

Abstract
Migraine attacks associated with throbbing (manifestation of peripheral sensitization) and cutaneous allodynia (manifestation of central sensitization) are readily terminated by intravenous administration of a non-selective cyclooxygenase (COX) inhibitor. Evidence that sensitization of rat central trigeminovascular neurons was also terminated in vivo by non-selective COX inhibition has led us to propose that COX inhibitors may act centrally in the dorsal horn. In the present study, we examined whether COX inhibition can also suppress peripheral sensitization in meningeal nociceptors. Using single-unit recording in the trigeminal ganglion in vivo, we found that intravenous infusion of naproxen, a non-selective COX inhibitor, reversed measures of sensitization induced in meningeal nociceptors by prior exposure of the dura to inflammatory soup (IS): ongoing activity of Aδ- and C-units and their response magnitude to mechanical stimulation of the dura, which were enhanced after IS, returned to baseline after naproxen infusion. Topical application of naproxen or the selective COX-2 inhibitor N-[2-(cyclohexyloxy)-4-nitrophenyl]-methanesulfonamide (NS-398) onto the dural receptive field of Aδ- and C-unit nociceptors also reversed the neuronal hyper-responsiveness to mechanical stimulation of the dura. The findings suggest that local COX activity in the dura could mediate the peripheral sensitization that underlies migraine headache.
Keywords: headache, inflammation, migraine, NSAID, pain, trigeminal
Introduction
Having studied the rat trigeminovascular system as a model for neural pathways underlying migraine pain, we have shown that chemical irritation of the cerebral dura mater can activate and sensitize meningeal nociceptors in the trigeminal ganglion and central trigeminovascular neurons in the medullary dorsal horn (Strassman et al., 1996; Burstein, 2001; Levy & Strassman, 2002). We proposed that sensitization of meningeal nociceptors mediates the throbbing nature of migraine headache, and that sensitization of central trigeminovascular neurons mediates cephalic allodynia and muscle tenderness that develop during migraine (Burstein, 2001). When we asked patients to distinguish between pain intensity and the throbbing of migraine headache, we found that triptan therapy consistently terminated the throbbing aspect of the pain; on the other hand, rendering the patient pain free was successful in the absence, but not in the presence, of well-established allodynia at the time of treatment (Burstein et al., 2004).
To understand this discrepancy, we studied the effects of triptans on the induction and maintenance of peripheral and central sensitization in the trigeminovascular pathway. We found that the treatment inhibited neither the sensitized meningeal nociceptors nor the sensitized central neurons, and concluded that triptan action is exerted through blockade of synaptic transmission between the two types of neurons (Burstein & Jakubowski, 2004; Levy et al., 2004). In search of drugs capable of directly inhibiting both the peripheral and central neurons, we focused on non-steroidal anti-inflammatory drugs (NSAIDs) because we found that one of them (ketorolac) was effective in terminating migraine pain in allodynic patients who had failed to respond to triptan therapy earlier in the same attack (Jakubowski et al., 2005).
The analgesic effect of NSAIDs in many inflammatory pain conditions is generally thought to be exerted through inhibition of the two cyclooxygenase (COX) isoforms (Zeilhofer & Brune, 2006). Our working hypothesis was that NSAIDs might inhibit meningeal nociceptors and central trigeminovascular neurons through local inhibition of COX activity in the meninges and dorsal horn, respectively. Hitherto, we have found that naproxen, a non-selective COX inhibitor, was extremely effective in suppressing ongoing activity as well as mechanical and thermal hypersensitivity in sensitized central trigeminovascular neurons. We concluded that the dorsal horn constitutes at least one critical site of action for this class of drugs (Jakubowski et al., 2007). The present study focused on NSAID action at the peripheral side of the trigeminovascular system, meningeal nociceptors. Using extracellular single-unit recording we tested whether ongoing sensitization of rat meningeal nociceptors can be reversed by administration of the non-selective COX inhibitor naproxen, or the more selective COX-2 inhibitor N-[2-(cyclohexyloxy)-4-nitrophenyl]-methanesulfonamide (NS-398).
Materials and methods
Animals
Sprague–Dawley male rats (250–300 g) were used in compliance with the experimental protocol approved by the institutional Animal Care and Use Committee of the Harvard Medical School and in accordance with the Ethical Guidelines of the International Association for the Study of Pain.
Electrophysiology
Rats were deeply anesthetized with an initial intraperitoneal dose of 1.8 g/kg urethane plus 0.2 g/kg supplemental doses as needed, and fitted with an intravenous plastic cannula for drug or saline infusions through the femoral vein. Single-unit recording of meningeal nociceptors in the trigeminal ganglion was recorded as described before (Strassman et al., 1996; Levy et al., 2005). Briefly, the rat's head was mounted in a stereotaxic apparatus (David Kopf), and a 2 × 2-mm craniotomy was performed to expose the dura over the mid-sagittal and left transverse sinuses. The exposed dura was bathed with a modified synthetic interstitial fluid (SIF; in mm: NaCl, 135; KCl, 5; MgCl2, 1; CaCl2, 5; glucose, 10; HEPES, 10; pH 7.2). A platinum-coated tungsten microelectrode (impedance 500 kΩ; FHC, Bowdoin, ME, USA) was lowered into the trigeminal ganglion through a small circular opening (2 mm wide) that was drilled in the left parietal bone, about 2 mm caudal to the Bregma suture and 2 mm left to the midline. Using repeated electrical stimulation of the dura overlying the ipsilateral transverse sinus (0.5 ms pulse, 5 mA, 0.5 Hz), a neuron exhibiting constant response latencies was identified as a meningeal nociceptor. The site on the dura from which electrical stimulation produced the shortest response latency was identified and used to determine the conduction velocity (CV) of the nociceptor, as described previously (Strassman & Raymond, 1999). Assuming a 12.5-mm conductance distance between the stimulation site on dura and the recording site in the trigeminal ganglion, a neuron was classified as a C-unit (CV ≤ 1.5 m/s) or an Aδ-unit (CV > 1.5 m/s). A waveform of the action potential evoked by the electrical stimuli was stored as a template using a real-time waveform discriminator (Spike 2, CED, Cambridge, UK), which was used to acquire experimental data and perform on- and off-line analyses. In all experiments, only one unit was tested in each animal.
Mechanical receptive fields of meningeal nociceptors were mapped initially by stroking the dura with blunt forceps. The lowest threshold point was determined using a series of calibrated von Frey monofilaments ranging from 0.39 to 58.82 mN (exerting pressure stimuli in the range of 38-443 kPa). Response magnitudes to mechanical stimulation were determined quantitatively, using a servo force-controlled mechanical stimulator (Aurora Scientific, Aurora, ON, Canada) fitted with a flat-ended plastic cylinder (0.5 mm diameter) aimed at the lowest threshold point on the dura. Stimulus trials for testing changes in mechanical sensitivity consisted of a graded series of three square-wave stimuli (100 ms rise time, 2 s width, 60 s interstimulus interval) delivered in ascending order, which included a threshold and two suprathreshold stimuli. Stimuli that evoked 1–2 Hz afferent discharge were considered as threshold. Suprathreshold stimuli were usually two and four times greater than threshold. Responses to these stimuli, as well as ongoing spontaneous activity, were recorded every 15 min throughout the experiment. Baseline measurements of spontaneous and mechanically evoked activity were obtained prior to drug administration. Only units that exhibited consistent responses (changes of less than 0.5 and 2.5 Hz for the threshold and suprathreshold responses, respectively, and 0.3 Hz for the ongoing activity level) in at least three consecutive baseline trials were tested further.
Experimental design
Activation and sensitization of meningeal nociceptors was induced by topically applying an inflammatory soup (IS) containing 0.1 mm prostaglandin E2, 1 mm histamine, serotonin, bradykinin, pH 5.5 (Steen et al., 1992, 1995, 1996). The exposed dura was bathed in IS for 15 min followed by wash with SIF as described before (Strassman et al., 1996; Levy & Strassman, 2002; Levy et al., 2004). Neurons were tested further only if they showed clear signs of activation and sensitization.
Given that sensitization induced by this method persists for at least 2 h (Levy et al., 2004; see also Fig. 2), the ability of the NSAIDs to reverse peripheral sensitization was tested 1 h after IS using naproxen sodium, a water-soluble, non-selective COX inhibitor (Sigma, St Louis, MO, USA), or NS-398, a selective COX-2 inhibitor (Cayman Chemicals, Ann Arbor, MI, USA). Naproxen was administered either systemically or directly onto the dural receptive field of the neuron. Systemic naproxen administration (1 mg/kg) was performed using 0.25–0.30 mL of a 1 mg/mL solution (made using a sterile saline solution) and infused intravenously over 5 min. Direct naproxen application was performed by bathing the exposed area of the dura for 1 h in 30 μL of a 0.1 mg/mL solution made in SIF. NS-398 was first dissolved in dimethylsulfoxide (DMSO) and then SIF (final concentration of DMSO 0.1%), and was applied topically as described for naproxen, by bathing the exposed area of the dura for 1 h in 30 μL of a 3 μg/mL solution. Drug doses chosen for this study were derived from previous reports (Morioka et al., 2002; Huntjens et al., 2005) and our own preliminary studies. A control group was treated with IS as described above, while receiving an infusion of 0.3 mL saline instead of naproxen plus SIF (which was the final vehicle for the topically applied naproxen and NS-398) on the dura. At the end of the experiment rats were killed with an intravenous bolus of 1 m KCl.
Fig. 2.
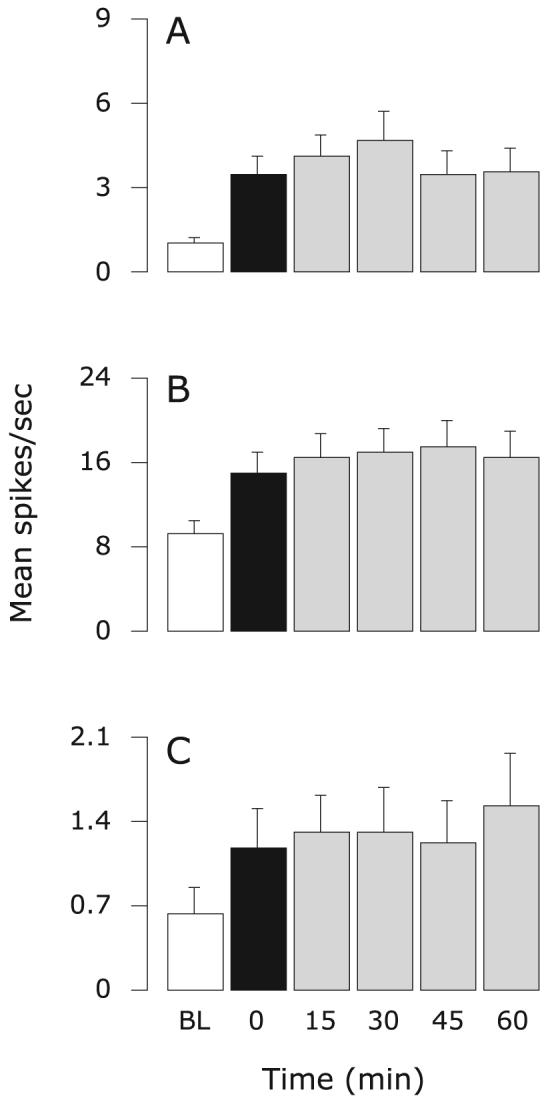
Control group: effects of intravenous infusion of saline and topical application of SIF on sensitized meningeal nociceptors. Mean ± SEM response magnitude to mechanical stimulation of the dura using threshold (A) and suprathreshold (B) pressure stimuli, and ongoing firing rate (C) before (white) and after IS (black), and again after control treatment (grey). Friedman test: comparing all six time points (α = 0.05): P ≤ 0.0001 (A–C); comparing five time points from 0 to 60 min (α = 0.025): P ≥ 0.05 (A), P ≥ 0.05 (B), P ≥ 0.3 (C). BL, baseline.
Data analysis
Data are presented as mean ± SEM. Aδ- and C-units showed comparable response profiles to the various treatments and were therefore combined for the purpose of statistical analysis. Neuronal ongoing activity and responses magnitude to mechanical stimulation of the dura were analysed using the Friedman test. The test was first applied on all six time points: baseline, 60 min after IS (= time 0) and 15, 30, 45 and 60 min after treatment with NSAID or vehicle. To dissect the effect of NSAID or vehicle treatment, the Friedman test was repeated using the five time points from 0 to 60 min. To determine whether the resulting measurements returned to the initial baseline values, the test was repeated using the 0, 30, 45 and 60 min time points. For the first overall analysis, the level of significance was set at 0.05. For the subsequent two post hoc repetitions of the test, the Bonferroni correction was applied to lower the level of significance to 0.025 (i.e. 0.05/2).
Results
Effects of systemic infusion of naproxen on sensitized meningeal nociceptors
Fourteen meningeal nociceptors were tested for the effects of naproxen infusion: six were Aδ-units (mean CV: 4.09 ± 1.37; range 1.67–10.42 m/s) and eight were C-units (mean CV: 0.93 ± 0.13; range 0.48–1.47 m/s; see example Fig. 1A). The control group consisted of 18 neurons: nine were Aδ-units (mean CV: 3.87 ± 0.86; range 1.76–10.00 m/s) and nine were C-units (mean CV: 0.83 ± 0.39; range 0.39–1.47 m/s).
Fig. 1.

Naproxen inhibition of sensitized C-unit meningeal nociceptor. (A) Identification and isolation of a C-unit meningeal nociceptor based on the response latency (16 ms) to electrical stimulation of the dura and a consistent waveform (insert). (B) Peri-stimulus time histograms depicting the responses to threshold and suprathreshold mechanical stimulation of the dura, as well as ongoing firing rate at baseline before IS, 60 min after IS, and then 15 and 30 min after intravenous administration of naproxen. The numbers in parentheses indicate mean spikes/s. Note the inhibition of responses to mechanical stimulation as well as ongoing firing rate.
In the control group, application of IS on the dura produced significant, long-lasting increases in neuronal responses to mechanical stimulation of the dura and ongoing firing rate that remained heightened after vehicle infusion (Friedman test: comparing baseline, 30, 45 and 60 min: P ≤ 0.002; Fig. 2A–C). In contrast, responses to mechanical stimuli as well as ongoing activity, which increased after IS, were significantly suppressed in the group that received naproxen infusion (Figs 1B and 3). The resulting threshold response and ongoing activity from 30 to 60 min after naproxen infusion were comparable to baseline values (Friedman test: comparing baseline, 30, 45 and 60 min: P ≥ 0.4; Fig. 3A; P ≥ 0.1; Fig. 3C). The resulting suprathreshold responses from 30 to 60 min were significantly lower than baseline (P ≤ 0.001; Fig. 3B; Friedman test).
Fig. 3.

Effects of intravenous infusion of naproxen on sensitized meningeal nociceptors. Mean ± SEM response magnitude to mechanical stimulation of the dura using threshold (A) and suprathreshold (B) pressure stimuli, and ongoing firing rate (C) before (white) and after IS (black), and again after treatment (grey). Friedman test: comparing all six time points (α = 0.05): P ≤ 0.0001 (A–C); comparing five time points from 0 to 60 min (α = 0.025): P ≤ 0.0001 (A–C). BL, baseline.
Effects of topical application of naproxen on sensitized meningeal nociceptors
Eight meningeal nociceptors were studied: four were Aδ-units (mean CV: 3.84 ± 0.81; range 2.84-6.50 m/s); four were C-units (mean CV: 0.82 ± 0.16; range 0.52–1.25 m/s). IS-induced enhanced threshold and suprathreshold response magnitudes were significantly suppressed after topical application of naproxen on the dura (Fig. 4A and B). The enhanced ongoing activity also dropped progressively over the same interval, though this change fell short of reaching statistical significance (Fig. 4C). The resulting threshold and suprathreshold values from 30 to 60 min after topical application of naproxen on the dura were either comparable to the baseline values (threshold: P > 0.1; Fig. 4A; Friedman test) or lower than baseline values (suprathreshold: P < 0.025; Fig. 4B; Friedman test).
Fig. 4.
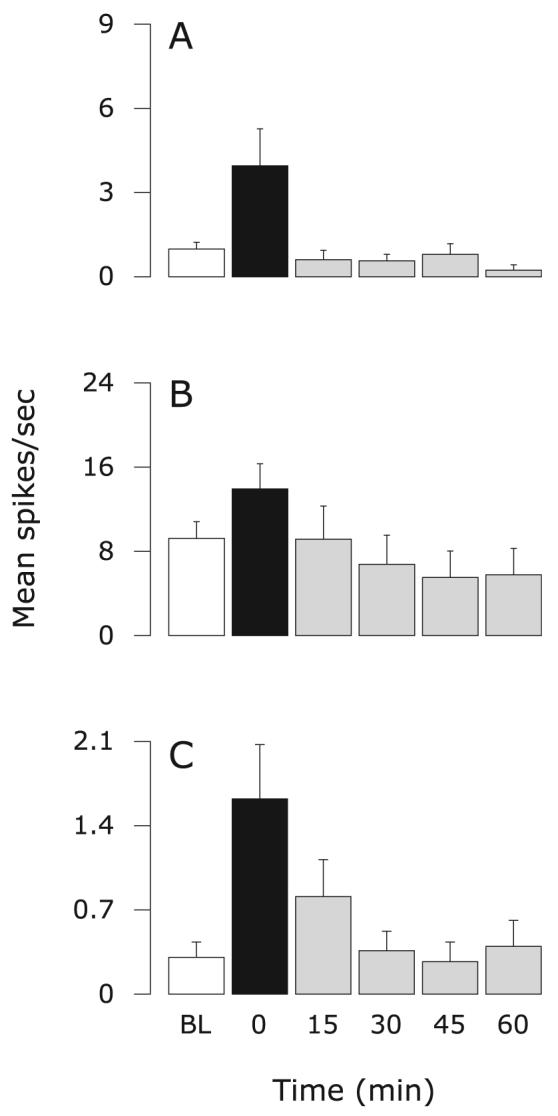
Effects of topical application of naproxen on sensitized meningeal nociceptors. Mean ± SEM response magnitude to mechanical stimulation of the dura using threshold (A) and suprathreshold (B) pressure stimuli, and ongoing firing rate (C) before (white) and after IS (black), and again after treatment (grey). Friedman test: comparing all six time points (α = 0.05): P ≤ 0.001 (A and B), P ≤ 0.05 (C); comparing five time points from 0 to 60 min (α = 0.025): P ≤ 0.001 (A and B), P = 0.031 (C). BL, baseline.
Effects of topical application of NS-398 on sensitized meningeal nociceptors
Ten meningeal nociceptors were studied: five were Aδ-units (mean CV: 2.17 ± 0.29; range 1.52–3.13 m/s); five were C-units (mean CV: 0.88 ± 0.18; range 0.46–1.47 m/s). All three measures of neuronal sensitization were significantly suppressed after topical application of NS-398 onto the dura (Fig. 5). The resulting values from 30 to 60 min after NS-398 infusion were comparable to the respective baseline values (P ≥ 0.2; Fig. 5A; P ≥ 0.1; Fig. 5B and C; Friedman test).
Fig. 5.
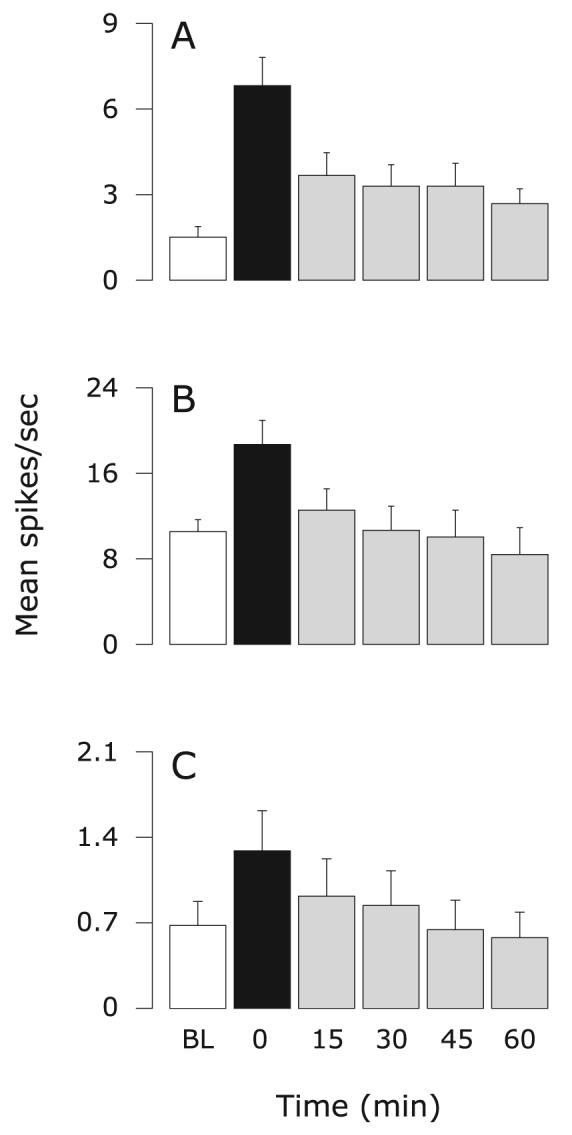
Effects of topical application of NS-398 on sensitized meningeal nociceptors. Mean ± SEM response magnitude to mechanical stimulation of the dura using threshold (A) and suprathreshold (B) pressure stimuli, and ongoing firing rate (C) before (white) and after IS (black), and again after treatment (grey). Friedman test: comparing all six time points (α = 0.05): P ≤ 0.0001 (A and B), P ≤ 0.05 (C); comparing five time points from 0 to 60 min (α = 0.025): P ≤ 0.0005 (A and B), P ≤ 0.015 (C). BL, baseline.
Discussion
Systemic infusion of naproxen, a non-selective COX inhibitor, reversed ongoing sensitization in meningeal nociceptors. Ongoing neuronal activity and response magnitudes to mechanical stimulation of the dura, which increased significantly after stimulating the dura with IS, returned to baseline within 60 min of naproxen infusion. Topical application of naproxen (a non-selective COX inhibitor) or NS-398 (a selective COX-2 inhibitor) onto the dural receptive field of the nociceptor also resulted in reversal of sensitization to mechanical stimulation of the dura. In the control group, all measures of sensitization remained elevated throughout the experiment, confirming that NSAID treatment indeed inhibited sensitization, which would have continued otherwise.
The finding that direct application of naproxen on the dura was as effective as systemic naproxen infusion in suppressing IS-induced neuronal hyper-responsiveness suggests that naproxen action was exerted, at least in part, at the level of the dura mater. That direct application of NS-398 (10 μm) on the dura produced similar suppression of neuronal hyper-responsiveness as topical application of naproxen (400 μm), likely indicates that naproxen action was mediated, at least in part, through local inhibition of COX-2 activity in the dura. Such local inhibition of the nociceptor could be mediated potentially through local blockade of COX-2 in adjacent components of the dura, such as in resident macrophages, blood vessels (Schiltz & Sawchenko, 2002) or even within the nociceptive nerve fibers themselves (Mayer et al., 2007; Levy, D., unpublished results), resulting in decreased levels of prostaglandins in the vicinity of the nociceptor terminals.
Although blockade of COX activity is generally believed to underlie the analgesic effect of NSAIDs, the relatively short latency (within 30 min) for inhibition of meningeal nociceptors mechanosensitivity by both naproxen and NS-398 in our study would be more consistent with a COX-independent mechanism, such as direct inhibition of neuronal excitability through the modulation of sodium currents (Lee et al., 2003; Acosta et al., 2007; Park et al., 2007). For example, both tetrodotoxin (TTX)-sensitive and TTX-resistant currents were shown to be inhibited in dissociated dorsal root ganglion neurons by 14 and 97 μm of diclofenac, respectively (Lee et al., 2003), and by 6 μm and 19 μm of celecoxib, respectively (Park et al., 2007). While these in vitro studies cannot be easily compared with our in vivo study, we cannot rule out that naproxen may act similarly by blocking both TTX-sensitive and TTX-resistant sodium channels in meningeal nociceptors.
Intravenous infusion of naproxen, as well as other NSAIDs, is extremely effective in suppressing both sensitized (Jakubowski et al., 2005, 2007) and non-sensitized (Kaube et al., 1993; Ellrich et al., 1999) trigeminovascular neurons in the dorsal horn. The present evidence for inhibition of sensitized meningeal nociceptors suggests that the inhibitory effect observed in the central neurons is mediated, at least in part, by a peripheral action of NSAIDs in the meninges. Thus, NSAID-mediated inhibition of sensitized peripheral and central trigeminovascular neurons is likely to account for their abortive action in migraine therapy.
Acknowledgements
This study was supported by NIH grants NS46502 (D.L.), NS051484 (R.B.), and grants from Glaxo-Smith-Kline (R.B.) and the National Headache Foundation (D.L.). We thank Dr John M. Kelly for his valuable comments on the statistical analysis.
Abbreviations
- COX
cyclooxygenase
- CV
conduction velocity
- DMSO
dimethylsulfoxide
- IS
inflammatory soup
- NSAID
non-steroidal anti-inflammatory drug
- NS-398
N-[2-(cyclohexyloxy)-4-nitrophenyl]-methanesulfonamide
- SIF
synthetic interstitial fluid
- TTX
tetrodotoxin
Footnotes
Publisher's Disclaimer: Copyright of European Journal of Neuroscience is the property of Blackwell Publishing Limited and its content may not be copied or emailed to multiple sites or posted to a listserv without the copyright holder's express written permission. However, users may print, download, or email articles for individual use.
References
- Acosta MC, Luna C, Graff G, Meseguer VM, Viana F, Gallar J, Belmonte C. Comparative effects of the nonsteroidal anti-inflammatory drug nepafenac on corneal sensory nerve fibers responding to chemical irritation. Invest. Ophthalmol. Vis. Sci. 2007;48:182–188. doi: 10.1167/iovs.06-0710. [DOI] [PubMed] [Google Scholar]
- Burstein R. Deconstructing migraine headache into peripheral and central sensitization. Pain. 2001;89:107–110. doi: 10.1016/s0304-3959(00)00478-4. [DOI] [PubMed] [Google Scholar]
- Burstein R, Collins B, Jakubowski M. Defeating migraine pain with triptans: a race against the development of cutaneous allodynia. Ann. Neurol. 2004;55:19–26. doi: 10.1002/ana.10786. [DOI] [PubMed] [Google Scholar]
- Burstein R, Jakubowski M. Analgesic triptan action in an animal model of intracranial pain: a race against the development of central sensitization. Ann. Neurol. 2004;55:27–36. doi: 10.1002/ana.10785. [DOI] [PubMed] [Google Scholar]
- Ellrich J, Schepelmann K, Pawlak M, Messlinger K. Acetylsalicylic acid inhibits meningeal nociception in rat. Pain. 1999;81:7–14. doi: 10.1016/s0304-3959(98)00267-x. [DOI] [PubMed] [Google Scholar]
- Huntjens DR, Danhof M, Della Pasqua OE. Pharmacokinetic-pharmacodynamic correlations and biomarkers in the development of COX-2 inhibitors. Rheumatology (Oxford) 2005;44:846–859. doi: 10.1093/rheumatology/keh627. [DOI] [PubMed] [Google Scholar]
- Jakubowski M, Levy D, Goor-Aryeh I, Collins B, Bajwa Z, Burstein R. Terminating migraine with allodynia and ongoing central sensitization using parenteral administration of COX1/COX2 inhibitors. Headache. 2005;45:850–861. doi: 10.1111/j.1526-4610.2005.05153.x. [DOI] [PubMed] [Google Scholar]
- Jakubowski M, Levy D, Kainz V, Zhang XC, Kosaras B, Burstein R. Sensitization of central trigeminovascular neurons: blockade by intravenous naproxen infusion. Neuroscience. 2007;148:573–583. doi: 10.1016/j.neuroscience.2007.04.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaube H, Hoskin KL, Goadsby PJ. Intravenous acetylsalicylic acid inhibits central trigeminal neurons in the dorsal horn of the upper cervical spinal cord in the cat. Headache. 1993;33:541–544. doi: 10.1111/j.1526-4610.1993.hed3310541.x. [DOI] [PubMed] [Google Scholar]
- Lee HM, Kim HI, Shin YK, Lee CS, Park M, Song JH. Diclofenac inhibition of sodium currents in rat dorsal root ganglion neurons. Brain Res. 2003;992:120–127. doi: 10.1016/j.brainres.2003.08.048. [DOI] [PubMed] [Google Scholar]
- Levy D, Burstein R, Strassman AM. Calcitonin gene-related peptide does not excite or sensitize meningeal nociceptors: implications for the pathophysiology of migraine. Ann. Neurol. 2005;58:698–705. doi: 10.1002/ana.20619. [DOI] [PubMed] [Google Scholar]
- Levy D, Jakubowski M, Burstein R. Disruption of communication between peripheral and central trigeminovascular neurons mediates the antimigraine action of 5HT 1B/1D receptor agonists. Proc. Natl Acad. Sci. USA. 2004;101:4274–4279. doi: 10.1073/pnas.0306147101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy D, Strassman AM. Distinct sensitizing effects of the cAMP-PKA second messenger cascade on rat dural mechanonociceptors. J. Physiol. (Lond.) 2002;538:483–493. doi: 10.1113/jphysiol.2001.013175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer S, Izydorczyk I, Reeh PW, Grubb BD. Bradykinin-induced nociceptor sensitisation to heat depends on cox-1 and cox-2 in isolated rat skin. Pain. 2007;130:14–24. doi: 10.1016/j.pain.2006.10.027. [DOI] [PubMed] [Google Scholar]
- Morioka N, Inoue A, Hanada T, Kumagai K, Takeda K, Ikoma K, Hide I, Tamura Y, Shiomi H, Dohi T, Nakata Y. Nitric oxide synergistically potentiates interleukin-1 beta-induced increase of cyclooxygenase-2 mRNA levels, resulting in the facilitation of substance P release from primary afferent neurons: involvement of cGMP-independent mechanisms. Neuropharmacology. 2002;43:868–876. doi: 10.1016/s0028-3908(02)00143-0. [DOI] [PubMed] [Google Scholar]
- Park SY, Kim TH, Kim HI, Shin YK, Lee CS, Park M, Song JH. Celecoxib inhibits Na+ currents in rat dorsal root ganglion neurons. Brain Res. 2007;1148:53–61. doi: 10.1016/j.brainres.2007.02.023. [DOI] [PubMed] [Google Scholar]
- Schiltz JC, Sawchenko PE. Distinct brain vascular cell types manifest inducible cyclooxygenase expression as a function of the strength and nature of immune insults. J. Neurosci. 2002;22:5606–5618. doi: 10.1523/JNEUROSCI.22-13-05606.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steen KH, Issberner U, Reeh PW. Pain due to experimental acidosis in human skin: evidence for non-adapting nociceptor excitation. Neurosci. Lett. 1995;199:29–32. doi: 10.1016/0304-3940(95)12002-l. [DOI] [PubMed] [Google Scholar]
- Steen KH, Reeh PW, Anton F, Handwerker HO. Protons selectively induce lasting excitation and sensitization to mechanical stimulation of nociceptors in rat skin, in vitro. J. Neurosci. 1992;12:86–95. doi: 10.1523/JNEUROSCI.12-01-00086.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steen KH, Steen AE, Kreysel HW, Reeh PW. Inflammatory mediators potentiate pain induced by experimental tissue acidosis. Pain. 1996;66:163–170. doi: 10.1016/0304-3959(96)03034-5. [DOI] [PubMed] [Google Scholar]
- Strassman AM, Raymond SA. Electrophysiological evidence for tetrodotoxin-resistant sodium channels in slowly conducting dural sensory fibers. J. Neurophysiol. 1999;81:413–424. doi: 10.1152/jn.1999.81.2.413. [DOI] [PubMed] [Google Scholar]
- Strassman AM, Raymond SA, Burstein R. Sensitization of meningeal sensory neurons and the origin of headaches. Nature. 1996;384:560–564. doi: 10.1038/384560a0. [DOI] [PubMed] [Google Scholar]
- Zeilhofer HU, Brune K. Analgesic strategies beyond the inhibition of cyclooxygenases. Trends Pharmacol. Sci. 2006;27:467–474. doi: 10.1016/j.tips.2006.07.007. [DOI] [PubMed] [Google Scholar]