain reaction.
Statistical Analyses
All values are presented as mean ± standard deviation. Statistical analyses were performed using the SPSS/PC statistical program (version 11.0.1 for Windows; SPSS, Inc., Chicago, IL). Differences between groups were compared by one-way ANOVA and subsequent independent Student’s t tests. Relationships between metabolic variables and gene expression were assessed by Pearson’s correlation analysis or by general linear modeling. The values for metabolic variables and mRNA expression levels were all normally distributed except for C/EBPβ, in which case a Spearman’s correlation analysis was performed.
Results
Anthropometric and metabolic characteristics of the subjects are described in Table 1 and in Table 3 for the differences observed between men and women. By experimental design, age, BMI, percentage body fat, sex, and race ratios were similar in the two groups (type 2 diabetic and non-diabetic subjects). By definition, fasting plasma glucose concentrations were higher in obese individuals with type 2 diabetes (145 ± 27 mg/dL) compared with obese non-diabetic subjects (100 ± 7 mg/dL, p < 0.001). Similarly, glucose disposal rates during the hyperinsulinemic glucose clamp were lower in obese subjects with type 2 diabetes [4.9 ± 1.9 mg/min per kilogram fat-free mass (FFM)] as compared with obese non-diabetic subjects (7.5 ± 2.8 mg/min per kilogram FFM, p < 0.001). The glucose disposal in diabetic subjects was significantly lower only in whites (p < 0.0001) but not in African Americans. The percentage of hemoglobin A1c (HbA1c) was higher in type 2 diabetic vs. non-diabetic subjects (6.9 ± 1.0 vs. 5.3 ± 0.7, p < 0.001); however, no difference in HbA1c percentage has been observed considering the sex or race phenotypes. Average fat cell size was larger in individuals with type 2 diabetes (0.90 ± 0.26 μL) as compared with obese non-diabetic subjects (0.78 ± 0.17 μL, p = 0.04), with a significant difference between men and women in type 2 diabetic subjects (0.76 ± 0.23 and 1.01 ± 0.24 μL respectively, p < 0.001) and in non-diabetic subjects (0.68 ± 0.14 in men vs. 0.84 ± 0.16 μL in women, p < 0.01). However, fat cell size was similar in whites and African Americans. Fat cell size was positively correlated with percentage body fat in the entire group of subjects (data not shown; r = 0.52, p < 0.0001), in whites (r = 0.52, p < 0.0001), and in African Americans (r = 0.83, p < 0.0001). When men and women were considered separately, the correlation between fat cell size and percentage body fat was significant only in men (r = 0.47, p < 0.01) but not in women (r = 0.15, p = not significant). Furthermore, fat cell size was negatively correlated with glucose disposal rate in the entire group (r = -0.31, p = 0.01; Figure 1), with no difference observed between men and women (r = -0.36, p = 0.04) (data not shown). However, this correlation was only significant in whites (r = -0.35, p = 0.01).
Table 3.
Clinical and metabolic characteristics of the subjects separate by sex
| Men | Women | |
|---|---|---|
| N | 30 | 37 |
| Race (white/black) | 27/3 | 27/10 |
| Age (years) | 58 ± 9 | 56 ± 8 |
| Weight (kg) | 104 ± 9 | 89 ± 8‡ |
| BMI (kg/m2) | 32.6 ± 2.2 | 33.6 ± 2.5 |
| Body fat (%)* | 30 ± 4 | 42 ± 3‡ |
| Ratio VAT/SAT | 1.09 ± 0.61 | 0.59 ± 0.28‡ |
| Fat cell size (μL) | 0.73 ± 0.20 | 0.95 ± 0.22‡ |
| Fat cell number/mg tissue | 3733 ± 1022 | 3460 ± 1085 |
| Fasting glucose (mg/dl) (mM) (SI units) |
128 ± 29 8 ± 1.7 |
128 ± 33 8 ± 2 |
| Fasting insulin (μU/mL) (pM) (SI units) |
10 ± 4 69 ± 28 |
13 ± 6† 90 ± 42† |
| HbA1c (%) | 6.2 ± 1.2 | 6.4 ± 1.2 |
| Clamp study (last 30 minutes) | ||
| Glucose concentration (mg/dl) (mM) (SI units) |
104 ± 6 6 ± 0.3 |
105 ± 6 6.2 ± 0.3 |
| Insulin concentration (μU/mL) (pM) (SI units) |
156 ± 38 1083 ± 264 |
178 ± 47† 1236 ± 326† |
| Glucose disposal (mg/min per kilogram FFM) | 5.8 ± 2.6 | 5.9 ± 2.7 |
VAT, visceral adipose tissue; SAT, subcutaneous adipose tissue; SI, systéme internationale; HbA1c, hemoglobin A1c; FFM, fat-free mass.
DXA was used to assess percent body fat; data are mean ± standard deviation.
p < 0.05.
p < 0.001.
Figure 1.
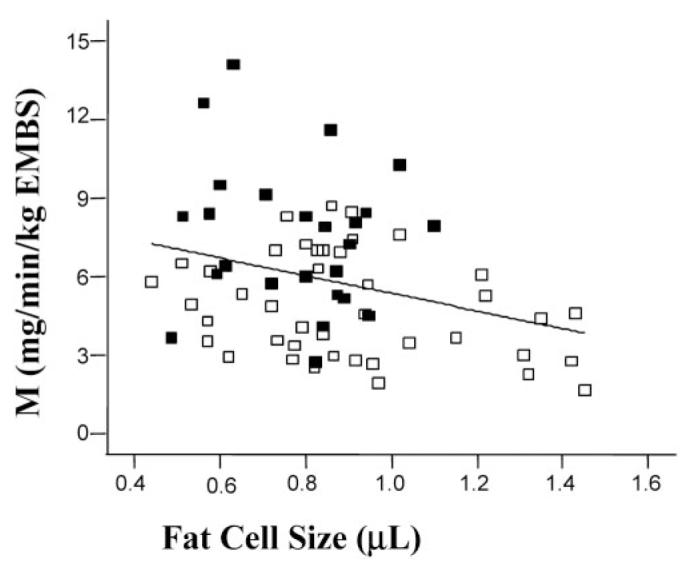
Insulin sensitivity negatively correlated to fat cells size: obese subjects with type 2 diabetes (open squares) vs. obese non-diabetic individuals (solid squares). Glucose disposal rate (M) calculated and normalized to estimate metabolic body size (estimated metabolic body size = FFM + 17.7 kg).
The ratio of visceral adipose tissue (VAT) vs. SAT (r-VAT/SAT) was not significantly different between type 2 diabetic and non-diabetic subjects but was higher in women compared with men either in all subjects or by considering separately type 2 diabetic or non-diabetic subjects (1.09 ± 0.61 in men vs. 0.59 ± 0.28 in women, p < 0.001 in the entire group). Indeed, we observed in our study that women show significant higher SAT percentage (64 ± 10%, p < 0.0001) compared with men (51± 12%). We observed in our group that white subjects have a higher r-VAT/SAT compared with black subjects (0.89 ± 53 vs. 0.52 ± 0.30, p = 0.02). On the other hand, we found a trend toward a negative correlation between r-VAT/SAT and glucose disposal rate and r-VAT/SAT and fat cell size in the entire group (r = -0.22, p = 0.7 and r = -0.23, p = 0.05). Interestingly, data showed in the entire group that gene expression levels of PPARγ2 and SREBP1c were negatively correlated with r-VAT/SAT (r = -0.27, p = 0.04; r = -0.25, p = 0.05, respectively) and that C/EBPβ gene expression level was positively correlated with r-VAT/SAT (r = 0.27, p = 0.04) (data not shown). Only C/EBPβ gene expression level showed significant positive correlation with r-VAT/SAT when analyzing type 2 diabetic (r = 0.42, p = 0.009, data not shown) compared with non-diabetic subjects.
Regarding the expression level of adipogenic genes, we found that PPARγ2 (p = 0.02), SREBP1c (p < 0.01), and STAT5A (p = 0.02) were significantly lower in obese individuals with type 2 diabetes as compared with non-diabetic obese subjects (Figure 2), whereas the expression of C/EBPβ only tended to be lower in obese type 2 diabetic individuals (p < 0.07; Figure 2). A weak positive correlation between glucose disposal rate and PPARγ2 mRNA level was observed in the entire group (r = 0.25, p = 0.05) but only significant in white subjects (r = 0.32, p = 0.02). Similar correlations existed between glucose disposal rate and SREBP1c mRNA expression. Finally, glucose disposal tended to correlate with STAT5a mRNA in whites only (r = 0.27, p = 0.06).
Figure 2.

Adipose tissue gene expression results in obese subjects with type 2 diabetic (hatched bars) compared with obese non-diabetic subjects individuals (solid bars).
No significant association has been observed between adipogenic genes mRNA levels and HbA1c percentage. Fat cell size correlated negatively with SREBP1c expression in men (r = -0.5, p < 0.01) but not in women (r = -0.1, p = not significant) and negatively with wnt-10b in the entire group (r = -0.26, p = 0.05) but only significantly in white subjects (r = -0.35, p = 0.015). There was no difference between groups for the other measured genes.
Discussion
Adipocytes are highly specialized cells that play an important role in energy metabolism and insulin sensitivity. People with enlarged subcutaneous adipocytes are more hyperinsulinemic and glucose intolerant and are at increased risk for the development of type 2 diabetes, relative to those with similar degrees of adiposity but smaller adipocytes (5,6). In the present study, we confirm that independent of total body fat and sex, subjects with type 2 diabetes have larger adipocytes but similar number of fat cells compared with non-diabetic individuals and that insulin sensitivity is inversely related to fat cell size. More importantly, we report that the expression level of some genes involved in adipogenesis is blunted in subjects with type 2 diabetes compared with obese non-diabetic individuals. Taken together, these results provide indirect support to the hypothesis that individuals with impaired potential for adipogenesis may develop larger fat cells, insulin resistance, and eventually type 2 diabetes (12).
The metabolic syndrome is known to affect races differently, especially with a high impact in African Americans (18–20). Because of our limited sample size, our study does not allow us to elucidate potential differences in the relationships among fat cell size, propensity of fat cell precursors to proliferate and differentiate, and insulin sensitivity in African American vs. whites. Because African-American subjects have clearly reduced insulin sensitivity and higher risks of developing diabetes (21,22), more mechanistic studies are needed to understand the reasons for such differences, including studies of their adipose tissue.
Adipogenesis is a complex process accompanied by coordinated changes in cell morphology and gene expression. SREBP1c, STAT5A, and PPARγ2 had significantly lower expression in obese individuals with type 2 diabetes as compared with non-diabetic obese subjects. Also, C/EBPβ expression tended to be lower. SREBP1c, STAT5A, and C/EBPβ are involved in early events of adipocyte differentiation, whereas PPARγ2 is responsible for terminal differentiation and the maintenance of the mature adipocyte phenotype (13,23). In support of our hypothesis, recent data by Yang et al. (24) showed that gene expression of PPARγ, SREBP1, and C/EBPβ was notably decreased in adipose tissue of insulin resistant first degree relatives of type 2 diabetic individuals.
Ducluzeau et al. (25) showed that insulin is able to induce a 2- to 3-fold increase in SREBP1c mRNA in skeletal muscle and adipose tissue of control subjects but not in type 2 diabetic patients. Furthermore, insulin infusion increases SREBP1c expression by 80% in lean healthy subjects but not in people with type 2 diabetes (26). Finally, Sewter et al. (27) showed that compared with lean subjects, both obese normoglycemic and obese type 2 diabetic patients have decreased expression of SREBP1c mRNA in SAT and that SREBP1c expression is inversely correlated to BMI. In our study, there was no significant relationship between BMI and SREBP1c but a negative correlation with r-VAT/SAT in the entire group and with fat cell size in men only. However, we identified a positive correlation between glucose disposal rate and SREBP1c mRNA level and in the entire group of subjects. Taken together, these results suggest a dysregulation of SREBP1c in insulin-resistant states such as obesity and type 2 diabetes. In summary, a dysregulation of SREBP1c gene expression in adipose tissue may play an important role in the induction and maintenance of the insulin resistance observed in obesity and type 2 diabetes by impairing adipogenesis.
STAT5A is part of a family of transcription factors that reside in the cytoplasm of preadipocytes. In response to different stimuli (cytokines, growth factors), STAT5 becomes tyrosine phosphorylated and translocates in the nucleus, where it mediates transcriptional regulation. It has been demonstrated that STAT5A is clearly involved in the regulation of adipocyte differentiation (28–30), probably by controlling the expression of PPARγ, C/EBPα, and SREBP1c (31). Interestingly, growth hormone but not insulin activates STAT5A in adipocytes in vitro and in vivo (26). Alternatively, mice lacking STAT5A have significantly smaller fat pads compared with wild-type animals (27).
PPARγ is recognized as a master regulator of adipogenesis and involved in the ability of fat cells to function normally (32). Ectopic expression of PPARγ is sufficient to induce the conversion of fibroblasts into adipocytes (33,34). Furthermore, in adipose tissue, different downstream genes are under the transcriptional control of PPARγ including lipoprotein lipase, acyl-CoA synthetase, fatty acid translocase (CD36), and fatty acid transport protein (35). The knockdown of PPARγ2 in white adipose tissue of C57BL/6J mice caused hypertrophic adipocytes in the subcutaneous white adipose tissue of homozygous mice (36). In the present study, we found that the decreased PPARγ2 mRNA expression in subjects with type 2 diabetes was accompanied by larger fat cells as compared with non-diabetic subjects and that PPARγ2 mRNA expression was positively correlated with glucose disposal rate, at least in white subjects.
C/EBPβ belongs to the basic leucine zipper family of transcription factors. Its role in adipogenesis has been well characterized. In 3T3-L1 cells, ectopic expression of C/EBPβ is sufficient to induce differentiation without the addition of hormonal inducers (37). Mice lacking C/EBPβ have defective adipocyte differentiation (38). These data are supportive of our results showing a trend toward decreased C/EBPβ expression in subjects with type 2 diabetes.
As for SREBP1c, C/EBPβ is implicated in the activation of PPARγ by triggering the production of ligands (39,40) and/or by inducing PPARγ promoter activity (41). In vitro, SREBP1c is able to enhance the transcriptional activity of PPARγ, increasing the proportion of cells undergoing adipocyte differentiation. In addition, using mRNA profiling techniques, Way et al. (42) showed that treating Zucker diabetic fatty rats with a non-glitazone PPARγ agonist resulted in up-regulation of several genes in adipose tissue, notably SREBP1c. These data suggest a feed-forward mechanism, in which PPARγ and SREBP1c activates each other promoting lipogenesis in adipose tissue. Although STAT5A, SREBP1c, C/EBPβ, and PPARγ2 can all independently induce adipocyte differentiation in vitro, they seem to act synergistically in vivo.
In conclusion, we identified that mRNA level of transcription factors involved in adipogenesis is decreased in SAT of obese subjects with type 2 diabetes compared with control obese subjects. Our data, therefore, support the hypothesis that impaired expression of adipogenic genes may result in impaired adipogenesis and larger fat cells in subjects with type 2 diabetes. However, because posttranslational and/or posttranscriptional events may occur, it would be of interest to measure in parallel the protein abundance of transcription factors involved in adipogenesis (note that in our study we did not dispose of enough adipose tissue material to do so). On the other hand, only prospective studies using a biological assay of adipocyte proliferation/differentiation will provide definitive evidence about the role of impaired adipogenesis in the development of type 2 diabetes and the relationship with fat cell size phenotype.
Impaired adipogenesis may play a pivotal role leading to hypertrophic obesity, increased ectopic fat deposition, insulin resistance, and eventually type 2 diabetes. Several factors linked to the development of type 2 diabetes might be involved directly or indirectly in the regulation of adipogenic genes. This includes the effect of insulin and the influence of the adipocyte size on gene expression of different transcription factors. One example is the FIRKO mouse (mice with a knockout of the insulin receptor specifically in the adipose tissue), where the knockout of the insulin receptor causes a change in adipocyte size, which differs in expression patterns of several genes (43,44). On the other hand, it has been shown in the literature that inflammatory cytokines might be linked to adipogenesis. Factors such as tumor necrosis factor-α and interleukin 6 for which plasma levels are elevated in type 2 diabetic patients are also able to interact with some of the transcription factors involved in adipogenesis such as SREBP1c or PPARγ (27,45). Recently, a protein inhibitor of activated STAT3 has been shown to play an inhibitory role in adipogenesis by modulating insulin-activated transcriptional activation events such as the inhibition of CEBPα, PPARγ,or ap2 gene expression (46). These data suggests that pro- and anti-adipogenic factors still remain to be discovered.
Overall, there are many potential mechanisms that could regulate adipogenic molecules as mentioned above. However, it is still unclear so far whether adipogenesis might represent a cause or a consequence of the development of type 2 diabetes, and characterization of the role of adipogenesis in the development of type 2 diabetes would be of interest for new therapeutic targets.
Acknowledgments
This work was supported by National Institute of Diabetes and Digestive and Kidney Diseases Grant DK 60412-02; and by the Look AHEAD grants at Pennington Biomedical Research Center [DK056990-04; Principal Investigator (PI), G. Bray], St. Luke’s Roosevelt Hospital (DK057178-06 and MO1RR000645; PI, X. Pi-Sunyer), and University of Pittsburgh (DK057002-06 and MO1RR00056; PI, D. Kelley). Part of the work was also supported by the Pittsburgh Obesity and Nutrition Research Center (P30 DK-046204). We are grateful to all for the support of the study from the Look AHEAD volunteers, PIs, study coordinators, and staff of the three Look AHEAD clinical sites: Baton Rouge (George Bray, PI and Kristi Rau and Allison Strate, study coordinators), New York (Xavier Pi Sunyer, PI and Jennifer Patricio, study coordinator), and Pittsburgh (David Kelley, PI and Jacqueline Wesche-Thobaben, study coordinator).
Appendix: LookAHEAD Adipose Research Group
Clinical Sites
Pennington Biomedical Research Center (Baton Rouge, LA). Eric Ravussin, Steven R. Smith, Leonie K. Heilbronn, Severine G. Dubois, Olga Sereda, Michele McNeil, Salman Balghian, Mandy Shipp.
University of Pittsburgh (Pittsburgh, PA). David Kelley, Therese McKolanis, Koichiro Azuma, Carol Kelley.
St. Luke’s Roosevelt Hospital Center (New York, NY). Jeanine Albu, Linda Haselman, Julia Johnson, Evan Berk.
Footnotes
- PPAR
- peroxisome proliferator-activated receptor
- C/EBP
- CCAAT/enhancer-binding protein
- SREBP1
- sterol regulatory element-binding protein 1c
- STAT
- signal transducer and activator of transcription
- SAT
- subcutaneous adipose tissue
- RT
- reverse transcription
- PCR
- polymerase chain reaction
- FFM
- fat-free mass
- HbA1c
- hemoglobin A1c
- VAT
- visceral adipose tissue
- r-VAT/SAT
- ratio of VAT vs. SAT
- PI
- principal investigator
References
- 1.King H, Aubert RE, Herman WH. Global burden of diabetes, 1995–2025: prevalence, numerical estimates, and projections. Diabetes Care. 1998;21:1414–31. doi: 10.2337/diacare.21.9.1414. [DOI] [PubMed] [Google Scholar]
- 2.Kopelman PG. Obesity as a medical problem. Nature. 2000;404:635–43. doi: 10.1038/35007508. [DOI] [PubMed] [Google Scholar]
- 3.Sims EA. Are there persons who are obese, but metabolically healthy? Metabolism. 2001;50:1499–504. doi: 10.1053/meta.2001.27213. [DOI] [PubMed] [Google Scholar]
- 4.Flier JS. The adipocyte: storage depot or node on the energy information superhighway? Cell. 1995;80:15–8. doi: 10.1016/0092-8674(95)90445-x. [DOI] [PubMed] [Google Scholar]
- 5.Paolisso G, Tataranni PA, Foley JE, Bogardus C, Howard BV, Ravussin E. A high concentration of fasting plasma non-esterified fatty acids is a risk factor for the development of NIDDM. Diabetologia. 1995;38:1213–7. doi: 10.1007/BF00422371. [DOI] [PubMed] [Google Scholar]
- 6.Weyer C, Foley JE, Bogardus C, Tataranni PA, Pratley RE. Enlarged subcutaneous abdominal adipocyte size, but not obesity itself, predicts type ii diabetes independent of insulin resistance. Diabetologia. 2000;43:1498–506. doi: 10.1007/s001250051560. [DOI] [PubMed] [Google Scholar]
- 7.Shulman GI. Cellular mechanisms of insulin resistance. J Clin Invest. 2000;106:171–6. doi: 10.1172/JCI10583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reitman ML, Mason MM, Moitra J, et al. Transgenic mice lacking white fat: models for understanding human lipoatrophic diabetes. Ann N Y Acad Sci. 1999;892:289–96. doi: 10.1111/j.1749-6632.1999.tb07802.x. [DOI] [PubMed] [Google Scholar]
- 9.Shimomura I, Hammer RE, Ikemoto S, Brown MS, Goldstein JL. Leptin reverses insulin resistance and diabetes mellitus in mice with congenital lipodystrophy. Nature. 1999;401:73–6. doi: 10.1038/43448. [DOI] [PubMed] [Google Scholar]
- 10.Kim JK, Gavrilova O, Chen Y, Reitman ML, Shulman GI. Mechanism of insulin resistance in A-ZIP/F-1 fatless mice. J Biol Chem. 2000;275:8456–60. doi: 10.1074/jbc.275.12.8456. [DOI] [PubMed] [Google Scholar]
- 11.Garg A. Lipodystrophies. Am J Med. 2000;108:143–52. doi: 10.1016/s0002-9343(99)00414-3. [DOI] [PubMed] [Google Scholar]
- 12.Danforth E., Jr Failure of adipocyte differentiation causes type II diabetes mellitus? Nat Genet. 2000;26:13. doi: 10.1038/79111. [DOI] [PubMed] [Google Scholar]
- 13.Rangwala SM, Lazar MA. Transcriptional control of adipogenesis. Annu Rev Nutr. 2000;20:535–9. doi: 10.1146/annurev.nutr.20.1.535. [DOI] [PubMed] [Google Scholar]
- 14.Rosen ED, Spiegelman BM. Molecular regulation of adipogenesis. Annu Rev Cell Dev Biol. 2000;16:145–71. doi: 10.1146/annurev.cellbio.16.1.145. [DOI] [PubMed] [Google Scholar]
- 15.Ryan DH, Espeland MA, Foster GD, et al. Look AHEAD (Action for Health in Diabetes): design and methods for a clinical trial of weight loss for the prevention of cardiovascular disease in type 2 diabetes. Control Clin Trials. 2003;24:610–28. doi: 10.1016/s0197-2456(03)00064-3. [DOI] [PubMed] [Google Scholar]
- 16.Harris RB, Ramsay TG, Smith SR, Bruch RC. Early and late stimulation of ob mRNA expression in meal-fed and overfed rats. J Clin Invest. 1996;97:2020–6. doi: 10.1172/JCI118637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–9. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 18.Lovejoy JC, de la Bretonne JA, Klemperer M, Tulley R. Abdominal fat distribution and metabolic risk factors: effects of race. Metabolism. 1996;45:1119–24. doi: 10.1016/s0026-0495(96)90011-6. [DOI] [PubMed] [Google Scholar]
- 19.Cossrow N, Falkner B. Race/ethnic issues in obesity and obesity-related comorbidities. J Clin Endocrinol Metab. 2004;89:2590–4. doi: 10.1210/jc.2004-0339. [DOI] [PubMed] [Google Scholar]
- 20.Tittelbach TJ, Berman DM, Nicklas BJ, Ryan AS, Goldberg AP. Racial differences in adipocyte size and relationship to the metabolic syndrome in obese women. Obes Res. 2004;12:990–8. doi: 10.1038/oby.2004.121. [DOI] [PubMed] [Google Scholar]
- 21.Brancati FL, Kao WH, Folsom AR, Watson RL, Szklo M. Incident type 2 diabetes mellitus in African American and white adults: The Atherosclerosis Risk in Communities Study. JAMA. 2000;283:2253–9. doi: 10.1001/jama.283.17.2253. [DOI] [PubMed] [Google Scholar]
- 22.Harris MI, Flegal KM, Cowie CC, et al. Prevalence of diabetes, impaired fasting glucose, and impaired glucose tolerance in U.S. Adults: The Third National Health and Nutrition Examination Survey, 1988–1994. Diabetes Care. 1998;21:518–24. doi: 10.2337/diacare.21.4.518. [DOI] [PubMed] [Google Scholar]
- 23.Rosen ED. The molecular control of adipogenesis, with special reference to lymphatic pathology. Ann N Y Acad Sci. 2002;979:143–58. doi: 10.1111/j.1749-6632.2002.tb04875.x. discussion 188–96. [DOI] [PubMed] [Google Scholar]
- 24.Yang X, Jansson PA, Nagaev I, et al. Evidence of impaired adipogenesis in insulin resistance. Biochem Biophys Res Commun. 2004;317:1045–51. doi: 10.1016/j.bbrc.2004.03.152. [DOI] [PubMed] [Google Scholar]
- 25.Ducluzeau PH, Perretti N, Laville M, et al. Regulation by insulin of gene expression in human skeletal muscle and adipose tissue: evidence for specific defects in type 2 diabetes. Diabetes. 2001;50:1134–42. doi: 10.2337/diabetes.50.5.1134. [DOI] [PubMed] [Google Scholar]
- 26.Koistinen HA, Forsgren M, Wallberg-Henriksson H, Zierath JR. Insulin action on expression of novel adipose genes in healthy and type 2 diabetic subjects. Obes Res. 2004;12:25–31. doi: 10.1038/oby.2004.5. [DOI] [PubMed] [Google Scholar]
- 27.Sewter C, Berger D, Considine RV, et al. Human obesity and type 2 diabetes are associated with alterations in SREBP1 isoform expression that are reproduced ex vivo by tumor necrosis factor-alpha. Diabetes. 2002;51:1035–41. doi: 10.2337/diabetes.51.4.1035. [DOI] [PubMed] [Google Scholar]
- 28.Stephens JM, Morrison RF, Pilch PF. The expression and regulation of STATs during 3T3—L1 adipocyte differentiation. J Biol Chem. 1996;271:10441–4. doi: 10.1074/jbc.271.18.10441. [DOI] [PubMed] [Google Scholar]
- 29.Harp JB, Franklin D, Vanderpuije AA, Gimble JM. Differential expression of signal transducers and activators of transcription during human adipogenesis. Biochem Biophys Res Commun. 2001;281:907–12. doi: 10.1006/bbrc.2001.4460. [DOI] [PubMed] [Google Scholar]
- 30.Floyd ZE, Stephens JM. STAT5a promotes adipogenesis in nonprecursor cells and associates with the glucocorticoid receptor during adipocyte differentiation. Diabetes. 2003;52:308–14. doi: 10.2337/diabetes.52.2.308. [DOI] [PubMed] [Google Scholar]
- 31.Nanbu-Wakao R, Morikawa Y, Matsumura I, et al. Stimulation of 3T3—L1 adipogenesis by signal transducer and activator of transcription 5. Mol Endocrinol. 2002;16:1565–76. doi: 10.1210/mend.16.7.0862. [DOI] [PubMed] [Google Scholar]
- 32.Rosen ED, Walkey CJ, Puigserver P, Spiegelman BM. Transcriptional regulation of adipogenesis. Genes Dev. 2000;14:1293–307. [PubMed] [Google Scholar]
- 33.Tontonoz P, Hu E, Graves RA, Budavari AI, Spiegelman BM. mPPAR gamma 2: tissue-specific regulator of an adipocyte enhancer. Genes Dev. 1994;8:1224–34. doi: 10.1101/gad.8.10.1224. [DOI] [PubMed] [Google Scholar]
- 34.Tontonoz P, Hu E, Spiegelman BM. Stimulation of adipogenesis in fibroblasts by PPAR gamma 2, a lipid-activated transcription factor. Cell. 1994;79:1147–56. doi: 10.1016/0092-8674(94)90006-x. [DOI] [PubMed] [Google Scholar]
- 35.Desvergne B, Wahli W. Peroxisome proliferator-activated receptors: nuclear control of metabolism. Endocr Rev. 1999;20:649–88. doi: 10.1210/edrv.20.5.0380. [DOI] [PubMed] [Google Scholar]
- 36.Koutnikova H, Cock TA, Watanabe M, et al. Compensation by the muscle limits the metabolic consequences of lipodystrophy in PPAR gamma hypomorphic mice. Proc Natl Acad Sci U S A. 2003;100:14457–62. doi: 10.1073/pnas.2336090100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yeh WC, Cao Z, Classon M, McKnight SL. Cascade regulation of terminal adipocyte differentiation by three members of the C/EBP family of leucine zipper proteins. Genes Dev. 1995;9:168–81. doi: 10.1101/gad.9.2.168. [DOI] [PubMed] [Google Scholar]
- 38.Tanaka T, Yoshida N, Kishimoto T, Akira S. Defective adipocyte differentiation in mice lacking the C/EBPbeta and/or C/EBPdelta gene. EMBO J. 1997;16:7432–43. doi: 10.1093/emboj/16.24.7432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hamm JK, Park BH, Farmer SR. A role for C/EBPbeta in regulating peroxisome proliferator-activated receptor gamma activity during adipogenesis in 3t3—l1 preadipocytes. J Biol Chem. 2001;276:18464–71. doi: 10.1074/jbc.M100797200. [DOI] [PubMed] [Google Scholar]
- 40.Kim JB, Wright HM, Wright M, Spiegelman BM. ADD1/SREBP1 activates PPARgamma through the production of endogenous ligand. Proc Natl Acad Sci U S A. 1998;95:4333–7. doi: 10.1073/pnas.95.8.4333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fajas L, Schoonjans K, Gelman L, et al. Regulation of peroxisome proliferator-activated receptor gamma expression by adipocyte differentiation and determination factor 1/sterol regulatory element binding protein: I. Implications for adipocyte differentiation and metabolism. Mol Cell Biol. 1999;19:5495–503. doi: 10.1128/mcb.19.8.5495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Way JM, Harrington WW, Brown KK, et al. Comprehensive messenger ribonucleic acid profiling reveals that peroxisome proliferator-activated receptor gamma activation has coordinate effects on gene expression in multiple insulin-sensitive tissues. Endocrinology. 2001;142:1269–77. doi: 10.1210/endo.142.3.8037. [DOI] [PubMed] [Google Scholar]
- 43.Bluher M, Michael MD, Peroni OD, et al. Adipose tissue selective insulin receptor knockout protects against obesity and obesity-related glucose intolerance. Dev Cell. 2002;3:25–38. doi: 10.1016/s1534-5807(02)00199-5. [DOI] [PubMed] [Google Scholar]
- 44.Bluher M, Patti ME, Gesta S, Kahn BB, Kahn CR. Intrinsic heterogeneity in adipose tissue of fat-specific insulin receptor knock-out mice is associated with differences in patterns of gene expression. J Biol Chem. 2004;279:31891–901. doi: 10.1074/jbc.M404569200. [DOI] [PubMed] [Google Scholar]
- 45.Chae GN, Kwak SJ. NF-kappab is involved in the TNF-alpha induced inhibition of the differentiation of 3T3—L1 cells by reducing PPARgamma expression. Exp Mol Med. 2003;35:431–7. doi: 10.1038/emm.2003.56. [DOI] [PubMed] [Google Scholar]
- 46.Deng J, Hua K, Caveney EJ, Takahashi N, Harp JB. Protein inhibitor of activated STAT3 inhibits adipogenic gene expression. Biochem Biophys Res Commun. 2006;339:923–31. doi: 10.1016/j.bbrc.2005.10.217. [DOI] [PubMed] [Google Scholar]