

Abstract

Abyssomicin C is a recently discovered antibiotic with promising antibacterial activity, high structural complexity, and a novel mechanism of action. We give an account of our abyssomicin campaign and some of the discoveries that were borne of our total synthesis efforts, including a new Lewis acid-templated Diels-Alder reaction, the previously undescribed atrop-abyssomicin C, a facile Brønsted acid-catalyzed isomerization of abyssomicin C, and clarification of the likely biosynthetic origin of abyssomicin D.
Keywords: Antibiotics, Atropisomerism, Diels-Alder reactions, Metathesis, Total synthesis
1. Introduction
In 2004, the groups of Fielder and Süssmuth disclosed the striking discovery of three novel natural products, abyssomicins B, C, and D from the marine actinomycete strain Verrocosispora AB18-032, one of which, abyssomicin C, inhibited the growth of methicillin-resistant Staphylococcus aureus (MRSA, MIC = 4 μg mL-1) and vancomycin-resistant S. aureus (VRSA, MIC = 13 μg mL-1).1 Their producing microbe was collected from the depth of 289 meters below the surface of the Sea of Japan, and its habitat, “the Abyss”, decisively influenced the naming of this natural product class. Curiously, the letter A was left out of the naming of these natural products, as if that privilege was reserved for an important compound yet to be discovered from the same source.
Abyssomicins B, C, and D were assigned structures 2, 1, and 3, respectively (Figure 1). Feeding experiments revealed that abyssomicin C (1) inhibited either 4-amino-4-deoxychorismate (ADC) synthase or ADC lyase, two enzymes responsible for the conversion of chorismate to p-aminobenzoic acid (pABA) in bacteria,2 making abyssomicin C the first naturally occurring substance to exhibit such activity.
Figure 1.
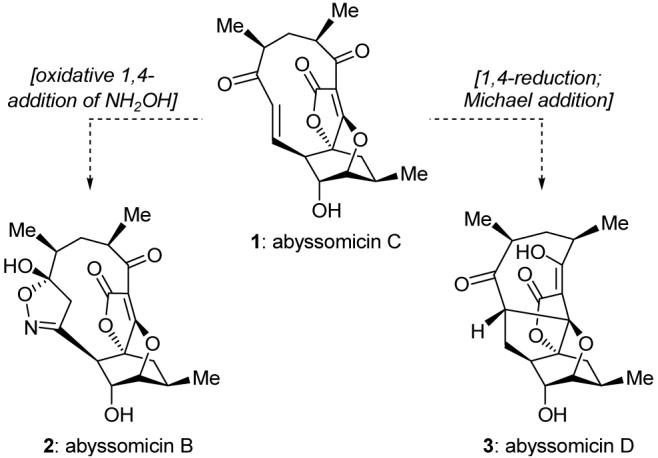
Molecular structures of abyssomicins B (2), C (1), and D (3) and the postulated biosynthetic origins of abyssomicins B and D.
Abyssomicins B (2) and D (3) were inactive, leading to the speculation that the enone moiety present in abyssomicin C (1), but lacking in 2 and 3, was involved in both the mechanism of action of abyssomicin C (1) and its biotransformation to abyssomicins B (2) (oxidative 1,4-addition of hydroxylamine and ring closure) and D (3) (1,4-hydride addition followed by intramolecular Michael addition onto the tetronate moiety).
Made of 19 carbon atoms, 6 oxygen atoms, and 22 hydrogen atoms, the molecule of abyssomicin C (1) constitutes an intriguing molecular assembly by virtue of its unique combination of structural motifs. Among them are the oxabicyclo[2.2.2]octane system, the tetronate moiety, and the 11-membered ring carrying an α,β-unsaturated ketone with a trans-configured olefin. Of particular interest is the novel fusion of the tetronate and oxabicyclo systems, as well as the intriguing possibility of atropisomerism around the rather constrained macrocycle, a phenomenon of which no mention was made in the structural elucidation report.
The exquisite molecular architecture of abyssomicin C combined with its unique biological activity as the first natural product to inhibit the essential biosynthesis of pABA in bacteria presented an attractive opportunity to synthetic chemists. Indeed, several groups responded immediately to the challenge posed by this target molecule. The first total synthesis of abyssomicin C (1) was disclosed in 2005 by the Sorensen group,3 who adopted a daring biomimetic strategy involving a late-stage, intramolecular Diels-Alder reaction4 of a monocyclic precursor (i.e. 5, generated in situ from 4) to cast the advanced tricyclic system 6 of their target (Scheme 1).
Scheme 1.

Highlights of the Sorensen total synthesis of abyssomicin C (1).
A short sequence thereafter involving regio- and stereoselective epoxidation of the cyclohexene double bond and intramolecular hydroxy epoxide opening completed their elegant total synthesis of abyssomicin C (1). Interestingly, this synthesis yielded another product in equal quantity which was isomeric in nature to abyssomicin C, and thus designated iso-abyssomicin C. The structure of this “mystery” compound was, however, left unassigned and would remain so until almost a year later when this compound resurfaced in our own synthetic investigations.
Soon after the Sorensen disclosure of the first total synthesis of abyssomicin C, Snyder and Zou reported their elegant formal total synthesis of this molecule by constructing advanced intermediate 6 through the same intramolecular Diels-Alder reaction (5→6, Scheme 1).5 And shortly after that report, the Couladouros group published their formal total synthesis of abyssomicin C that also proceeded through the same reaction (5→6, Scheme 1).6 Model studies on the abyssomicins were also published from the Maier7 and Georgiadis8 laboratories. Below we unravel the story of our independent work in the field that, after a number of twists and turns, led to a few interesting discoveries in chemistry and biology, some expected and some not.9
2. Retrosynthetic Analysis
Figure 2 shows our initial retrosynthetic analysis of the abyssomicin C structure (1) that led to two distinct approaches to the molecule. The late-stage key operations of route a entailed an intramolecular Dieckmann condensation7b, 8, 10 and epoxide opening cascade as a means to forge the tetronate oxabicyclo[2.2.2]octane core, whereas route b called upon the olefin metathesis reaction11 to form the macrocycle. Each route had the other’s late-stage key reaction reserved for an early-stage step, a choice that serves as a reminder of the power of these enabling processes. Both strategies [route a: 8→7→1; route b: 13→12→11→10→1] traced the target molecule to intermediate 9, whose origin from simple starting materials through a Diels-Alder cycloaddition4 was easy to discern. Our abyssomicin campaign, therefore, began with a search for an efficient construction of common intermediate 9.
Figure 2.
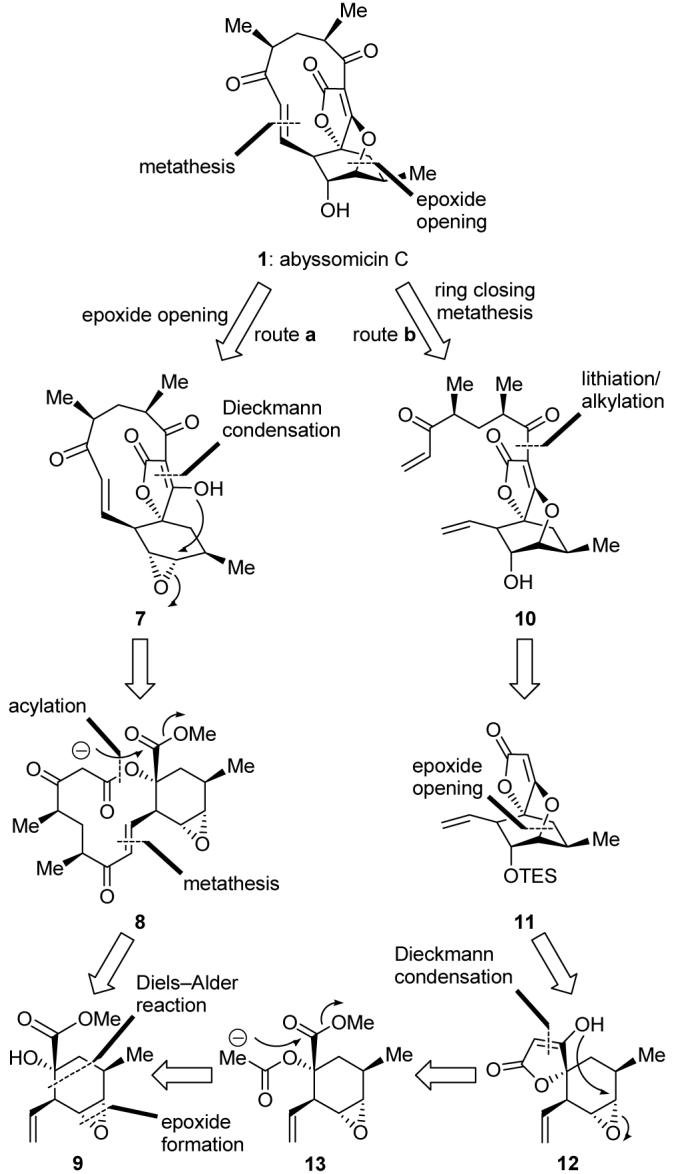
Retrosynthetic analysis of abyssomicin C (1).
3. Initial Forays
In 2000, Ward and Abaee published an interesting Lewis acid-templated Diels-Alder reaction that appeared to be perfectly suited for our purposes to prepare cyclohexene 16 (Scheme 2), the first key building block required for both our strategies as a precursor to 9.12 Thus, and according to the published procedure, diene 14 was first treated with MeMgBr, and the resulting alkoxide was reacted with methyl acrylate at ambient temperature to afford bicyclic lactone (±)-16 in high yield (90%), presumably through magnesium-templated complex 15. Although this intermediate was racemic, the fact that it was obtained in such a high yield, and as a single diastereoisomer, encouraged us to press on in the hope of first working out the chemistry along the chartered pathway before returning to address its enantioselective synthesis.
Scheme 2.
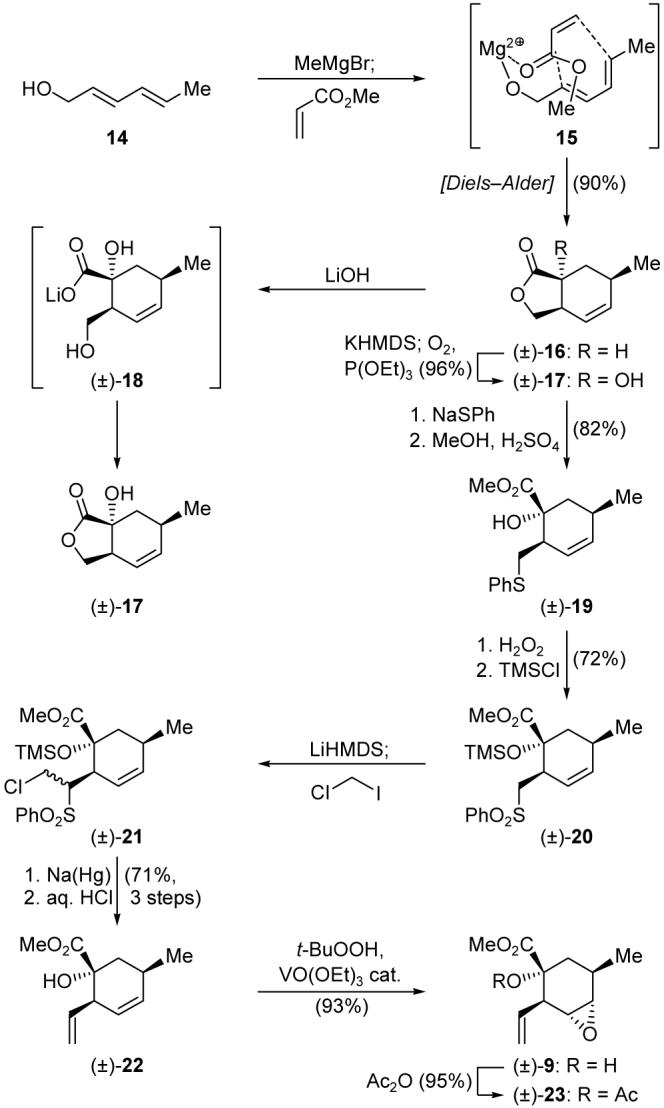
Initial synthesis of key building blocks (±)-9 and (±)-23.
Thus, enolate formation (KHMDS), followed by quenching with O2 and P(OEt)3, converted (±)-16 to the corresponding α-hydroxy lactone [(±)-17] in 96% yield. Attempts to open the γ-lactone ring of (±)-17 with aqueous alkaline solutions failed, with the initially formed hydroxy carboxylate [e.g. (±)-18] shutting back into the starting lactone upon acidification. A solution to this problem was found by opening the γ-lactone ring at a different site as initiated by nucleophilic attack with thiophenoxide ion. Thus, treatment of (±)-17 with PhSNa, generated in situ from PhSH and NaH in DMF, produced, after methylation [(MeO)3CH, H2SO4, MeOH], phenyl thioether methyl ester (±)-19 (82% overall yield). Molybdenum-catalyzed (ammonium molybdate cat.) oxidation with H2O2 and subsequent silylation (TMSCl, Et3N, 4-DMAP cat.) led to phenyl sulfone (±)-20 in 72% overall yield. Having achieved the ring opening, it was now time to introduce the required olefinic bond. This was accomplished by reacting the anion of (±)-20 (generated by treatment with LiHMDS in THF:DMPU at -78 °C) with ICH2Cl to produce a mixture of diastereomeric chloromethylene sulfones [(±)-21], which was converted to the targeted intermediate (±)-22 in 71% overall yield by sequential treatment with sodium amalgam and aqueous HCl in MeOH. Next in our quest for the oxabicyclo fragment was the regioand stereoselective epoxidation of the internal olefin within structure (±)-22, a task that was admirably accomplished through the action of t-BuOOH in the presence of the rarely used VO(OEt)3 catalyst.13 The latter reaction yielded epoxide (±)-9 as the sole product, and in 93% yield. Acetylation of (±)-9 led, in 95% yield, to tertiary acetate (±)-23.
With epoxides (±)-9 and (±)-23 in hand, we were now in a position to test the two strategies delineated in Figure 2. We first explored the possible conversion of epoxide (±)-9 to a macrocycle such as 8. Towards this goal, we attempted to prepare compounds such as diester 24 (by coupling with 25) or methyl ketone 26 through esterification or cross metathesis,11 but unfortunately to no avail (Scheme 3).
Scheme 3.
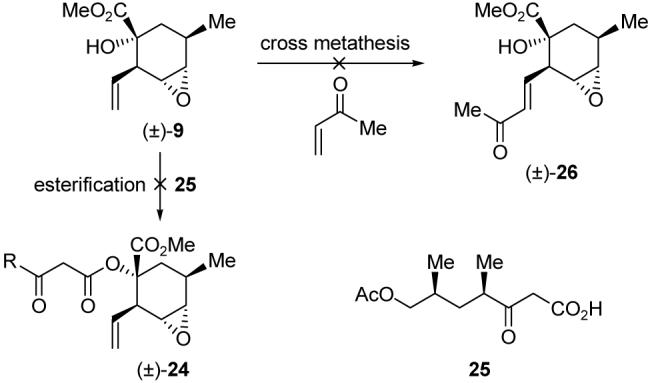
Attempted fragment couplings with epoxide 9.
As a temporary relief from these initial frustrations, we then turned our attention to the potential construction of the oxabicyclo[2.2.2]octane tetronate moiety from acetate epoxide (±)-23. As depicted in Scheme 4, the construction of such a system [i.e. (±)-27] proved both highly successful and uplifting. Thus, tertiary acetate (±)-23 served as an excellent substrate for the ensuing cascade sequence to produce, after silylation, compound (±)-11 through intermediate (±)-27 in 97% overall yield. Involving a Dieckmann condensation7b, 8, 10 and a hydroxy epoxide opening, and presumably proceeding through intermediates 13 and 12, this transformation was brought about by the action of LiHMDS followed by quenching the resulting alkoxy epoxide with saturated aqueous NH4Cl solution and warming to 45 °C. A similar sequence to a related model system had been reported by the Georgiadis laboratory in 2005.8 Interestingly, and in contrast to substrate 9, terminal olefin(±)-27 under-went smooth cross metathesis with methyl vinyl ketone, inspiring confidence in our planned strategy. This was well and good, but we still needed an enantioselective process for the synthesis of the oxabicyclo tetronate core and a suitable means for its elaboration to its destination, abyssomicin C. As we shall see below, the crucial clue came, once again, from the Ward camp.
Scheme 4.

Dieckmann condensation/epoxide opening cascade entry to the abyssomicin C core.
4. A Novel Diels-Alder Reaction and Process Development
It was in 2005, and after completion of our synthesis of racemic tetronate compound 11 through bicyclic lactone (±)-16, that Ward and Souweha reported their enantioselective approach to the latter compound.14 Their new synthesis proceeded through the same Diels-Alder process employed in the Ward group’s original preparation of racemic 16, but now was templated by zinc, magnesium, and BINOL, as shown in Scheme 5. Putative complexes 28 and 29 were tentatively proposed to explain this highly efficient (95% yield) and enantioselective (93% ee) reaction that proceeded under carefully defined conditions. Despite its requirement for stoichiometric amounts of Me2Zn and BINOL, this reaction proved inspirational in our search for an improved synthesis of our intermediates in enantioselective form. Although our ultimate route did not employ an enantioselective DielsAlder reaction, the concept of organizing the transition state around an organometallic template was instrumental in allowing our synthetic plans to advance, ultimately leading to a successful process towards abyssomicin C, as delineated below.
Scheme 5.
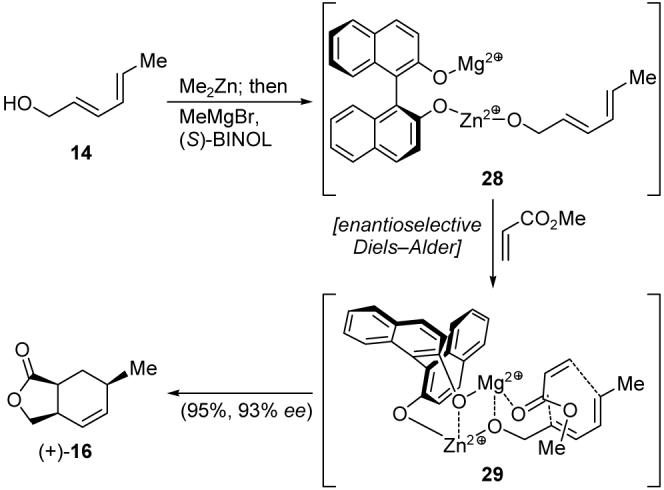
Ward’s enantioselective Diels-Alder synthesis of lactone 16.
Thus, back at the drawing board in search of an enantioselective synthesis of our tetronate fragment, we wondered if the employment of a more functionalized diene (e.g. 31, Figure 3) could allow easier installation of the terminal olefin in our desired intermediate 11 and, at the same time, amount to an enantioselective synthesis through early procurement of absolute stereochemistry and subsequent stereocontrol of the Diels-Alder reaction. Our new approach looked, more or less, as the one depicted in Figure 3.
Figure 3.
Revised retrosynthetic analysis of the abyssomicin core structure 11.
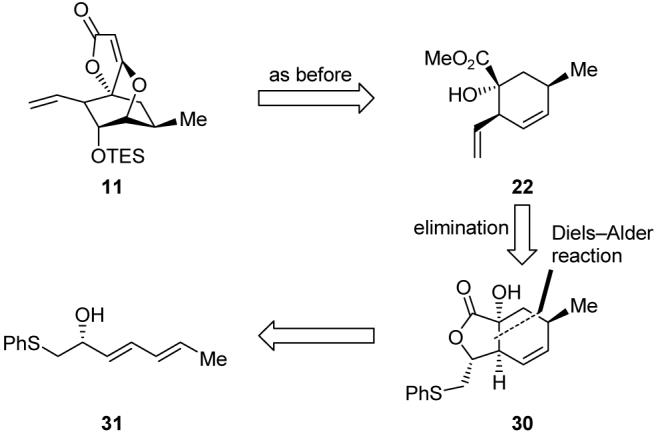
For expedience, our initial explorations to optimize the desired Diels-Alder reaction were carried out with racemic diene 31 (obtained by addition of LiCH2SPh to aldehyde 32, Scheme 6) and methyl acrylate. The best we could do at the outset with MeMgBr as the sole templating agent was a 30% yield of bicyclic lactone 34 as a single stereoisomer, presumably obtained through complex 33. Disappointing as these results were, the switch to the Ward and Souweha conditions [Me2Zn, MeMgBr, (±)-BINOL] proved even more devastating, as only a trace of the desired product was obtained (see Table 1). Further explorations with other phenol-type bidentate ligands, however, soon led to exciting results. Thus, we found that catechol acted as a better ligand than BINOL in the presence of Me2Zn and MeMgBr, bringing the yield of the Diels-Alder reaction up to 35%. Although not a dramatic improvement, we recognized this result as a new avenue for investigation in our quest to improve this Diels-Alder process. Thus, experimenting with the structurally similar 2-aminophenol in the presence of MeMgBr (but not Me2Zn) was rewarded with a markedly better yield (49%), and extensive screening of other 2-aminophenols led to the ultimate discovery of 2-(pyrrolidin-1-yl)phenol (35, Scheme 7) as the optimum ligand, facilitating, in the presence of MeMgBr, the formation of the desired Diels-Alder adduct (34) in a satisfying 80% yield and as a single diastereoisomer (Table 1). This fascinating reaction is presumed to proceed through templated transition state 38 (see Scheme 7). Although three equivalents of 35 were required to drive the reaction to completion, the ligand was readily recovered from the crude reaction mixture by acid-base extraction.
Scheme 6.
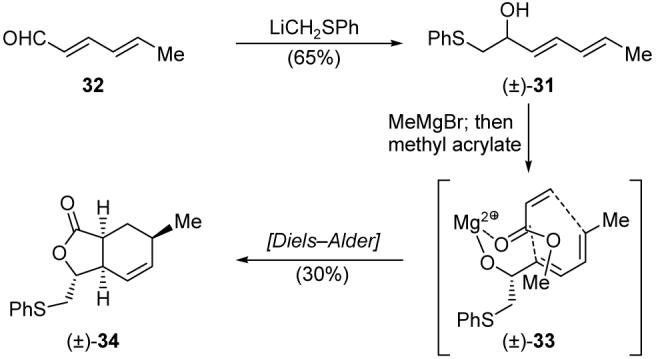
Initial Diels-Alder-based construction of lactone 34.
Table 1.
Optimization of the Diels-Alder construction of lactone 34
 | |||
|---|---|---|---|
| auxiliary | base | time | yield |
| none | MeMgBr (1 equiv) | 24 h | 30% |
| (±)-BINOL | Me2Zn (1 equiv), MeMgBr (1 equiv) | 24 h | < 5% |
| catechol | Me2Zn (1 equiv), MeMgBr (1 equiv) | 36 h | 35% |
| 2-aminophenol | MeMgBr (2 equiv) | 24 h | 49% |
| 35 | MeMgBr (2 equiv) | 48 h | 55% |
| 35 (2 equiv) | MeMgBr (3 equiv) | 48 h | 70% |
| 35 (3 equiv) | MeMgBr (4 equiv) | 12 h | 80% |
Scheme 7.

Enantioselective synthesis of abyssomicin core structure 11.
Applying the newly developed synthetic technology, and following steps from our initial route, we soon arrived at enantiomerically pure tetronate fragment 11, as shown in Scheme 7. Thus, Weinreb amide 36 reacted with the lithium derivative of thioanisole to afford conjugated ketone 37 in 81% yield. CBS reduction15 [Me-(R)-CBS, catecholborane] of the latter compound furnished enantiomerically enriched alcohol 31 (95% yield, 90% ee), which entered the optimized Diels-Alder reaction with methyl acrylate in the presence of MeMgBr and 2-(pyrrolidin-1-yl)phenol (35) at ambient temperature to give the expected enantioenriched bicyclic lactone 34 in 80% yield. Subsequent α-hydroxylation of this lactone [LiHMDS, O2, P(OEt)3] proceeded smoothly as before, to afford hydroxy lactone 30 in 74% yield, and setting the stage for the rupture of the γ-lactone moiety towards the terminal olefin installation. To this end, it was pleasing to find that lithium 4,4′-di-tert-butylbiphenyl (LDBB) anion caused reduction of the phenylthio group to the corresponding anion, which suffered spontaneous β-elimination/lactone rupture to afford, upon methylation in the same pot, the desired olefinic hydroxy methyl ester 22 in 99% overall yield. This intermediate was then converted to the enantiopure tetronate core 11 as was previously done with the racemic compound.
With the enantiomerically pure core domain of abyssomicin C secured, all that was now missing was the tether that would allow the casting of the macrocycle and completion of the synthesis. By now, our first choice for generating the macrocycle was the ring closing metathesis reaction, and, therefore, the olefinic aldehyde 42 (Scheme 8) became our logical building block required for the task. Its construction began with the meso 1,5-diol 39, whose enzymatic desymmetrization and further elaboration led to intermediate 40.16 Oxidation of the latter, followed by vinylmagnesium bromide addition to the resulting aldehyde, furnished allylic alcohol 41. Standard manipulation of 41 delivered aldehyde 42, whose diastereomeric nature was of no consequence since, in the final act, the center of isomerism would be eradicated through oxidation as required for the target molecule.
Scheme 8.

Synthesis of aldehyde fragment 42.
5. Fragment Coupling and Ring Closing Metathesis Studies
With our strategy firmly established and the requisite fragments in hand, we were ready to explore the tactics that would allow us to reach our target. Our first forays towards this goal are summarized in Scheme 9. Thus, lithiation of tetronate fragment 11 (t-BuLi, -78 °C)17 followed by addition of aldehyde 42 resulted, after removal of the PMB group, in the formation of diol 43 as a mixture of diastereomers. Our rapid progress, however, would come to an abrupt halt when we attempted the much anticipated ring closing metathesis11 of substrate 43. Indeed, only dimeric materials of type 45 were observed in this reaction when performed with Grubbs catalyst II (44).18 Faced with this hurdle, we removed the rather bulky TES group from 43 to obtain triol 46 as a mixture of isomers. While the ring closing metathesis on the latter substrate proceeded smoothly in the presence of catalyst 44 (78% yield, mixture of diastereomers), attempted oxidation of the resulting product (47) under a variety of conditions failed to produce abyssomicin C (1). The alternative pathway of first oxidizing (46→10, IBX, 64% yield), and then exposing the so generated diketone to catalyst 44 in the presence of 1,4-benzoquinone,19 however, proved more promising in that it led to abyssomicin C (1), albeit in low yield (14%).
Scheme 9.

Initial ring closing metathesis route to abyssomicin C (1).
An explanation for the failure of the bis-allylic alcohol 47 to yield abyssomicin C under the many conditions employed may be found in the propensity of the initially formed hydroxy ketone to lock itself in its hemiketal form. It was further reasoned that the reluctance of the diketone 10 to enter into a smooth cyclization under metathesis conditions as compared to 46 was attributable to its two additional strain-inducing sp2 centers. Combining these two observations, we reasoned that a cyclization substrate with the same hybridization structure as 46 but devoid of the intramolecular hemiketalization option may provide a solution to our problem. Aiming for ring closing metathesis substrate 50 (Scheme 10) as the most likely to succeed due to the remoteness of the dithiolane moiety from the cyclization vicinity, we reacted the lithio derivative of 11 (t-BuLi) with known lactone 48 and exposed the resulting hydroxy ketone to 1,2-ethanedithiol and TMSOTf to afford hydroxy dithiane 49, in 76% overall yield. Selective generation of the corresponding aldehyde from 49 with IBX followed by addition of vinylmagnesium bromide furnished allylic alcohol 50, in 65% overall yield as a 3:2 diastereomeric mixture. Pleasantly, and as planned, substrate 50 underwent smooth ring closing metathesis upon exposure to Grubbs catalyst II (44) to afford two diastereomeric products in high yield (85%, ca. 3:2 ratio). We were pleased even more to confirm, by 1H NMR spectroscopic analysis (NOE studies), that both isomers possessed the desired C8-C9 trans geometry and, furthermore, despite the rather constrained 11-membered ring and the potential for atropisomerism, no such isomers were detected.
Scheme 10.
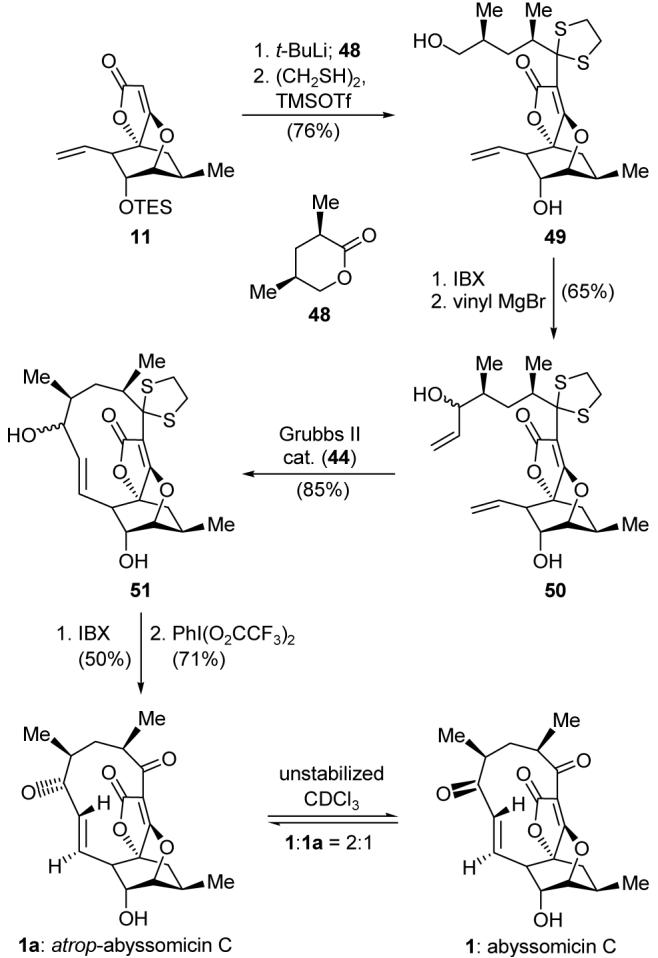
Improved synthesis of the abyssomicin C skeleton by ring closing metathesis.
With compound 51 in hand, it seemed that the coveted prize, abyssomicin C (1), was but a short stone’s throw away. Indeed, IBX oxidation of the mixture of diastereoisomers led to a single ketone product. However, subsequent removal of the dithiane group from the latter compound with PhI(O2CCF3)2 led to a puzzle. Thus, although the reaction proceeded as expected to cleave the dithiolane moiety and successfully generated the second ketone, the spectroscopic data of the resulting product did not quite match those reported for the natural product. The mystery became even more intriguing upon realizing that the new compound was isomeric in nature to abyssomicin C (1). Indeed, attempts to assign the structure by NMR, including NOE studies, strongly pointed toward the structure assigned to 1, despite the fact that the data did not match. It was up to serendipity to defog this picture, for while collecting additional NMR spectroscopic data for this compound in deuterated chloroform, we noticed signals of a second compound appearing; this last compound turned out to be identical in all respects to abyssomicin C (1)! This joyous observation led to an additional quandary. What was the identity of the final intermediate in our total synthesis of abyssomicin C? Our wild speculations were put to rest after X-ray crystallographic analysis demonstrated that the mystery compound was, in fact, an unanticipated conformational isomer of abyssomicin C (1), which we designated atrop-abyssomicin C (1a). To our surprise, early attempts to isomerize either 1a or 1 thermally were unsuccessful up to temperatures of 180 °C,20 yet the traces of HCl that must have been present in our CDCl3 were responsible for its facile isomerization to the natural product (1:1a ca. 2:1) at room temperature. Although several mechanistic pathways may be written for this fascinating rearrangement, one possible explanation for this acid-promoted isomerization from 1 to 1a is that shown in Figure 4. Thus, protonation of the tetronate olefinic bond would lead to the oxocarbenium species 52, in which the strain demonstrated by the badly deformed C2 carbon is released by its conversion to a pyramidal center as shown in structure 52. This would allow the necessary rotations of the σ-bonds flanking the C7 carbonyl group within the macrocycle to take place, so as to allow equilibration of the atropisomers.
Figure 4.
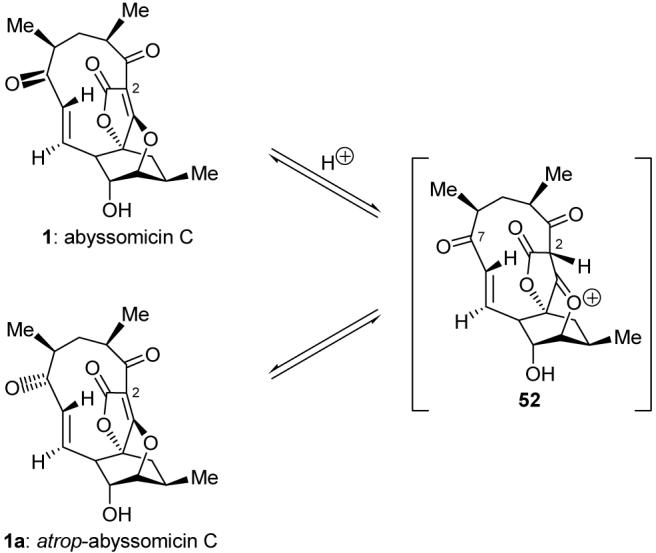
Proposed mechanism of acid-mediated interconversion of abyssomicin C (1) and atrop-abyssomicin C (1a).
The interconversion of 1 and 1a can also be brought about under a variety of other controlled conditions, including those employed by Sorensen and his team (p-TsOH, LiCl, MeCN, 50 °C, 2 h) in their final step yielding abyssomicin C and what they termed “iso-abyssomicin C”.3 These experiments made it clear that iso-abyssomicin C and atrop-abyssomicin C are one and the same compound.21 Recalling that, for whatever reason, the name abyssomicin A was left open by the isolation chemists, it seems that this newcomer compound was destined for this designation!
6. The Structures of the Abyssomicins: Implications for Their Synthesis, Biosynthesis, and Biological Activity
The X-ray structures of the two atropisomeric abyssomicins C (1)1 and (1a) (see Figure 5) are revealing. Thus, the enone moiety (C7-C9) of abyssomicin C (1) adopts a transoid conformation, whereas that of atrop-abyssomicin C (1a) resides in a cisoid conformation. Both structures show significant deviations from planarity for the sp2 carbon at position 2, reflecting the considerable strain associated with these molecules.22 Interestingly, the cisoid enone of atrop-abyssomicin C (1a) exhibits a higher degree of planarity as compared with the transoid enone of abyssomicin C (1), suggesting that 1a should exhibit increased reactivity in conjugate addition reactions. As discussed below, this proposed difference in reactivity was confirmed in subsequent experiements, including comparisons of antibacterial potency and direct measurements of stability under reducing conditions.
Figure 5.

Overlay of the X-ray crystal structures of abyssomicin C (1, blue) and atrop-abyssomicin C (1a, yellow). [Reprinted with permission from the Journal of the American Chemical Society 2007, 129, 429-440. Copyright (2007) American Chemical Society.]
With both synthetic abyssomicin C (1) and atrop-abyssomicin C (1a) in hand, we decided to study their chemistry with an eye on the proposed biosynthesis of abyssomicin D (3) from abyssomicin C (1) through nucleophilic attack at the enone moiety, followed by transannular intramolecular Michael addition of the incipient enolate upon the tetronate unit across the macrocycle. Specifically, and in order to mimic this hypothetical bioconversion, we reacted abyssomicin C (1) with L-Selectride in the expectation that we could obtain abyssomicin D (3). The reaction proceeded well, presumably through E-enolate 52 (Scheme 11), generated through conjugate addition of hydride to the transoid enone moiety, but did not furnish abyssomicin D. Instead, compound 54 was obtained in 44% yield. Differing from abyssomicin D at C8 [8(R) instead of 8(S)] and the fact that C2 is pyramidalized and C3 is fully in its keto form, this product was named iso-abyssomicin D. Its formation may be rationalized by a transannular Michael addition of 52, followed by epimerization of C8 to relieve strain. Alternatively, a series of bond rotations of enolate 52 might give rise to conformer 53, with the C3 ketone syn to the tetronate carbonyl and the enolate oriented such that the ensuing Michael addition generates the observed C8 stereochemistry of iso-abyssomicin D (54). Interestingly, attempted crystallization of iso-abyssomicin D from EtOH at room temperature resulted in its quantitative conversion to abyssomicin D (3). In contrast to this result, L-Selectride reduction of atrop-abyssomicin C (1a) under the same conditions furnished abyssomicin D (3) directly and in 60% yield. Conjugate addition of hydride to atrop-abyssomicin C (1a) presumably formed the Z-enolate 55, and rotation of the C3 ketone to be syn to the tetronate carbonyl afforded its conformer (56). Transannular Michael addition then cast abyssomicin D (3). Thus, we concluded that abyssomicin D (3) likely is a product of a reductive process of atrop-abyssomicin C (1a), rather than of abyssomicin C (1) as previously suggested.1 Furthermore, we suggested9 that atrop-abyssomicin C (1a) was a previously unidentified secondary metabolite of Verrucosispora strain AB18-032.
Scheme 11.
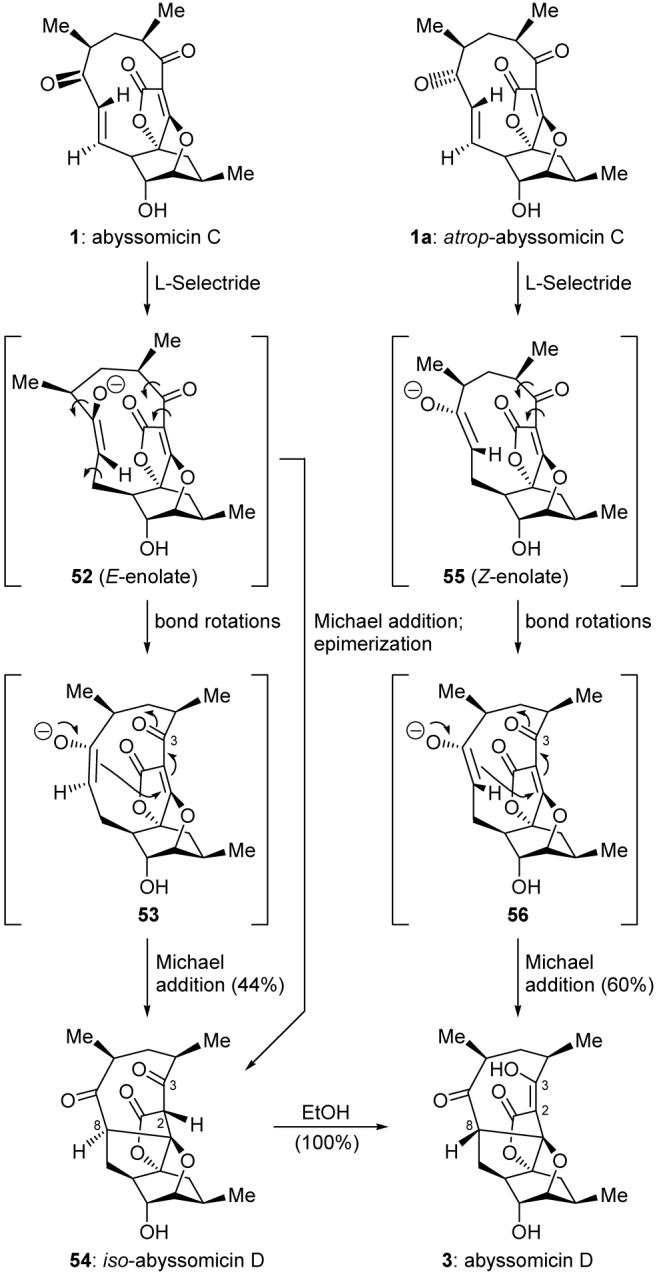
Biomimetic synthesis of abyssomicin D (3).
Our expectation that the cisoid enone moiety of atrop-abyssomicin C (1a) enjoyed a higher degree of conjugation and, therefore, higher reactivity than abyssomicin C (1) was borne out in kinetics experiments studying the reduction of 1 and 1a with NADH analog 57 (Figure 6). Thus, it was found that incubation of 1 and 1a with 57 in CDCl3 at 55 °C for 48 h resulted in complete consumption of atrop-abyssomicin C (1a), but only 30% consumption of abyssomicin C (1). Furthermore, while atrop-abyssomicin C delivered abyssomicin D (3) as the predominant product in this reaction, the reduction of abyssomicin C under these conditions did not yield any detectable (by 1H NMR spectroscopy) amounts of abyssomicin D (3) or iso-abyssomicin D (54), lending further support to our hypothesis of the existence of atrop-abyssomicin C (1a) in nature and its possible role as a biosynthetic precursor to abyssomicin D (3).
Figure 6.

Kinetics of the reduction of abyssomicin C (1) and atrop-abyssomicin C (1a) by NADH analog 57. [Reprinted with permission from the Journal of the American Chemical Society 2007, 129, 429-440. Copyright (2007) American Chemical Society.]
The abyssomicin campaign was concluded with the biological evaluation of a number of abyssomicin analogs and fragments as potential antibacterial agents against MRSA. From the several compounds synthesized in this program, abyssomicin C (1, MIC = 20 μM), atrop-abyssomicin C (1a, MIC = 15 μM), acetyl abyssomicin C (structure not shown) (MIC = 20 μM), and 3-dithiolane atrop-abyssomicin C (structure not shown) (MIC = 70 μM) exhibited significant activity. The slightly improved activity of atrop-abyssomicin C (1a) was attributed to its increased reactivity toward nucleophiles.
At the end of our story on the abyssomicins, we are pleased to note that atrop-abyssomicin C (1a) was eventually discovered as a naturally occurring substance from Verrucosispora strain AB18-032.23 Indeed, and surprisingly so, atrop-abyssomicin C was found to be the major abyssomicin metabolite produced by this bacterial strain, while abyssomicin C (1) is but a minor product. Furthermore, Süssmuth and coworkers recently demonstrated that atrop-abyssomicin C (1a) is a chorismate mimic, irreversibly binding at the PabB subunit of 4-amino-4-deoxychorismate (ADC) synthase at Cys263.24
7. Conclusion
The abyssomicin project demonstrates a number of important aspects of the essence of total synthesis—some common, some rare. The strategies and tactics deployed in this campaign led to interesting discoveries ranging from improvements in the Diels-Alder reaction to a new bioactive natural product whose discovery in nature had not been reported at the time, from expanding the scope of the olefin metathesis reaction to new insights into the biosynthesis and mechanism of action of the abyssomicins, and from novel aspects of atropisomerism in medium-sized rings to a mechanistic understanding of the chemical reactivity of these novel structures. This story also underscores the virtues of weaving together aspects of chemical synthesis and chemical biology and the importance of persistence to improve existing synthetic technologies and innovate new ones in the cause of pursuing the total synthesis of a target molecule. Indeed, that this campaign yielded so much new science is a testament to the value of such total synthesis endeavors and the importance of courage to pursue the unknown.
Acknowledgment
We are grateful to the Skaggs Institute for Research (predoctoral fellowships to S. T. H. and J. S. C.) and the National Institutes of Health (USA) for financial support for this work.
References
- (1).(a) Riedlinger J, Reicke A, Zähner H, Krismer B, Bull AT, Maldonado LA, Ward AC, Goodfellow M, Bister B, Bischoff D, Süssmuth RD, Fiedler H-P. J. Antibiot. 2004;57:271. doi: 10.7164/antibiotics.57.271. [DOI] [PubMed] [Google Scholar]; (b) Bister B, Bischoff D, Ströbele M, Riedlinger J, Riecke A, Wolter F, Bull AT, Zähner H, Fiedler H-P, Süssmuth RD. Angew. Chem. Int. Ed. 2004;43:2574. doi: 10.1002/anie.200353160. [DOI] [PubMed] [Google Scholar]
- (2).Walsh CT, Liu J, Rusnak F, Sakaitani M. Chem. Rev. 1990;90:1105. [Google Scholar]
- (3).Zapf CW, Harrison BA, Drahl C, Sorensen EJ. Angew. Chem. Int. Ed. 2005;44:6533. doi: 10.1002/anie.200502119. [DOI] [PubMed] [Google Scholar]
- (4).For selected reviews on the Diels-Alder reaction and its applications to total synthesis, see:Corey EJ. Angew. Chem. Int. Ed. 2002;41:1650. doi: 10.1002/1521-3773(20020517)41:10<1650::aid-anie1650>3.0.co;2-b.Nicolaou KC, Snyder SA, Montagnon T, Vassilikogiannakis G. Angew. Chem. Int. Ed. 2002;41:1668. doi: 10.1002/1521-3773(20020517)41:10<1668::aid-anie1668>3.0.co;2-z.
- (5).Snider BB, Zou Y. Org. Lett. 2005;7:4939. doi: 10.1021/ol0518941. [DOI] [PubMed] [Google Scholar]
- (6).Couladouros EA, Bouzas EA, Magos AD. Tetrahedron. 2006;62:5272. [Google Scholar]
- (7).(a) Rath J-P, Eipert M, Kinast S, Maier ME. Synlett. 2005:314. [Google Scholar]; (b) Rath J-P, Kinast S, Maier ME. Org. Lett. 2005;7:3089. doi: 10.1021/ol0511068. [DOI] [PubMed] [Google Scholar]
- (8).Zografos AL, Yiotakis A, Georgiadis D. Org. Lett. 2005;7:4515. doi: 10.1021/ol051872e. [DOI] [PubMed] [Google Scholar]
- (9).(a) Nicolaou KC, Harrison ST. Angew. Chem. Int. Ed. 2006;45:3256. doi: 10.1002/anie.200601116. [DOI] [PubMed] [Google Scholar]; (b) Nicolaou KC, Harrison ST. J. Am. Chem. Soc. 2007;129:429. doi: 10.1021/ja067083p. [DOI] [PubMed] [Google Scholar]
- (10).For examples of similar Dieckmann condensation reactions, see:Roush WR, Reilly ML, Koyama K, Brown BB. J. Org. Chem. 1997;62:8708.Takeda K, Shibata Y, Sagawa Y, Urahata M, Funaki K, Hori K, Sasahara H, Yoshii E. J. Org. Chem. 1985;50:4673.
- (11).For a review on metathesis reactions in total synthesis, see:Nicolaou KC, Bulger PG, Sarlah D. Angew. Chem. Int. Ed. 2005;44:4490. doi: 10.1002/anie.200500369.
- (12).Ward DE, Abaee MS. Org. Lett. 2000;2:3937. doi: 10.1021/ol0067235. [DOI] [PubMed] [Google Scholar]
- (13).Evans JM, Kallmerten J. Synlett. 1992:269.This catalyst was recommended to us by Prof. K. B. Sharpless.
- (14).Ward DE, Souweha MS. Org. Lett. 2005;7:3533. doi: 10.1021/ol051262e. [DOI] [PubMed] [Google Scholar]
- (15).Corey EJ, Helal CJ. Angew. Chem. Int. Ed. 1998;37:1986. doi: 10.1002/(SICI)1521-3773(19980817)37:15<1986::AID-ANIE1986>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- (16).Prusov E, Röhm H, Maier ME. Org. Lett. 2006;8:1025. doi: 10.1021/ol052917e. [DOI] [PubMed] [Google Scholar]
- (17).Takeda K, Kawanishi E, Nakamura H, Yoshii E. Tetrahedron Lett. 1991;32:4925. [Google Scholar]
- (18).(a) Scholl M, Ding S, Lee CW, Grubbs RH. Org. Lett. 1999;1:953. doi: 10.1021/ol990909q. [DOI] [PubMed] [Google Scholar]; (b) Trnka TM, Morgan JP, Sanford MS, Wilhelm TE, Scholl M, Choi T-L, Ding S, Day MW, Grubbs RH. J. Am. Chem. Soc. 2003;125:2546. doi: 10.1021/ja021146w. [DOI] [PubMed] [Google Scholar]
- (19).Hong SH, Sanders DP, Lee CW, Grubbs RH. J. Am. Chem. Soc. 2005;127:17160. doi: 10.1021/ja052939w. [DOI] [PubMed] [Google Scholar]
- (20).In ref. 9b, we disclosed that isomerization between 1 and 1a occurs at 180 °C in 1,2-dichlorobenzene. However, it is not clear whether this is occurring thermally or mediated by trace amounts of HCl released by the solvent at elevated temperature.
- (21).The identity of iso-abyssomicin C was later confirmed: personal communication with Prof. E. J. Sorensen.
- (22).For selected examples of other stable atropisomers in strained medium-sized rings lacking obvious steric barriers, see:Shea KJ, Gilman JW, Haffner CD, Dougherty TK. J. Am. Chem. Soc. 1986;108:4953.Anastasiou D, Campi EM, Chaouk H, Jackson WR. Tetrahedron. 1992;48:7467.
- (23).Keller S, Nicholson G, Drahl C, Sorensen EJ, Fiedler H-P, Süssmuth RD. J. Antibiot. 2007;60:391. doi: 10.1038/ja.2007.54. [DOI] [PubMed] [Google Scholar]
- (24).Keller S, Schadt HS, Ortel I, Süssmuth RD. Angew. Chem. Int. Ed. 2007;46:8284. doi: 10.1002/anie.200701836. [DOI] [PubMed] [Google Scholar]