


Abstract
Rapid and reliable virus subtype identification is critical for accurate diagnosis of human infections, effective response to epidemic outbreaks and global-scale surveillance of highly pathogenic viral subtypes such as avian influenza H5N1. The polymerase chain reaction (PCR) has become the method of choice for virus subtype identification. However, designing subtype-specific PCR primer pairs is a very challenging task: on one hand, selected primer pairs must result in robust amplification in the presence of a significant degree of sequence heterogeneity within subtypes, on the other, they must discriminate between the subtype of interest and closely related subtypes. In this article, we present a new tool, called PrimerHunter, that can be used to select highly sensitive and specific primers for virus subtyping. Our tool takes as input sets of both target and nontarget sequences. Primers are selected such that they efficiently amplify any one of the target sequences, and none of the nontarget sequences. PrimerHunter ensures the desired amplification properties by using accurate estimates of melting temperature with mismatches, computed based on the nearest neighbor model via an efficient fractional programming algorithm. Validation experiments with three avian influenza HA subtypes confirm that primers selected by PrimerHunter have high sensitivity and specificity for target sequences.
INTRODUCTION
RNA viruses, such as avian influenza, hepatitis C virus and human immunodeficiency virus are characterized by a extensive genetic heterogeneity, primarily due to the lack of proofreading mechanisms in their RNA polymerase. As a result, most RNA viruses can be subdivided into distinct taxonomic subunits referred to as genotypes or subtypes. For example, over 100 avian influenza subtypes have been identified in wild birds as the result of independent assortment of 16 subtypes of the RNA segment encoding the hemagglutinin (HA) protein with nine subtypes of the segment encoding for neuraminidase (NA). Rapid virus subtype identification is critical for accurate diagnosis of human infections, effective response to epidemic outbreaks and global-scale surveillance of highly pathogenic subtypes such as avian influenza H5N1 (1).
The polymerase chain reaction (PCR) has become the method of choice for virus subtype identification, largely replacing traditional immunological assays due to its high sensitivity and specificity, fast response time and affordable cost (2). However, designing subtype-specific PCR primer pairs is a very challenging task (3): on one hand, selected primer pairs must result in robust amplification in the presence of a significant degree of sequence heterogeneity within subtypes, on the other, they must discriminate between the subtype of interest and closely related subtypes.
Unfortunately, existing primer design tools are not well suited for designing PCR primers for subtype identification. Commonly used packages such as Primer3 (4,5) seek to amplify a single known target nucleic acid sequence, and cannot guarantee amplification sensitivity in the presence of high sequence heterogeneity within a subtype. A widely used approach to primer design for virus identification relies on first constructing a ‘consensus gestalt’ from a multiple alignment of target virus sequences (6). After masking regions that also appear in the genome of related viruses, remaining ‘unique’ regions are mined for primers using standard tools such as Primer3. This approach can be quite successful at finding species-specific primers, since virus genomes often include highly conserved genes and noncoding regions that serve critical roles in replication, transcription and packaging. However, the approach has limited applicability when the goal is to discriminate between virus subtypes, since most highly conserved regions are shared by all subtypes. The same limitation applies to several suffix-tree-based algorithms (7–9) that search for long substrings that appear exactly or with a small number of mutations in all (or a large percentage) of the sequences of a given target set, and in none of the sequences of a given nontarget set.
Another common approach to ensuring amplification of heterogenous sets of nucleic acid sequences is the use of primers with degenerate bases. Several methods have been proposed for selecting degenerate primers, including various greedy algorithms (10–12) and heuristics based on multiple alignments of nucleic acid (13) and protein sequences (14). Unfortunately, all these methods ignore primer specificity (i.e. preventing amplification of related virus subtypes) which prevents their use for direct viral subtyping assays.
A comparison of the main features provided by a selection of most relevant existing primer and probe selection tools (4,8,9,13–19) is presented in Table 1. As it can be seen from the table, most existing tools miss key features that make them inappropriate for use in designing PCR primers for virus subtyping. Of the surveyed methods, only OligoSpawn (16) and SLICSel (17) were successful at finding subtype-specific probes when run on a large set of avian influenza HA sequences. The other methods were either not available, could not handle multiple target/nontarget sequences or simply did not find any subtype-specific primers or probes.
Table 1.
Features comparison between primer and probe selection tools most similar to PrimerHunter
| Design tool | Multiple targets | Nontargets | TM Model | Salt correction | Output |
|---|---|---|---|---|---|
| Primer3 (4) | No | Yes (DB) | NN | Yes | Multiple primer pairs |
| Insignia (9) | Yes (DB) | Yes (DB) | None | No | Multiple signatures |
| QPrimer (15) | No (DB) | No | NN | No | Multiple primers |
| DePict (14) | Yes (MSA) | No | None | No | Best primer |
| PROBEMer (8) | Yes | Yes | NN | No | Multiple probes |
| Greene SCPrimer (13) | Yes (MSA) | No | NN | Yes | Multiple primer pairs |
| OligoSpawn (16) | Yes | Yes | NN | No | Multiple probes |
| SLICSel (17) | Yes | Yes | NN | Yes | Multiple probes |
| Primaclade (18) | Yes (MSA) | No | NN | No | Multiple primers |
| OligoArray (19) | Yes | Yes (DB) | NN | No | Multiple probes |
| PrimerHunter (this article) | Yes | Yes | NN w/mismatches | Yes | Multiple primer pairs |
DB: user can select targets from a preconstructed database; MSA: input must be provided as a multiple sequence alignment; NN: nearest neighbor model.
In this article, we present a new tool, called PrimerHunter, that can be used for selecting highly sensitive and specific primers for virus subtyping and is likely to find applications in other contexts that require discriminative probes/primers. As in (8,9,16), our tool takes as input sets of both target and nontarget sequences. To guarantee high sensitivity, primers are selected such that they efficiently amplify any one of the target sequences representing different isolates of the subtype of interest. High specificity is ensured by requiring that none of the nontarget sequences be amplified by selected primers; nontargets typically being sequences representing isolates of closely related virus subtypes. Unlike previous methods, which restrict the primer search space to the set of substrings shared by all target sequences or to highly conserved regions in a multiple alignment, PrimerHunter achieves a higher design success rate by generating an exhaustive set of candidate primers from the target sequences and using accurate melting temperature computations to ensure the desired amplification/nonamplification properties. Melting temperature computation is performed based on the state-of-the-art nearest neighbor model of (20). Of critical importance in selective target amplification is accurate prediction of primer-template hybridization with mismatches. Melting temperature with mismatches is efficiently computed in PrimerHunter by using the fractional programming approach of (21), modified to incorporate the salt correction model of (20).
PrimerHunter has been used to design specific primer pairs for all avian influenza HA and NA subtypes from complete sequences of North American origin in the NCBI flu database (22). Validation experiments confirm that primers selected by PrimerHunter are both specific and robust in the PCR amplification of target sequences. The PrimerHunter web server, as well as the open source code released under the GNU General Public License, are available at http://dna.engr.uconn.edu/software/PrimerHunter/.
MATERIALS AND METHODS
Problem formulation
Unless stated otherwise, we assume that all sequences are over the DNA alphabet, {A, C, G, T}, and are given in 5′–3′ orientation. For a sequence s, we denote by |s| its length, and by s(l, i) the subsequence of length l ending at position i, i.e. s(l,i) = si−l+1 …si-1si. We denote by T(p,t,i) the melting temperature of the duplex formed by a primer p and the Watson–Crick complement of t(|p|,i). In order to ensure sensitive amplification of target sequences, we require for each selected primer p to have at least one position i within each target t such that T(p,t,i) is greater than or equal to a user-specified threshold Tmintarget. Since mismatches at the 3′-end of the primer can significantly reduce amplification efficiency (23), we additionally require that the 3′-end of p match perfectly t(|p|,i) at a set of bases specified using a 0–1 perfect match mask M. For example, a mask M=3′-1101-5′ specifies that the first, second and fourth 3′ most bases of the primer must be matched exactly. For a primer p and a target sequence t, we denote by ℐ (p, t, M) the set of positions i of t at which the 3′ end of p matches t(|p|,i) according to M. Thus, in order to ensure sensitive PCR amplification of target sequences, we require that a selected primer p have, for every target t, at least one position i ∈ ℐ (p, t, M) for which T(p, t, i) ≥ Tmintarget.
To avoid nonspecific amplification, we further require for each selected primer to have a melting temperature T(p,t,i) below a user-specified threshold Tmaxnontarget at every position i of every nontarget sequence t. The problem of selecting target-specific forward PCR primers is therefore formulated as follows:
Discriminative primer selection problem
Given: sets TARGETS and NONTARGETS of 5′–3′ DNA sequences, perfect match mask M, melting temperature thresholds Tmintarget and Tmaxnontarget and constraints on primer length, GC content, self-complementarity, etc.
Find: primers p satisfying given constraints on primer length, GC content, self-complementarity, etc., such that:
For every t ∈ TARGETS, there exists i ∈ ℐ (p, t, M) such that T(p, t, i) ≥ Tmintarget, and
For every t ∈ NONTARGETS, T(p, t, i) ≤ Tmaxnontarget for every i ∈ {|p|, …, |t|}.
Melting temperature calculation
PrimerHunter estimates the melting temperature of primer-target and primer-nontarget duplexes using the nearest neighbor model of (20), which is considered to be the most accurate melting temperature model to date (24). However, unlike most other primer design packages, which only require estimates of the melting temperature between a primer and its perfectly complementary template, PrimerHunter critically relies on accurate estimates of the melting temperature for noncomplementary duplexes. This requires finding the optimum thermodynamic alignments for all evaluated duplexes, i.e. the alignments with minimum Gibbs free energy. As in (21), optimum alignments are computed using the fractional programming algorithm of (25). In this section, we describe our modification of the algorithm to incorporate SantaLucia's; correction for the concentration of salt cations in the PCR mix (20). As shown below, incorporating this correction yields significantly improved estimates compared to (21).
In SantaLucia's; nearest neighbor model (20), the melting temperature of a specific alignment x between a 5′–3′ primer p with concentration cp and a 3′–5′ template t with concentration ct is given by
| 1 |
where Δ H(x) and Δ S(x) are enthalpy and entropy changes for the annealing reaction resulting in a duplex with Watson–Crick pairings given by alignment x, N is the total number of phosphates in the duplex, R is the gas constant, C is the total DNA concentration calculated as cp − ct/2 if cp > ct and (cp/2) if cp = ct (20) and Na+ is the concentration of salt cations. For a given alignment x, the enthalpy and entropy changes Δ H(x) and Δ S(x) are computed by summing experimentally estimated contributions of constitutive dimer duplexes (including internal mismatches and gaps), with additional terms for duplex initiation/termination and (when applicable) symmetry correction.
The melting temperature between p and t is given by the most stable alignment x, i.e. it is taken to be the maximum TM(x) over all possible alignments x. This maximum can be found using Dinkelbach's; fractional programming algorithm (25), which relies on a simple iterative procedure to maximize the ratio between two functions when linear combinations of the two functions can be maximized efficiently. More specifically, given a finite set S and two functions f,g: S → ℝ with g > 0, the maximum ratio t*=maxx ∈ S (f(x)/g(x)) can be approximated arbitrarily close via the following algorithm:
Choose t1 ≤ t*; i ← 1
Find xi∈ S maximizing F(x) := f(x) − ti g(x)
If F(xi) ≤ ɛ for some tolerance ɛ > 0, output ti
Else, set ti+1 ← f(xi)/g(xi) and i ← i+1, and then go to step 2
As shown by Dinkelbach, this algorithm produces values t1 < t2 < t3 < … converging to t*. When using Dinkelbach's; algorithm to maximize equation (1) over the set of alignments x, the function to be maximized in Step 2 is −Δ G(x) = ti[Δ S(x) + (0.368) × N/2 × ln (Na+) + R × ln (C)] − Δ H(x). Since −Δ G(x) is additively decomposable, the alignment x maximizing it can be found efficiently by a standard dynamic programming algorithm, similar to (21). As shown in (21), the algorithm typically converges in a small number of iterations.
Algorithm
PrimerHunter works in two stages: in the first stage, forward and reverse primers are selected according to the problem formulation given above, while in the second stage, feasible primer pairs are formed using the primers selected in first stage.
The first stage starts with a preprocessing step that builds a hash table storing all occurrences in the target sequences of ‘seed’ nucleotide patterns consistent with the given mask M. This is done by aligning the mask M at every position i of every target sequence t, and storing in the hash table an occurrence of the seed pattern created by extracting from t(|M|, i) the nucleotides that appear at positions aligned with the 1's; of M. For example, if M=3′-1101-5′ and t(4, i) = 5′-GATC-3′, we store in the hash table an occurrence of seed GTC at position i of t.
Once the hash table is constructed, candidate primers are generated by taking substrings with lengths within a user-specified interval [lm, lM] from one or more of the target sequences. Similar to the Primer3 package (4), PrimerHunter filters the list of primer candidates by enforcing user-specified bounds on GC Content, 3′-end GC clamp, maximum number of consecutive mononucleotide repeats and self-complementarity. For each surviving candidate p, PrimerHunter uses the hash table to recover for each target t the list ℐ (p, t, M) of positions at which p matches t according to M. It then computes the melting temperature of p with the Watson–Crick complement of t at each of these positions, retaining p only if 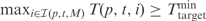 . Finally, PrimerHunter computes the maximum melting temperature between p and the Watson–Crick complements of nontarget sequences, retaining p only if
. Finally, PrimerHunter computes the maximum melting temperature between p and the Watson–Crick complements of nontarget sequences, retaining p only if 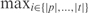 T(p, t, i) ≤ Tmaxnontarget for every nontarget sequence t.
T(p, t, i) ≤ Tmaxnontarget for every nontarget sequence t.
The above process is repeated on the reverse complements of target and nontarget sequences to generate reverse primers. Then, in the second stage of the algorithm, the lists of selected forward and reverse primers are used to create feasible primer pairs by enforcing the following constraints:
Product length: for each target sequence, the total product length must fall between user-specified bounds.
Melting temperature similarity: for every target sequence, the difference between the maximum and the minimum melting temperature of the two primers must not exceed a user defined value.
Primer dimers: a criteria similar to that used for preventing primer self-complementarity is used to avoid hybridization between the two primers of the pair; the test is identical to that implemented in Primer3 (4).
Algorithm extensions
Since degenerate bases at specific primer positions yield perfect matches at these positions regardless of target variability, the use of degenerate primers is an effective technique for ensuring robust amplification of heterogenous targets. However, degenerate primer design is a difficult problem due to the large space from which degenerate primers can be selected (10–12). To overcome this difficulty, we adopted a simple pattern-based approach to degenerate primer design, based on the observation that most of a virus' sequence is coding for proteins and that the vast majority of sequence heterogeneity is observed at synonymous positions. PrimerHunter uses a user-specified degeneracy mask, specifying the positions at which fully degenerate nucleotides should be incorporated in candidate primers. Formally, the degeneracy mask is a vector D of integers 1 or 4 in 3′–5′ orientation. In each position i where Di = 4, a degenerate base N will be included in every primer. For example, if D=3′-114114-5′, every primer will end with the pattern 5′-NxxNxx-3′. A degeneracy mask may be used in conjunction with a complementary perfect match mask (M=3′-110110-5′ for the above D), although this is not required. The only required change to the primer selection algorithm is in the computation of melting temperatures: the range of melting temperatures for a degenerate primer is obtained by computing the melting temperatures against the given template for all compatible nondegenerate primers.
For target sets exhibiting a very high degree of heterogeneity, or for overly stringent design constraints, it may be impossible to find specific primer pairs that amplify all targets. When detecting this situation, PrimerHunter automatically seeks and reports a small set of primer pairs that collectively amplify all targets. The set of pairs is constructed using the classic greedy set cover algorithm (26,27), where the elements to be covered are target sequences and the sets correspond to pairs of compatible primers that amplify at least one of the target sequences and none of the nontargets. From the well-known approximation guarantee in (26,27), it follows that the greedy algorithm yields a number of primer pairs within a factor of 1+ln mt of optimum for mt target sequences.
When multiple primer pairs are needed to cover all targets, the number of primer pairs can be further reduced by relaxing the constraint that forward and reverse primer candidates must amplify all targets. As in (8), this is achieved in PrimerHunter by specifying a minimum percentage of target sequences to which selected primers must hybridize. Similarly, the nontargets filtering can be relaxed, allowing selected primers to hybridize to a small percentage of nontargets. However, to maintain specificity, primer pairs that feasibly amplify one of the nontarget sequences are discarded before running the greedy set cover algorithm.
HA fragment cloning and quantitative PCR
In order to assess the specificity and selectivity of designed primers, HA-coding region fragments from isolates of H3, H5 and H7 avian influenza viruses were cloned into pTOPO (Invitrogen). The subtype identity of each cloned fragment was confirmed by sequencing and confirmed plasmids were diluted and used as on-target and off-target templates for quantitative PCR (Q-PCR) using selected primer pairs.
Q-PCR was performed on an Applied Biosystems 7500 using ABI SYBR green master mix. PCR conditions were as follows: 1 cycle, 95°C × 10 min; 40 cycles, 95°C × 15 sec, 40°C × 15 sec, 60°C × 1 min. Following amplification and detection, melt curves from 60–95°C were performed to confirm specificity of the amplicons.
RESULTS
Accuracy of melting temperature predictions
We compared the accuracy of estimates obtained based on equation (1) to those obtained as in (21) by using a simplified formula that does not include the salt correction term 0.368 × N/2 × ln(Na+) in the denominator. Figure 1 shows the mean and standard deviation of the difference between the melting temperature determined experimentally and that predicted by the two models for a set of 812 duplexes of perfectly complementary oligonucleotides with lengths between 9 and 30 bp, GC content between 8% and 80% and salt concentrations between 0.069 M and 1.02 M (24,28). The data has been stratified in four categories of salt concentration, with ranges given in Table 2. Table 2 also includes the mean squared error (MSE) for each model and each salt concentration category. The results show that predictions given by (1) have much lower MSE values for all salt concentration categories except 1–1.02 M. Although the two models result in identical predictions at 1 M concentration, for salt concentrations >1 M applying the salt correction produces slightly worse estimates. The difference between the two models is statistically significant: within each salt concentration category the null hypothesis that prediction errors of the two models have the same mean is rejected by the Wilcoxon signed-rank test with a P-value <10−16.
Figure 1.

Average and standard deviation of the difference (in degrees Celsius) between experimental melting temperatures and predictions obtained by fractional programming without salt correction (21) and with salt corrections performed using the SantaLucia model equation (1) for 812 duplexes of perfectly complementary oligonucleotides with lengths between 9 and 30 bp and GC content between 8% and 80%.
Table 2.
MSE for residuals calculated as the difference (in degrees Celsius) between experimental melting temperatures and predictions obtained by fractional programming without salt correction (21) and with salt corrections performed using the SantaLucia model equation (1)
| Salt Conc. | Primer length 9–30 | Primer length 20–25 | ||||
|---|---|---|---|---|---|---|
| (M) | GC content 8%–80% |
GC content 25%–75% |
||||
| Number of duplexes | MSE w/o salt correction | MSE with salt correction | Number of duplexes | MSE w/o salt correction | MSE with salt correction | |
| 0.069–0.15 | 351 | 150.03 | 2.30 | 158 | 148.91 | 2.25 |
| 0.22 | 152 | 47.44 | 2.71 | 72 | 43.14 | 3.18 |
| 0.62–0.621 | 152 | 8.98 | 2.52 | 72 | 6.90 | 1.38 |
| 1–1.02 | 157 | 4.75 | 4.97 | 74 | 2.61 | 2.76 |
Since duplexes involving primers with atypical length or GC content could potentially skew the results, we repeated the above comparison by considering only duplexes consisting of primers with length between 20 and 25 bp and GC content between 25% and 75%, which are typical values used in primer design and the default ranges for PrimerHunter. The results shown in Supplementary Figure 1 and Table 2 show that the predictions given by equation (1) remain more accurate than predictions based on (21) for salt concentrations below 1 M even when disregarding primers with extreme GC content or length. In all categories, the null hypothesis that prediction errors of the two models have the same mean is still rejected by the Wilcoxon signed-rank test, with a P-value <10−14.
Unfortunately, experimental data on melting temperature of duplexes with mismatches is much more limited. We could collect only 110 duplexes with one mismatch and 28 duplexes with two mismatches from (29–32). Duplexes with one mismatch have lengths between 9 and 16 bp and GC content between 21% and 78%, while duplexes with two mismatches have lengths between 12 and 14 bp and GC content between 50% and 75%. Except for 12 duplexes with 1 mismatch, the melting temperature of all these duplexes was experimentally calculated at 1 M of salt concentration. Since both prediction models produce exactly the same answer for a salt concentration of 1 M, we did not have enough information to compare them for duplexes with mismatches. Table 3 gives the mean and standard deviation for the prediction errors made by the SantaLucia model equation (1). The results suggest that, although less accurate than in the case of perfectly complementary duplexes, melting temperature estimates for duplexes with mismatches still provide good approximations. (We have also implemented the salt correction model of (28), but found the SantaLucia model to be slightly more accurate.)
Table 3.
Average and standard deviation for the difference (in degrees Celsius) between experimental melting temperature and predictions made by the SantaLucia model equation (1) on duplexes with one and two mismatches
| Number of mismatches | Length range | GC content range | Number of duplexes | Average difference | Standard deviation |
|---|---|---|---|---|---|
| 1 | 9–16 | 21%–78% | 110 | 0.56 | 2.06 |
| 2 | 12–14 | 50%–75% | 28 | −1.25 | 2.70 |
Design success rate
Primer Hunter has been implemented in C++ on a standard Linux platform. We designed primer pairs for 14 HA subtypes using the complete avian influenza HA sequences from North America available in the NCBI flu database (22) as of March 2008 (a total of 574 HA sequences). Figure 2 shows the unrooted phylogenetic tree generated using the TREEVIEW program (33) from a multiple alignment of a subset of these sequences constructed using ClustalW (34).
Figure 2.

Phylogenetic tree of avian influenza HA sequences of North American origin from the NCBI flu database (five complete sequences selected at random for each subtype).
When designing primers for each subtype Hi we used all available HA sequences classified as Hi as targets, and all NCBI HA sequences labeled with different subtypes as nontargets. Primer selection was performed using the following parameters:
Primer length between 20 and 25
Amplicon length between 75 and 200
GC content between 25% and 75%
Maximum mononucleotide repeat of 5
3′-end perfect match mask M = 11
No required 3′ GC clamp
Primer concentration of 0.8 μM
Salt concentration of 50 mM
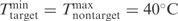
We also attempted to design primer pairs for the nine known NA subtypes based on the 668 avian Influenza NA sequences available in (22), using the same set of parameters as for HA subtypes. An initial PrimerHunter run resulted in primer pairs selected for all subtypes except N4 and N1. Upon inspection of the phylogenetic tree (Supplementary Figure 2) we detected an N1 sequence (GI:115278096) that was mislabeled as N4. After correcting the label of the sequence, PrimerHunter was able to select discriminative primer pairs for all NA subtypes (Supplementary Table 1).
The numbers of identified primer pairs using these parameters are summarized in Table 4. For comparison, we also include in Table 4 the number of probes reported by OligoSpawn (16) and SLICSel (17). These were the only methods among those listed in Table 1 that were available and could run successfully on the HA dataset. OligoSpawn and SLICSel were run using similar settings as PrimerHunter for the common parameters. Using these settings, all three methods were able to identify discriminative primers/probes for each subtype represented in the NCBI flu database. The number of discriminative primers found by PrimerHunter is consistently larger than the number of probes found by OligoSpawn and SLICSel. PrimerHunter identified at least a few tens of forward and reverse primers for each subtype. With an amplicon length constrained to be between 75 and 200 bp, PrimerHunter was able to always identify feasible primer pairs, i.e. pairs of primers predicted to amplify all target sequences and none of the nontarget sequences when using an annealing temperature of 40° C in the PCR reaction. Typically, for identified primers minimum primer-target melting temperature is significantly >40°C, and maximum primer-nontarget melting temperature is significantly <40°C (Supplementary Data). The large number of feasible primers enables further optimizations such as selecting most discriminative primers (based on the difference between minimum primer-target TM and maximum primer-nontarget TM) and TM matching the primers within selected primer pairs.
Table 4.
Primers found for each subtype of avian influenza HA and comparison with number of probes generated by related tools
| Subtype | Number of targets | Number of Nontargets | Avg. percentage of diss. | Number of FP | Number of RP | Number of PP | Number of probes SlicSel | Number of probes OligoSpawn |
|---|---|---|---|---|---|---|---|---|
| H1 | 48 | 526 | 8.4 | 51 | 52 | 70 | 20 | 2 |
| H2 | 41 | 533 | 9.1 | 42 | 43 | 187 | 14 | 2 |
| H3 | 72 | 502 | 11.1 | 41 | 61 | 135 | 7 | 1 |
| H4 | 67 | 507 | 7.4 | 265 | 225 | 3724 | 18 | 2 |
| H5 | 69 | 505 | 9.1 | 68 | 66 | 160 | 17 | 1 |
| H6 | 100 | 474 | 15.4 | 36 | 27 | 3 | 4 | 3 |
| H7 | 55 | 519 | 8.9 | 77 | 81 | 260 | 2 | 1 |
| H8 | 9 | 565 | 6.3 | 489 | 482 | 14415 | 100 | 1 |
| H9 | 23 | 551 | 8.7 | 140 | 152 | 1222 | 58 | 1 |
| H10 | 16 | 558 | 6.8 | 243 | 302 | 3712 | 35 | 1 |
| H11 | 45 | 529 | 5.9 | 267 | 262 | 4117 | 32 | 1 |
| H12 | 15 | 559 | 7.1 | 472 | 494 | 12895 | 52 | 1 |
| H13 | 10 | 564 | 14.4 | 41 | 33 | 98 | 1 | 2 |
| H16 | 4 | 570 | 9.5 | 367 | 352 | 7629 | 68 | 1 |
The dissimilarity within a subtype is calculated as the average pairwise Hamming distance in the multiple sequence alignment expressed as percentage of the average sequence length. (FP: forward primers; RP: reverse primers; PP: primer pairs)
Primer validation
A total of nine randomly selected primer pairs specific to H3, H5 and H7 subtypes (three pairs per subtype, see the Supplementary Data) were ordered from Integrated DNA Technologies (IDT). In a first experiment, triplicate Q-PCR reactions were performed for each primer pair with 1:103 dilutions of each of the three plasmid types as template. Triplicate reactions with no template (no template controls, or NTC) were also performed. Figure 3 gives the amplification curves for a typical experiment where three on-target and six off-target Q-PCR reactions were performed with one of the H3-specific primer pairs. For each reaction, the threshold cycle Ct is defined as the PCR cycle in which the fluorescent signal intensity passes the self-calibrated detection threshold. When no detectable fluorescent signal is present (e.g. in a NTC reaction), Ct is set to 40.
Figure 3.

Amplification curves using an H5-specific primer pair and H3, H5, H7 plasmids or no template (three replicates each).
For each reaction, Δ Ct is computed as the difference between the respective threshold cycle and the average threshold cycle of the three NTC reactions. The minimum, maximum and average Δ Ct values for all nine primer pairs and both on- and off-target templates are given in Figure 4. The results show a large difference (15 cycles or more) between the average on- and off-target Δ Ct values.
Figure 4.

Average ΔCt for on-target (T), off-target (OT1 and OT2) and NTC Q-PCR amplification with nine primer pairs (three subtype-specific pairs for each of H3, H5 and H7; error bars indicate minimum/maximum values).
To assess the discriminative power over a range of template concentrations, three primer pairs (one specific to each of the three cloned subtypes) were used in triplicate Q-PCR reactions performed using each of the on- and off-target plasmids at 10 different dilutions. As can be seen from these graphs, PrimerHunter primer pairs showed template-specific amplification over 5 to 7 orders of magnitude. Figure 5 shows Δ Ct values of these reactions plotted against approximate plasmid copy numbers.
Figure 5.

ΔCt for triplicate Q-PCR reactions performed with H3-, H5- and H7-specific primer pairs at 10 different dilutions of on- and off-target templates. Lines connect triplicate means at each dilution. The legend in each graph indicates the color for the primer (numerator) and target (denominator) combination.
DISCUSSION
PrimerHunter is a new tool to design primers for subtype identification using PCR. Compared to existing tools based on exact matches or multiple sequence alignment, PrimerHunter achieves a higher design success rate by relying on accurate melting temperature computations allowing for mismatches based on the nearest neighbor model of (20) and the fractional programming approach of (21). Using this approach, PrimerHunter can design primers that will selectively amplify target sequences from a complex background of related targets.
We demonstrate the performance of PrimerHunter by designing thousands of primer pairs specific to 14 HA and 9 NA avian influenza subtypes. For the HA subtypes, the number of primers found by PrimerHunter is consistently larger than the number of probes found by two probe design tools with closely related functionality (16,17). The number of discriminative primers and primer pairs found for a subtype is positively correlated with the amount of variability within the subtype and negatively correlated with the average similarity to closely related subtypes. Indeed, for pairs of subtypes such as (H3, H4), (H7, H10), (H8, H12) and (H13, H16) which are nearest neighbors in the NA phylogenetic tree in Figure 2, the subtype with lower within-subtype dissimilarity (included in Table 4) always yields a larger number of primer pairs. For our design parameters, the number of suitable primer pairs varies from three for the highly variable H6 subtype, which has an average within-subtype dissimilarity of 15.4%, to 14 415 for the H8 subtype, which has an average within-subtype dissimilarity of 6.3%. Degenerate primers were not needed by PrimerHunter when designing primer pairs based on avian influenza originating from North America. We expect that degenerate primers will become useful when designing discriminative primer pairs based on world-wide subtype isolates, and we plan to experiment with degenerate primers in the future.
In order to assess the specificity of these primers we tested three primer pairs designed to amplify HA fragments from H3, H5 and H7. To avoid the possibility of contaminated or nonclonal primary viral samples, fragments of the HA gene from one isolate of each subtype were cloned into a plasmid vector. This allowed us to test both the specificity of the PrimerHunter primers on defined on- and off-target sequences, and to assess the performance of the primers over a very large range of template concentrations. We found that in each of these experiments, PrimerHunter primers selectively amplified the targeted HA subtype over 5–7 orders of magnitude of target concentrations and that the target sequence was first detected at 104- to 106-fold lower concentrations than nontarget templates. When template concentrations of both targets are raised to detectable levels, the target is typically amplified to concentrations >215-fold greater than the off-target sequence.
In a typical field or clinical assay, target and off-target nucleic acid sequences are likely to be present at low concentrations. In the case of retroviruses such as influenza, the target nucleic acid will be viral RNA and any PCR assay will perforce be preceded by a reverse transcription (RT) step resulting in a linear DNA template. While the sensitivity of such an assay will be heavily dependent upon the efficiency of the RT step, we have shown that PrimerHunter primers are functional and specific under a wide range of template concentrations and thus are likely to be robust under a variety of experimental conditions including viral subtyping by RT–PCR in the clinic and in the field (35–38).
The PrimerHunter web server, as well as the open source code released under the GNU General Public License, are available at http://dna.engr.uconn.edu/software/PrimerHunter/. By default, PrimerHunter seeks to select primer pairs predicted to amplify all target sequences and none of the nontarget sequences under specified reaction conditions. When targets exhibit extremely large dissimilarity and such primer pairs cannot be found, PrimerHunter automatically seeks and reports a small set of primer pairs that collectively amplify all targets and none of the nontargets. If the number of primer pairs required to cover all targets is large, the pairs may need to be portioned into multiple multiplex PCR reactions due to limitations on the number of primers that can be used in a single reaction.
Complete classification of unknown viral samples into subtypes can be achieved by using PrimerHunter to design a specific primer pair (or set of primers) for each subtype, then running n parallel PCR reactions where n is the number of subtypes. The number of PCR reactions can be further reduced by designing primer pairs specific to sets of subtypes (e.g. superclades in the phylogenetic tree). By employing such nonspecific primer pairs and group testing methods similar to those in (39) the number of reactions can potentially be reduced to log n, and we plan to explore such methods in future work.
We also plan to explore the potential application of PrimerHunter to designing PCR assays for identification and subtyping of pathogens other than influenza, including bacteria, parasites and fungi. Another potential application for PrimerHunter is designing specific probes for gene expression and genome enrichment microarrays. For large eukaryotic genomes, these applications would require very large numbers of melting temperature computations which can be feasibly performed by parallelizing the testing of candidate primers.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
US National Science Foundation [0543365 and 0546457]. Funding for open access charge: US National Science Foundation [0543365 and 0546457].
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank the authors of (21) for allowing us to distribute a modified version of their melting temperature computation code as part of the PrimerHunter package.
REFERENCES
- 1.Curran M, Ellis J, Wreghitt T, Zambon M. Establishment of a UK national influenza H5 laboratory network. J. Med. Microbiol. 2007;56:1263–1267. doi: 10.1099/jmm.0.47336-0. [DOI] [PubMed] [Google Scholar]
- 2.Suarez D, Das A, Ellis E. Review of rapid molecular diagnostic tools for avian influenza. Avian Dis. 2007;51:201–208. doi: 10.1637/7732-101006-REGR.1. [DOI] [PubMed] [Google Scholar]
- 3.Gardner S, Kuczmarski T, Vitalis E, Slezak T. Limitations of TaqMan PCR for detecting divergent viral pathogens illustrated by hepatitis A, B, C, and E viruses and Human Immunodeficiency Virus. J. Clin. Microbiol. 2003;41:2417–2427. doi: 10.1128/JCM.41.6.2417-2427.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rozen S, Skaletsky H. Primer3 on the WWW for general users and for biologist programmers. In: Krawetz S, Misener S, editors. Bioinformatics Methods and Protocols: Methods in Molecular Biology. Totowa, NJ: Humana Press; 2000. pp. 365–386. [DOI] [PubMed] [Google Scholar]
- 5.Koressaar T, Remm M. Enhancements and modifications of primer design program Primer3. Bioinformatics. 2007;23:1289–1291. doi: 10.1093/bioinformatics/btm091. [DOI] [PubMed] [Google Scholar]
- 6.Fitch J, Gardner S, Kuczmarski T, Kurtz S, Myers R, Ott L, Slezak T, Vitalis E, Zemla A, McCready P. Rapid development of nucleic acid diagnostics. Proc. of the IEEE. 2002;90:1708–1721. [Google Scholar]
- 7.Angelov S, Harb B, Kannan S, Khanna S, Kim J. Efficient enumeration of phylogenetically informative substrings. J. Comput. Biol. 2007;14:701–723. doi: 10.1089/cmb.2007.R011. [DOI] [PubMed] [Google Scholar]
- 8.Emrich S, Lowe M, Delcher A. PROBEmer: a web-based software tool for selecting optimal DNA oligos. Nucleic Acids Res. 2003;31:3746–3750. doi: 10.1093/nar/gkg569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Phillippy A, Mason J, Ayanbule K, Sommer D, Taviani E, Huq A, Colwell R, Knight I, Salzberg S. Comprehensive DNA signature discovery and validation. PLOS Comput. Biol. 2007;3:0887–0894. doi: 10.1371/journal.pcbi.0030098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Balla S, Rajasekaran S, Mandoiu I. Efficient algorithms for degenerate primer search. Int. J. Found. Comput. Sci. 2007;18:899–910. [Google Scholar]
- 11.Linhart C, Shamir R. The degenerate primer design problem. Bioinformatics. 2002;18:S172–S181. doi: 10.1093/bioinformatics/18.suppl_1.s172. [DOI] [PubMed] [Google Scholar]
- 12.Souvenir R, Buhler J, Stormo G, Zhang W. Proceedings of the Third International Workshop on Algorithms in Bioinformatics (WABI). Vol. 2812. Verlag: Lecures Notes in Computer Science, Springer; 2003. Selecting degenerate multiplex PCR primers; pp. 512–526. [Google Scholar]
- 13.Jabado O, Palacios G, Kapoor V, Hui J, Renwick N, Zhai J, Briese T, Lipkin W. Greene SCPrimer: a rapid comprehensive tool for designing degenerate primers from multiple sequence alignments. Nucleic Acids Res. 2006;34:6605–6611. doi: 10.1093/nar/gkl966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wei X, Khun D, Narasimhan G. Degenerate primer design via clustering; IEEE Computer Society Press; 2003. pp. 75–83. In Proceedings of the IEEE Computer Society Bioinformatics Conference. [PubMed] [Google Scholar]
- 15.Kim N, Lee C. QPRIMER: a quick web-based application for designing conserved PCR primers from multi-genome alignments. Bioinformatics. 2007;23:2331–2333. doi: 10.1093/bioinformatics/btm343. [DOI] [PubMed] [Google Scholar]
- 16.Zheng J, Svensson T, Madishetty K, Close T, Jiang T, Lonardi S. OligoSpawn: a software tool for the design of overgo probes from large unigene datasets. [(accessed March 2008)];BMC Bioinformatics. 2006 7 doi: 10.1186/1471-2105-7-7. Available at http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1361790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.University of Tartu, Department of Bioinformatics Slicsel 1.1. [(accessed March 2008)]. http://bioinfo.ut.ee/slicsel/
- 18.Gadberry M, Malcomber S, Doust A, Kellogg E. Primaclade–a flexible tool to find conserved PCR primers across multiple species. Bioinformatics. 2005;21:1263–1264. doi: 10.1093/bioinformatics/bti134. [DOI] [PubMed] [Google Scholar]
- 19.Rouillard J, Zuker M, Gulari E. OligoArray 2.0: design of oligonucleotide probes for DNA microarrays using a thermodynamic approach. Nucleic Acids Res. 2003;31:3057–3062. doi: 10.1093/nar/gkg426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.SantaLucia J, Hicks D. The thermodynamics of DNA strucutural motifs. Annu. Rev. Biophys. Biomol. Struct. 2004;33:415–440. doi: 10.1146/annurev.biophys.32.110601.141800. [DOI] [PubMed] [Google Scholar]
- 21.Leber M, Kaderali L, Schonhuth A, Schrader R. A fractional programming approach to efficient DNA melting temperature calculation. Bioinformatics. 2005;21:2375–2382. doi: 10.1093/bioinformatics/bti379. [DOI] [PubMed] [Google Scholar]
- 22.Bao Y, Bolotov P, Dernovoy D, Kiryutin B, Zaslavsky L, Tatusova T, Ostell J, Lipman D. The influenza virus resource at the national center for biotechnology information. J. Virol. 2008;82:596–601. doi: 10.1128/JVI.02005-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kwok S, Chang S, Sninsky J, Wong A. A guide to the design and use of mismatched and degenerate primers. PCR Methods Appl. 1994;3:S539–S547. doi: 10.1101/gr.3.4.s39. [DOI] [PubMed] [Google Scholar]
- 24.Panjkovich A, Melo F. Comparison of different melting temperature calculation methods for short DNA sequences. Bioinformatics. 2005;21:711–722. doi: 10.1093/bioinformatics/bti066. [DOI] [PubMed] [Google Scholar]
- 25.Dinkelbach W. On nonlinear fractional programming. Manage. Sci. 1967;13:492–498. [Google Scholar]
- 26.Chvátal V. A greedy heuristic for the set covering problem. Math. Oper. Res. 1979;4:233–235. [Google Scholar]
- 27.Johnson D. Approximation algorithms for combinatorial problems. J. Comput. Syst. Sci. 1974;9:256–278. [Google Scholar]
- 28.Owczarzy R, You Y, Moreira B, Manthey J, Huang L, Behlke M, Walder J. Effects of sodium ions on DNA duplex oligomers: improved predictions of melting temperatures. Biochemistry. 2004;43:3537–3554. doi: 10.1021/bi034621r. [DOI] [PubMed] [Google Scholar]
- 29.Allawi H, SantaLucia J. Nearest neighbor thermodynamic parameters for internal G-A mismatches in DNA. Biochemistry. 1998;37:2170–2179. doi: 10.1021/bi9724873. [DOI] [PubMed] [Google Scholar]
- 30.Allawi H, SantaLucia J. Nearest-neighbor thermodynamics of internal A-C mismatches in DNA: sequence dependence and pH effects. Biochemistry. 1998;37:9435–9444. doi: 10.1021/bi9803729. [DOI] [PubMed] [Google Scholar]
- 31.Allawi H, SantaLucia JJ. Thermodynamics of internal C-T mismatches in DNA. Nucleic Acids Res. 1998;26:2694–2701. doi: 10.1093/nar/26.11.2694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Peyret N, Seneviratne P, Allawi H, SantaLucia J. Nearest-neighbor thermodynamics and NMR of DNA sequences with internal A-A, C-C, G-G, and T-T mismatches. Biochemistry. 1999;38:3468–3477. doi: 10.1021/bi9825091. [DOI] [PubMed] [Google Scholar]
- 33.Page R. TREEVIEW: an application to display phylogenetic trees on personal computers. Comput. Appli. Biosci. 1996;12:357–358. doi: 10.1093/bioinformatics/12.4.357. [DOI] [PubMed] [Google Scholar]
- 34.Larkin M, Blackshields G, Brown N, Chenna R, McGettigan P, McWilliam H, Valentin F, Wallace I, Wilm A, et al. ClustalW2 and ClustalX version 2. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 35.Chang H, Park J, Song M, Oh T, Kim S, Kim C, Kim H, Sung M, Han H, et al. Development of multiplex rt-PCR assays for rapid detection and subtyping of influenza type A viruses from clinical specimens. J. Microbiol. Biotechnol. 2008;18:1164–1169. [PubMed] [Google Scholar]
- 36.Spackman E, Senne D, Bulaga L, Trock S, Suarez D. Development of multiplex real-time RT-PCR as a diagnostic tool for avian influenza. Avian Dis. 2003;47:1087–1090. doi: 10.1637/0005-2086-47.s3.1087. [DOI] [PubMed] [Google Scholar]
- 37.Wu C, Cheng X, He J, Lv X, Wang J, Deng R, Long Q, Wang X. A multiplex real-time RT-PCR for detection and identification of influenza virus types A and B and subtypes H5 and N1. J. Virol. Methods. 2008;148:81–88. doi: 10.1016/j.jviromet.2007.10.023. [DOI] [PubMed] [Google Scholar]
- 38.Xie Z, Pang Y, Liu J, Deng X, Tang X, Sun J, Khan M. A multiplex RT-PCR for detection of type A influenza virus and differentiation of avian H5, H7, and H9 hemagglutinin subtypes. Mol. Cell Probes. 2006;20:245–249. doi: 10.1016/j.mcp.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 39.DasGupta B, Konwar K, Mandoiu I, Shvartsman A. DNA-BAR: distinguisher selection for DNA barcoding. Bioinformatics. 2005;21:3424–3426. doi: 10.1093/bioinformatics/bti547. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.