


Abstract
Sequencing technologies and new bioinformatics tools have led to the complete sequencing of various genomes. However, information regarding the human transcriptome and its annotation is yet to be completed. The Human Cancer Genome Project, using ORESTES (open reading frame EST sequences) methodology, contributed to this objective by generating data from about 1.2 million expressed sequence tags. Approximately 30% of these sequences did not align to ESTs in the public databases and were considered no-match ORESTES. On the basis that a set of these ESTs could represent new transcripts, we constructed a cDNA microarray. This platform was used to hybridize against 12 different normal or tumor tissues. We identified 3421 transcribed regions not associated with annotated transcripts, representing 83.3% of the platform. The total number of differentially expressed sequences was 1007. Also, 28% of analyzed sequences could represent noncoding RNAs. Our data reinforces the knowledge of the human genome being pervasively transcribed, and point out molecular marker candidates for different cancers. To reinforce our data, we confirmed, by real-time PCR, the differential expression of three out of eight potentially tumor markers in prostate tissues. Lists of 1007 differentially expressed sequences, and the 291 potentially noncoding tumor markers were provided.
INTRODUCTION
Understanding the genetic basis of human development and the mechanisms implicated in the physiopathology of diseases has improved dramatically after the disclosure of the human genome sequence, and its encoded genes (1–3). It is now widely accepted that, in mammals, there is no linear correlation between the number of genes, transcripts, and functionally diverse proteins. In the human transcriptome, a myriad of controlling mechanisms involving alternative splicing and a diversity of 5′ and 3′ ends contribute to, a yet unknown universe of transcripts (4). It is known that most of the genome is transcribed in complex patterns of interacting and overlapping transcripts from both strands (5–9), and most mammalian genes also have antisense transcripts (7,9–11). We currently have a great deal of information (4,5,12–14) arising from modern technologies, such as tiling arrays, that confirm the genome to be pervasively transcribed, and that the noncoding regions, such as the introns and intergenic regions, play an important role in human genome regulation by cis-acting at the transcriptional level (4,15,16). These approaches have resulted in the discovery of many novel transcribed sequences, and provide a new perspective on the number and extent of transcripts.
Noncoding RNAs (ncRNAs) are emerging as key players in transcriptional and translational control, and represent a new level of complexity (17,18). Available data shows that the ratio of noncoding versus coding RNAs increases from prokaryotes to mammals (6,19). Furthermore, ncRNAs appear to have cell- or condition-restricted expression, and at lower levels compared with the well-characterized coding genes (20–22). In addition, although cross-species conservation of many ncRNA transcribed regions is weak, promoters of these transcripts are generally much more evolutionarily conserved, and the conserved regions extend further than in the promoters of protein coding RNAs (5 kb versus 500 bp) (5,22,23). In recent years, the use of bioinformatics tools allied to experimental studies, particularly for the whole genome, has become a common and promising means to predict and screen novel ncRNAs and antisense RNAs (10,14,22,24,25).
Although sequencing efforts based on generating cDNA fragments had a major impact on gene discovery, the unspliced human transcripts that map exclusively to introns, and with no similarity to known expressed genes from any organism, were not fully appreciated. Most investigators selected transcripts with evidence of splicing, or ESTs only where both a polyadenylation signal and a poly(A) tail were present (18). It is now accepted that only a small fraction of the sequences generated through EST methods represent mitochondrial transcripts, reverse transcribed copies of rRNA, bacterial contaminants or immature mRNA molecules (26,27). Large fractions of what were, until recently, considered ‘junk’ DNA are indeed transcribed, and may play a fundamental role in understanding genomes (5,15,28). In addition, the results presented by Ravasi et al. (29) show that most of the cloned, noncoding sequences in the RIKEN cDNA collection, are expressed and are not, on the whole, derived from genomic, or pre-mRNA (premature mRNA), contamination.
A large contribution toward identifying ESTs was the Human Cancer Genome Project (HCGP) (3,26,27,30), performed by the ORESTES (open reading frame EST sequences) methodology. ORESTES is a technique to generate ESTs encompassing midpoints of genes, unlike conventional EST methodologies (5′ and 3′) that cover the ends of transcripts. This characteristic results from the cDNA synthesis using arbitrarily selected, nondegenerate primers under low-stringency conditions, that permits sequence analysis of less abundant gene transcripts, and therefore, lead us to access genes with lower levels of expression (26). Thus, the HCGP, through ORESTES methodology, generated 1 190 044 open reading frame EST sequences using RNA extracts from 24 types of normal or tumor tissues (3,27). From this total, almost 30% (341 680 sequences) showed no similarity with known transcripts and were considered no-match ORESTES (27). With the aim to explore the potential of ORESTES with no similarities with ESTs in the public databases as tumor markers, we constructed a cDNA microarray. This platform, containing ORESTES with a high probability of representing actively transcribed regions not associated with annotated transcripts, was hybridized against 12 different normal and tumor human tissues. The differential expression observed among distinct tissues or pathological conditions demonstrates that this strategy was very useful for identifying tissue-specific, or tumor-specific RNAs that do not correspond to previously annotated transcripts. These hitherto-uncharacterized transcripts may represent new human genes, splice variants, ncRNAs or natural antisense transcripts (NATs) with a restricted pattern of gene expression. As prostate tumor is the most prevalent cancer in the Brazilian male population (http://www.inca.gov.br), we have explored some of these sequences as potential prostate tumor markers.
MATERIALS AND METHODS
Selection of ORESTES and genome mapping
To construct the array, 4356 ORESTES with higher probability to represent actively transcribed regions of the human genome not associated with annotated transcripts, were randomly selected from the data generated by Fonseca et al. (31), resulted from the exploration of the 341 680 ORESTES from the Human Cancer Genome Project that showed no similarity to known transcripts (27). In this work, a bioinformatics pipeline was constructed for the sequences mapped on the human genome that were annotated as no-match in the Human Cancer Genome Project, starting with the removal of sequences derived from libraries containing genomic DNA or immature mRNA contamination, according to Sorek & Safer, 2003 (32), followed by selection of clusters containing at least one no-match sequence derived from prostate or breast tissues and that were formed by ESTs originating from at least two distinct libraries, and the singletons that showed gaps upon genomic alignment. Also, clusters aligned with full-length transcripts or ESTs of other projects were removed.
Genome mapping was done through a local database composed of data downloaded from the UCSC Genome Bioinformatics database (http://genome.ucsc.edu). ORESTES were classified according to their mapping on the human genome using three different gene tracks (Ensembl, KnownGene and RefSeq), and sequences mapped once on the genome were further classified as exonic, intronic and intergenic sequences.
cDNA microarrays
Glass arrays with 4356 elements were prepared in our lab with the aid of the Flexys Robot (Genomic Solutions, Ann Arbor, MI, USA), as described by Brentani et al., 2005 (33). Microarray data are deposited at Gene Expression Omnibus (GEO) under accession number GSE12737. Detailed information is provided in Supplementary Data.
RNA extraction and amplification
The institutional research ethics committee approved the current study (REC number 970/07), which was performed in accordance with the principles expressed in the Declaration of Helsinki. All samples kept in the A.C. Camargo Hospital BioBank, have signed informed consent for use in research, provided and approved by patients.
Total RNA derived from 56 normal or tumor tissues, obtained from the A.C. Camargo Hospital BioBank, was extracted with TRIzol (Invitrogen, Carlsbad, CA, USA) (Supplementary Data, Table S1). As a reference, we used a pool of RNAs obtained from 15 distinct human cell lines (Table S2). RNA samples were linearly amplified using a T7-based protocol (34,35). cDNA was prepared with aminoallyl-dUTP (Sigma–Aldrich, St. Louis, MO, USA) (36). Detailed information is provided in Supplementary Data.
Labeling, hybridization and data extraction
cDNA samples were submitted to indirect labeling (36) using Alexa Fluor 555 or Alexa Fluor 647 labels (Invitrogen). Hybridizations were performed in duplicate using the dye-swap method (35,37) in the GeneTAC Hybridization Station (Genomic Solutions). Slides were scanned on a confocal laser scanner (ScannArray Express, PerkinElmer, Waltham, MA, USA), using identical parameters for all slides and data was extracted with ScanArray Express software (PerkinElmer). The histogram method was used to estimate signal and local background intensities. Detailed information is provided in Supplementary Data.
Selection of bonafide transcripts
After subtracting local background, data was normalized by Lowess (38). For each sample, we determined the correlation between replica hybridizations and the number of spots with signal greater than local background. We also determined, for each sample, the differences between average signal intensity for elements representing intergenic or intragenic (exonic and intronic) sequences and for exonic or intronic sequences. To define a sequence as expressed, and to minimize the risk of a false-positive call, we applied a second level of cutoff for low-intensity spots. First, we determined, for each element, the lowest background-corrected intensity value among the 112 reads (main and swap slides) in each channel. Then, for each channel, we considered, as threshold, the highest value among the 112 lowest reads in each slide. Next, we eliminated, for each channel, all elements with median intensity below this threshold. Elements that survived these criteria were considered bonafide transcripts. For all expression data we applied log2 to the values.
Prediction of structured ncRNA candidates
Genomic sequences corresponding to ORESTES were analyzed to predict structured ncRNAs candidates. First, we separated the sequences into three groups: fully exonic, partially exonic, and nonexonic, according to the annotation systems KnownGene and RefSeq (UCSC Genome Bioinformatics). For each group, we combined searches for three features: (i) putative ORF, (ii) coding/noncoding potential of sequences and (iii) sequence and secondary structure conservation. To determine if a sequence is entirely an ORF, we used the getorf program (EMBOSS program suite, http://www.ebi.ac.uk/Tools/emboss), which analyzes if the three reading frames of both strands of the sequence could generate a coding sequence, and checked if the longest ORF identified by this software corresponded to the whole sequence (or its trimmed version of up to 2 bases from each end). Also, we used the Coding Potential Calculator (CPC) software (39), with default parameters, which classifies sequences in coding and noncoding (weak-coding, coding, weak noncoding and noncoding), to refine our initial ORF prediction. This software takes into account six features, being three of them based on the predicted ORF extension, quality and integrity, and the other three derived from BLASTX searches (UniRef90, BLAST Assembled Genomes; http://blast.ncbi.nlm.nih.gov/Blast.cgi): the number, quality and frame of the hits. We grouped sequences classified as noncoding or weak noncoding and sequences classified as coding and weak coding. To detect sequence and secondary structure conservation, we searched for multispecies alignments (16 vertebrate genomes with human http://hgdownload.cse.ucsc.edu/goldenPath/hg18/multiz17way) that overlapped the ORESTES sequence locations. These alignments were analyzed using the RNAz software (40) with default parameters, to detect evidence of secondary structure conservation, like compensatory base substitution.
Validation by RT-PCR
To select sequences for validation by RT-PCR, we first determined the average intensity value for each element in all slides. Using an MA plot (intensity ratios versus average intensities), we randomly selected elements with intensity 20-fold higher than the background (cutoff value of log212 for A, average intensities), since we intended to validate highly expressed sequences. Primers for 12 selected sequences were designed using Primer3 software (http://frodo.wi.mit.edu) (Table S3). RNAs from 23 normal or tumor tissues were obtained from the A.C. Camargo Hospital BioBank (Table S1), extracted with TRIzol (Invitrogen) and DNase treated (Illustra RNAspin Mini Isolation Kit, GE Healthcare, Buckinghamshire, ENG, UK). RT-PCR reactions were carried on Gene Amp PCR System 9700 (Applied Biosystems, Foster City, CA, USA) and the amplicons were fractionated by electrophoresis thorough a 3% NuSieve GTG (Cambrex, East Rutherford, NJ, USA) and stained with ethidium bromide. Detailed information is provided in Supplementary Data.
Differential expression analysis
To select differentially expressed sequences to be considered as tumor marker candidates we constructed MA plots showing, for each spot, fold differences and median signal intensity for tumor versus normal tissues. For these analyses, three (placenta, lung and testis) out of 12 tissues that were used in cDNA microarray experiments were discarded because we had only normal samples from them, and therefore, we could not perform differential expression analyses with the aim to identify tumor markers for these tissues.
Validation by quantitative real-time PCR
To select sequences to validate by real-time PCR, we determined, for each element, fold differences between median signal intensity for: (i) prostate tumor versus normal prostate tissue and (ii) prostate tumor versus all normal tissues analyzed on cDNA microarray experiments. Using MA plots, we selected elements expressed at least 4-fold more or 4-fold less in prostate tumor relative to normal prostate, and at least 2-fold more or 2-fold less in prostate tumor relative to all normal tissues (values converted to log2). Primers were constructed for nine sequences differentially expressed in prostate tissue, using Primer Express software (Applied Biosystems) and Oligo Tech program (http://www.oligosetc.com/analysis.php) (Table S5). Real-time PCR reactions were optimized using a pool of RNAs from three tumor prostate cell lines (PC-3, DU 145 and LNCaP), provided by the São Paulo branch of the Ludwig Institute for Cancer Research, and cultivated by the Laboratório de Investigação Médica/24 from Universidade de São Paulo. Real-time PCR validation was performed in seven paired samples from prostate (prostate adenocarcinoma and its surrounding non-neoplasic tissue), obtained from the A.C. Camargo Hospital BioBank (Table S6), extracted with TRIzol (Invitrogen) and DNase treated (RQ1 RNase-Free DNase, Promega, Madison, WI, USA). Real-time PCR experiments were carried out in duplicate using the SYBR Green detection method (Applied Biosystems). The housekeeping gene HPRT was selected through literature review (41). We used a previously described molecular marker for prostate carcinoma (AMACR) (42) as positive control for real-time PCR reactions. Real-time PCR was performed on a 7900HT Fast Real-Time PCR System (Applied Biosystems). The relative expression ratio was calculated according to Pfaffl formula (43). For all expression data we applied log2 to the values. Detailed information is provided in Supplementary Data.
Sequencing of validated ORESTES
ORESTES validated as real-transcripts by RT-PCR had their PCR products sequenced to verify their correspondence to the immobilized sequences and differentially expressed ORESTES validated by real-time PCR had their original clones sequenced to verify their correspondence to the sequences with which we expected that they were. Sequencing was carried on the 3130 Genetic Analyzer (Applied Biosystems). Detailed information is provided in Supplementary Data.
RESULTS
Genomic mapping of the cDNA microarray sequences
An analysis comparing the genomic location of ORESTES and non-ORESTES ESTs, with respect to coordinates of coding genes, was performed. As expected, we found that both non-ORESTES ESTs, as well as ORESTES, were preferentially mapped in transcribed regions of the human genome, using three different gene tracks (RefSeq, Ensembl and KnownGene, UCSC Genome Bioinformatics; http://genome.ucsc.edu) (Figure 1A). The proportion of ORESTES sequences that overlapped annotated exons of coding genes was somewhat reduced in the ORESTES data set (Figure 1B). The preferential mapping of ORESTES to transcriptional units suggests that fully intronic ORESTES may represent valid transcripts instead of genomic DNA contamination of ORESTES libraries.
Figure 1.

Mapping of ESTs on the human genome according to three different data sets. (A) ESTs mapped onto human transcript regions. (B) ESTs mapped onto human exonic regions. Black bar, ESTs; gray bar, ORESTES (open reading frame expressed sequence tags); and white bar, ORESTES that compound the cDNA microarray.
We constructed a cDNA microarray containing 4356 distinct ORESTES, selected using a previously described pipeline developed to maximize the probability of identifying new expressed sequences (31). Our data showed that most ORESTES that compounded the array was mapped to transcribed regions of the genome (Figure 1A), and had a fully intronic location (Figure 1B). Only a small fraction of spotted sequence overlapped annotated exons of coding genes or had intergenic mapping (Figure 1). For further analysis, we considered 3872 sequences that map once to the human genome. We divided these sequences into exonic (335 sequences), intronic (3178 sequences) and intergenic sequences (359 sequences), representing 8.6%, 82.1% and 9.3% of the sequences respectively. A large proportion of these ORESTES (3767) are unspliced relative to the genome.
Analysis and identification of actively transcribed regions not associated with annotated transcripts, and their evaluation as potential ncRNAs
Many low expression transcripts, splicing isoforms and ncRNAs are involved in specialized biological functions, and show a tissue-specific or even a pathological-specific expression patterns. To survey new transcripts associated with ORESTES, 24 tumor and 32 normal RNA samples from 12 different tissues (Table S1) were hybridized with the microarray platform.
Some preliminary analyses were performed to determine the overall quality of data. The Pearson correlation between two replicate slides showed a median value of 0.86, and 76% of the elements that compounded this platform had signal greater than local background. We investigated if there was any bias that could be associated with the different types of sequences immobilized on the array, according to the previous classification: exonic, intronic or intergenic sequences. Using the Wilcoxon test, there were no statistically significant differences in either case, i.e. in the comparison of average signal intensity for elements representing intergenic or intragenic sequences, as well as in the comparison of only intragenic (exonic or intronic) sequences. The spotted sequences showed no systematic bias associated with their classification, corroborating the likelihood of those sequences mapped on nonexonic regions as being transcribed sequences. To be more accurate in defining true hybridization signals we created a more stringent criterion, described in ‘Materials and methods’ section, with signal intensity cutoff values of 196 and 65 for channels 1 and 2, respectively. Thus, for each channel, we eliminated all elements with a median signal intensity below these thresholds. For channels 1 and 2 we had 86.6% and 91.1% of slides with more than 3000 valid elements, respectively. Therefore, the total number of actively transcribed regions not associated with annotated transcripts was 3421 (3079 out of 3178 intronic and 342 out of 359 intergenic sequences). The additional number of 319 out of 335 exonic elements identified as valid elements, corroborated the potential of our approach to identify new real, transcribed regions, since these sequences were deposited by others in public databases while this work was being performed. From this final number of valid elements (3740), 96 sequences (80 intronic, 6 intergenic and 10 exonic sequences) had intensity above our established cutoff value (20-fold higher than the background) and were eligible for RT-PCR validation. From this 96 sequences, we arbitrarily selected nine intronic sequences (roughly 11% of the total of intronic sequences), and three intergenic sequences (50% of intergenic group) and validated the existence of all of them as actively transcribed regions not associated with annotated transcripts, in RNAs derived from 10 different tissues (Tables S1 and S3, Figure S1). PCR products of validated sequences were submitted to sequencing and their correspondence to the immobilized sequences on the array was confirmed.
Evidence of secondary structures coupled with some sequence conservation at the RNA level can provide important clues that a given ‘locus’ is probably transcribed, and that this transcript may have a biological role (14,40,44,45). RNA secondary structures are known to play an important functional role, not only in many noncoding transcripts, but also in the context of protein-coding mRNAs (46). To analyze the proportion of spotted sequences that may represent structurally conserved putative ncRNAs, we searched for three features: (i) putative ORF, (ii) coding/noncoding potential and (iii) sequence and secondary structure conservation. For this analysis, sequences that did not overlap to known exons (intronic and intergenic sequences) were grouped together (3537 sequences) and the exonic sequences were further classified to fully exonic (131) and partially exonic (166). As for this analysis we only considered the KnownGene and RefSeq gene tracks to classify analyzed sequences, we discarded 38 sequences, previously classified as exonic according to the initial mapping, using the RefSeq, Ensembl and KnownGene gene tracks (UCSC Genome Bioinformatics) (Figure S2). We considered as putative ncRNAs sequences which presented all following features: partially exonic or nonexonic mapping, CPC software prediction of noncoding potential and evidence of secondary structure conservation according to the RNAz software. From the partially exonic sequences, we found 38 putative ncRNAs and from the nonexonic sequences, we found 1040 ncRNAs candidates (Table 1, Figure S2). It is noteworthy that some known ncRNAs possess a subsequence that is not as short as is usual, and resembles an ORF (46). In summary, about 28% (1078 of 3834) of our transcribed regions, not associated with annotated transcripts, are potential ncRNAs (Table 1, Figure S2).
Table 1.
Putative noncoding RNAs and their distribution with respect to differential expression
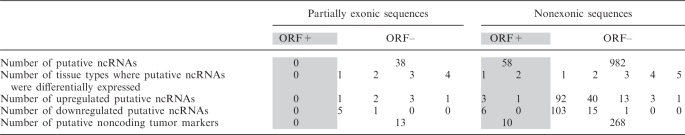 |
ncRNAs (noncoding RNAs).
Differential expression analyses and validation by quantitative real-time PCR
We constructed MA plots (intensity ratios versus average intensities) showing, for each spot, fold differences and median signal intensity for tumor versus normal tissues, for all the different tissues used in the cDNA microarray (Figure 2). We observed in all tissues, a large number of differentially expressed (at least 2-fold) sequences between tumor and normal samples, suggesting the potential to explore uncharacterized molecular markers (about 28% of the intronic and intergenic sequences mapped once on the genome). The total number of differentially expressed sequences, with fold differences between tumor and normal samples of at least two, in one or more different tissues and in agreement in respect to these sequences being up- or downregulated in all tissues in which they were expressed, were 1007, being 111 out of 335 exonic sequences, 885 out of 3178 intronic sequences and 111 out of 359 intergenic sequences (a list of all 1007 differentially expressed sequences is provided in our website, http://www.lbhc.hcancer.org.br/orestes_tumor_markers). Considering the same criteria of differentially expressed sequences described above, 291 transcripts were classified as differentially expressed putative ncRNAs by our pipeline. Four percent of these putative noncoding tumor markers were in the NONCODE database (47), or were predicted as an antisense pair by Galante et al. (48), again corroborating the validity of our approach, but have never been identified as differentially expressed in tumors. In Table 1 we assessed whether these candidates were expressed in one or more tumor tissues and found that at least five putative noncoding tumor markers were upregulated in at least four different tumors (AW803984, BE161676, CV358552, AW814925 and AW935941), compared with normal tissues. This is a very promising result for the search for tumor markers. A list of all putative noncoding tumor markers is provided in Table S4.
Figure 2.

MA plot (intensity ratios versus average intensities) showing the fold differences and median signal intensity for tumor versus normal tissues for each spot on microarray. (A) Prostate tissue. (B) Other tissues used on cDNA microarray. Gray circles, the sequences from prostate selected for real-time PCR validation (with fold value in prostate tumor 4-fold more or 4-fold less relative to normal prostate and 2-fold more or 2-fold less, relative to all normal tissues). Dotted line, 2-fold line; dashed line, 4-fold line.
We constructed MA plots to present an overview of the sequence expression distribution in prostate tissue (Figure 2A). For each spot, we observed the fold differences and median signal intensity for prostate tumor versus normal prostate (Figure 2A), and for prostate tumor versus all normal tissues (Figure S3). The nine sequences from prostate selected for validation by real-time PCR (Table S5) had at least a 4-fold variation in prostate tumor relative to normal prostate, and had at least 2-fold variation in prostate tumor relative to all normal tissues. We observed that, in general, the selected sequences were differentially expressed only in prostate when compared with other tissues (Figure 2, gray circles).
Using real-time PCR, we validated eight of the nine sequences as real transcripts. We considered valid differentially expressed sequences as those that presented a 3-fold difference in at least three out of seven paired samples. Using this criterion, three sequences were considered to be potential prostate tumor markers (Table 2). One of the potential tumor markers (BQ373258) was previously described as a ncRNA (DD3PCA3) by Bussemakers et al. (49). Its differential expression was confirmed in five of our seven paired prostate samples, and was upregulated in prostate cancer, serving as a positive control for our real-time PCR experiments. The overexpression of AW793062 ORESTES in prostate tumor was confirmed in four paired tissues. Genome mapping of this sequence showed its alignment to the first intron of a putative isoform of the RNF217 gene. The sequence BF910617 was validated in three samples and showed overexpression in prostate cancer. It is an intronic sequence of the KIAA1432 gene. Considering our criteria of valid differentially expressed transcripts (3-fold difference in at least three out of seven paired samples), we validated the overexpression of the AMACR gene. This molecular marker for prostate carcinoma was previously described as having high sensitivity and specificity for prostate carcinoma from different grades and types, being its mRNA overexpressed in about 30% (microarray) to 60% (real-time PCR) of prostate tumors and is low to undetectable in normal tissues (42,50,51).
Table 2.
Results of quantitative real-time PCR validating paired prostate samples with cDNA microarray results
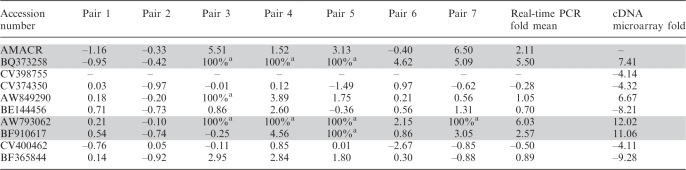 |
a100% values represent expression only in tumor samples (no detectable signal in normal samples) and were converted 10-fold to calculate fold mean.
All values represent log2 of expression values, considering tumor/normal ratios.
A summary of all sequences and samples sets used in each performed assay, as well as obtained results, is provided in Supplementary Data (Table S7).
DISCUSSION
Since a significant set of ORESTES remains unassociated with annotated transcripts, and could potentially represent actively transcribed regions of the human genome, we constructed a cDNA microarray containing ORESTES with a high probability of representing actively transcribed regions of the human genome, and not associated with annotated transcripts. Most of the sequences immobilized on the array map on intronic regions and are unspliced. After hybridization using 12 different tissues, we identified 3421 actively transcribed regions not associated with annotated transcripts. With RT-PCR we validated 100% of actively transcribed regions not associated with annotated transcripts that were evaluated (12 sequences).
Based on an ORF detector program (getorf, http://www.ebi.ac.uk/Tools/emboss), only 9% of the sequences mapped once on the genome may represent coding genes, leading us to search for potential noncoding sequences. In spite of the ORESTES methodology being biased to cover transcript midpoints with high probability of representing open reading frames, our data showed that from the sequences mapped to intronic or intergenic location (nonexonic group) only 7.6% presented a putative ORF. In contrast, 47.3% of fully exonic sequences had a putative ORF (Figure S2).
Our next step was to look for sequences that could be tumor, tissue or tumor/tissue associated. We observed in all tissues, a large number of differentially expressed (at least 2-fold) sequences between tumor and normal samples, suggesting the potential to explore uncharacterized molecular markers (about 28% of the intronic and intergenic sequences mapped once on the genome). The total number of differentially expressed sequences, with fold differences between tumor and normal samples of at least two and in agreement in respect to these sequences being up- or downregulated in all tissues in which they were expressed, in one or more different tissues, were 1007. We investigated the number of intronic ORESTES that mapped in a cancer gene list, compounded by 382 genes for which mutations have been causally implicated in cancer. This catalog of cancer genes is available on the Sanger Institute (Cancer Gene Census, http://www.sanger.ac.uk/genetics/CGP/Census) and it is based on a previously published review (52). We found 189 intronic ORESTES mapped to 97 cancer genes. The number of the differentially expressed ORESTES, considering the same criteria described above, located within introns of these cancer genes were 47. Using a list of cellular signal pathways curated by NCI-Nature (http://pid.nci.nih.gov), we expanded the original list of cancer genes for 1003 cancer pathway related genes. We found that 287 ORESTES mapped to 170 cancer-pathway related genes. From these 287 ORESTES related to cancer pathways, 70 were differentially expressed, considering the same criteria described above.
De novo computational prediction of ncRNA genes is difficult, since these transcripts lack most of the signatures that make protein-coding gene prediction possible (45). However, ncRNA genes produce a functional RNA rather than a translated protein, and often display a conserved, base-paired secondary structure instead of primary sequence similarity. These features can be combined in analyses and result in profiles of a multiple sequence alignment of ncRNAs that can be captured by statistical models (14,53). There are several approaches that are used to successfully predict ncRNAs based on the idea that functionally significant RNA structures will be conserved in related species, even when primary sequence is not conserved (54). The secondary structure base pairings are maintained by compensatory base mutations. These changes can be used as statistical evidence of evolutionary pressure to keep the base pairs at those positions (14,40,44,45). Pedersen et al. (44) predicted, from an initial set of more than 48 000 structured regions, ∼10 000 structured RNA transcripts in the human genome. Washietl et al. (40) estimated that 35 000 structured RNAs are conserved in mammals. The annotation of ncRNAs on a genome-wide scale is currently restricted to searching for homologs of known RNA families. More than 1500 homologs of known classical RNA genes can be annotated in the human genome sequence, and automatic, homology-based methods predict up to 5000 related sequences (45). Major databases containing thousands of annotated ncRNA sequences are RNAdb (10) and NONCODE (47). Thus, using a combination of methods (see ‘Materials and methods’ section), we identified about 28% (1078 of 3834) of our transcripts as potential ncRNA. These sequences showed a small overlap (4%) to sequences deposited on these ncRNA databases. One of them, CV372409 ORESTES, aligns with a sequence in the NONCODE database and was downregulated in three different tumors, compared with normal tissues in our cDNA microarray experiments. A common theme seems to be that many ncRNA genes have a very restricted expression. Often, they have low, or no, EST coverage, but this does not necessarily mean that they are not expressed and are nonfunctional (14,55).
Microarray technology has dramatically enhanced the discovery of molecular markers for cancer. Prostate cancer is the most prevalent cancer in Brazilian males (http://www.inca.gov.br) as well in men wordwide (http://www.cancer.gov), and investigators have searched for molecular markers of the disease. The first gene identified by cDNA microarray to be suitable for clinical practice, and to potentially improve the diagnosis of prostate cancer was AMACR (42). AMACR was suggested as a new molecular marker for prostate carcinoma by Xu et al. (42) in 2000, and confirmed by Jiang et al., (51). This protein is already used clinically as an aid in distinguishing prostate cancer from benign disease (56), and discriminating different grades and types of prostate cancer (50). Another potential molecular marker for prostate cancer, identified through cDNA microarray analysis, is the polycomb gene, EZH2. The expression of EZH2 indicates poor survival, and could be used as a marker for prostate cancer progression and metastasis (57–59). Also identified as a molecular marker is the TMPRSS2-ERG gene fusion, which is involved in the development of prostate cancer (60).
Increasing evidence shows a relationship between changes in expression levels of ncRNAs and cancer (18,61–63), emphasizing the potential role of ncRNAs in tumorigenesis, and the potential of this type of transcript as a tumor molecular marker (62). For example, in breast carcinoma, BC1 is deregulated (64), and the overexpression of BC200 RNA was recently evaluated as a new molecular marker for a poor prognosis (65). In lung cancer, increased expression of the MALAT-1 gene indicates a poor clinical outcome (66), and in hepatocellular carcinoma, HULC ncRNA is one of the most upregulated genes (62). In prostate cancer, there is overexpression of PCGEM (67), and DD3PCA3 (49) is implicated in tumorigenesis (68). These findings present a strong argument for the inclusion of noncoding transcripts into the arsenal of markers used for molecular diagnostics, which, thus far, has been almost exclusively populated by assays of protein-coding transcripts (11).
We validated three differentially expressed sequences in paired prostate samples as potential tumor markers. Validation of the BQ373258 sequence enhanced the value of our approach to identify molecular markers, since this sequence is mapped on the last exon of a described ncRNA (DD3PCA3) (49). DD3PCA3 has been described as highly overexpressed in prostate cancer tissue when compared with adjacent nonmalignant prostatic tissue, and its expression is restricted to the prostate (49). An unusually high density of stop codons has been identified along the entire DD3PCA3 cDNA sequence (49,69), which, in addition to the lack of an extended open frame and, after several years of analyzing putative proteins from predicted small ORFs, has resulted in the classification of DD3PCA3 as a polyadenylated ncRNA (69–71). Its function is unknown, although there is speculation that DD3PCA3 functions to regulate gene expression or participates in gene splicing (69). Both our cDNA microarray and real-time PCR show that this sequence is upregulated in prostate cancer relative to normal prostate (fold mean of 5.50 for real-time PCR and 7.41 for cDNA microarray).
An interesting observation arises from the data of two ORESTES, BF910617 and AW793062. ORESTES BF910617 is aligned with an intron of the KIAA1432 gene. From the analyses performed through Oncomine Research (http://www.oncomine.org) of the Lapointe et al. (72) data set, we observed that, in prostate cancer relative to normal prostate, the BF910617 ORESTES has diametrically opposite expression compared with the KIAA1432 gene. In the data set provided by Lapointe et al. (72) using cDNA microarray, the KIAA1432 gene was highly expressed in normal prostate, decreasing as the aggressiveness of prostate cancer increased. According to this data set, it was least expressed in metastatic prostate cancer in the lymph node (72). Therefore, our hypothesis is that BF910617 ORESTES may play a role in regulating the KIAA1432 gene, inhibiting its expression in prostate cancer when it is expressed at high levels. ORESTES AW793062 was validated with high fold values in almost 70% of the paired samples. This sequence is located in the first intron of a putative isoform of the RNF217 gene. Once again, the differential expression of AW793062 in prostate cancer was opposite to that observed for the RNF217 gene, with respect to primary and metastatic prostate cancer (Oncomine Research analyses) (73).
Although the differential expression of –4.32-fold in prostate tumor, showed by cDNA microarray experiments, of the CV374350 ORESTES was not confirmed by real-time PCR, we observed that this sequence maps to the last intron of the SGK1 gene, an inducible Ser/Thr kinase activated via phosphoinositide 3-kinase (PI3K) signal pathway (74,75). It is worth to note that there is an mRNA sequence (BX649005), also mapped to the SGK1 locus, which shows an extensive intron retention that includes the SGK1 last intron. It has been suggested that SGK1 may regulate androgen receptor activity, affecting androgen-mediated prostate cancer growth through a positive-feedback mechanism (76). Oncomine Research analysis of the SGK1 gene (73) suggests that this gene is expressed in normal prostate and benign prostate hyperplasia and its expression is fairly reduced among primary prostate carcinoma samples but is significantly reduced in metastatic prostate cancer. Further analysis of metastatic tumor samples could reveal if CV374350 expression follows the pattern of SGK1 gene expression and if this sequence may represents an SGK1 intron retention event or may be associated with other gene-regulation mechanism. This ORESTES was found in the list of cellular signal pathways related to cancer, analyzed as described above.
The power of our data to explore uncharacterized molecular markers was demonstrated with the large number of differentially expressed sequences, between tumor and normal samples from all tissues (about 28% of the intronic and intergenic sequences mapped once on the genome). Also, 291 of these differentially expressed transcripts have ncRNA potential, as predicted by our analysis. It is also very promising that at least five putative noncoding tumor markers are upregulated in at least four different tumors, compared with normal tissues. On the basis of these results, we believe in the value of our approach to identify uncharacterized molecular markers. Our data set contains a large number of actively transcribed regions of the human genome not associated with annotated transcripts not yet widely explored. These may represent new genes, splice variants, NATs or ncRNAs, which could be used as molecular markers for other cancers.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq; 142330/2007-8 to B.P.M.); and Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP; 04/11774-8, 07/55791-1 to B.P.M.; 07/01549-5 to A.M.-L.). Funding for open access charge: Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq).
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr Alex Carvalho for construction of the cDNA microarray. We thank Dra. Anamaria Camargo for providing the prostate cancer cell lines and Dra. Maria Mitzi Brentani and Dra. Rose Roela for cultivating them.
REFERENCES
- 1.Maxam AM, Gilbert W. A new method for sequencing DNA. Proc. Natl Acad. Sci. USA. 1977;74:560–564. doi: 10.1073/pnas.74.2.560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sanger F, Nicklen S, Coulson AR. DNA sequencing with chain-terminating inhibitors (classical article: 1977) Biotechnology. 1992;24:104–108. [PubMed] [Google Scholar]
- 3.Brentani H, Caballero OL, Camargo AA, da Silva AM, da SWA, Jr, Dias NE, Grivet M, Gruber A, Guimaraes PE, Hide W, et al. The generation and utilization of a cancer-oriented representation of the human transcriptome by using expressed sequence tags. Proc. Natl Acad. Sci. USA. 2003;100:13418–13423. doi: 10.1073/pnas.1233632100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Birney E, Stamatoyannopoulos JA, Dutta A, Guigo R, Gingeras TR, Margulies EH, Weng ZP, Snyder M, Dermitzakis ET, Thurman RE, et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447:799–816. doi: 10.1038/nature05874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carninci P, Kasukawa T, Katayama S, Gough J, Frith MC, Maeda N, Oyama R, Ravasi T, Lenhard B, Wells C, et al. The transcriptional landscape of the mammalian genome. Science. 2005;309:1559–1563. doi: 10.1126/science.1112014. [DOI] [PubMed] [Google Scholar]
- 6.Frith MC, Pheasant M, Mattick JS. The amazing complexity of the human transcriptome. Eur. J. Hum. Genet. 2005;13:894–897. doi: 10.1038/sj.ejhg.5201459. [DOI] [PubMed] [Google Scholar]
- 7.Katayama S, Tomaru Y, Kasukawa T, Waki K, Nakanishi M, Nakamura M, Nishida H, Yap CC, Suzuki M, Kawai J, et al. Antisense transcription in the mammalian transcriptome. Science. 2005;309:1564–1566. doi: 10.1126/science.1112009. [DOI] [PubMed] [Google Scholar]
- 8.Mattick JS, Makunin IV. Non-coding RNA. Hum. Mol. Genet. 2006;15:R17–R29. doi: 10.1093/hmg/ddl046. [DOI] [PubMed] [Google Scholar]
- 9.Mehler MF, Mattick JS. Non-coding RNAs in the nervous system. J. Physiol. 2006;575:333–341. doi: 10.1113/jphysiol.2006.113191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pang KC, Stephen S, Dinger ME, Engstrom PG, Lenhard B, Mattick JS. RNAdb 2.0-an expanded database of mammalian non-coding RNAs. Nucleic Acids Res. 2007;35:D178–D182. doi: 10.1093/nar/gkl926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Reis EM, Nakaya HI, Louro R, Canavez FC, Flatschart AVF, Almeida GT, Egidio CM, Paquola AC, Machado AA, Festa F, et al. Antisense intronic non-coding RNA levels correlate to the degree of tumor differentiation in prostate cancer. Oncogene. 2004;23:6684–6692. doi: 10.1038/sj.onc.1207880. [DOI] [PubMed] [Google Scholar]
- 12.Johnson JM, Edwards S, Shoemaker D, Schadt EE. Dark matter in the genome: evidence of widespread transcription detected by microarray tiling experiments. Trends Genet. 2005;21:93–102. doi: 10.1016/j.tig.2004.12.009. [DOI] [PubMed] [Google Scholar]
- 13.Kapranov P, Drenkow J, Cheng J, Long J, Helt G, Dike S, Gingeras TR. Examples of the complex architecture of the human transcriptome revealed by RACE and high-density tiling arrays. Genome Res. 2005;15:987–997. doi: 10.1101/gr.3455305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Weile C, Gardner PP, Hedegaard MM, Vinther J. Use of tiling array data and RNA secondary structure predictions to identify noncoding RNA genes. BMC Genomics. 2007;8:244. doi: 10.1186/1471-2164-8-244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Soares LMM, Valcarcel J. The expanding transcriptome: the genome as the ‘Book of Sand’. EMBO J. 2006;25:923–931. doi: 10.1038/sj.emboj.7601023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Seidl CIM, Stricker SH, Barlow DP. The imprinted Air ncRNA is an atypical RNAPII transcript that evades splicing and escapes nuclear export. EMBO J. 2006;25:3565–3575. doi: 10.1038/sj.emboj.7601245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Goodrich JA, Kugel JF. Non-coding-RNA regulators of RNA polymerase II transcription. Nat. Rev. Mol. Cell Biol. 2006;7:612–616. doi: 10.1038/nrm1946. [DOI] [PubMed] [Google Scholar]
- 18.Nakaya HI, Amaral PP, Louro R, Lopes A, Fachel AA, Moreira YB, El Jundi TA, da Silva AM, Reis EM, Verjovski-Almeida S. Genome mapping and expression analyses of human intronic noncoding RNAs reveal tissue-specific patterns and enrichment in genes related to regulation of transcription. Genome Biol. 2007;8:R43. doi: 10.1186/gb-2007-8-3-r43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mattick JS. RNA regulation: a new genetics? Nat. Rev. Genet. 2004;5:316–323. doi: 10.1038/nrg1321. [DOI] [PubMed] [Google Scholar]
- 20.Numata K, Kanai A, Saito R, Kondo S, Adachi J, Wilming LG, Hume DA, Hayashizaki Y, Tomita M. Identification of putative noncoding RNAs among the RIKEN mouse full-length cDNA collection. Genome Res. 2003;13:1301–1306. doi: 10.1101/gr.1011603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kampa D, Cheng J, Kapranov P, Yamanaka M, Brubaker S, Cawley S, Drenkow J, Piccolboni A, Bekiranov S, Helt G, et al. Novel RNAs identified from an in-depth analysis of the transcriptome of human chromosomes 21 and 22. Genome Res. 2004;14:331–342. doi: 10.1101/gr.2094104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gustincich S, Sandelin A, Plessy C, Katayama S, Simone R, Lazarevic D, Hayashizaki Y, Carninci P. The complexity of the mammalian transcriptome. J. Physiol. 2006;575:321–332. doi: 10.1113/jphysiol.2006.115568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sun H, Skogerbo G, Chen RS. Conserved distances between vertebrate highly conserved elements. Hum. Mol. Genet. 2006;15:2911–2922. doi: 10.1093/hmg/ddl232. [DOI] [PubMed] [Google Scholar]
- 24.Babak T, Blencowe BJ, Hughes TR. A systematic search for new mammalian noncoding RNAs indicates little conserved intergenic transcription. BMC Genomics. 2005;6:104. doi: 10.1186/1471-2164-6-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Washietl S, Pedersen JS, Korbel JO, Stocsits C, Gruber AR, Hackermuller J, Hertel J, Lindemeyer M, Reiche K, Tanzer A, et al. Structured RNAs in the ENCODE selected regions of the human genome. Genome Res. 2007;17:852–864. doi: 10.1101/gr.5650707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dias NE, Correa RG, Verjovski-Almeida S, Briones MR, Nagai MA, da SW, Jr, Zago MA, Bordin S, Costa FF, Goldman GH, et al. Shotgun sequencing of the human transcriptome with ORF expressed sequence tags. Proc. Natl Acad. Sci. USA. 2000;97:3491–3496. doi: 10.1073/pnas.97.7.3491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Camargo AA, Samaia HP, Dias-Neto E, Simao DF, Migotto IA, Briones MR, Costa FF, Nagai MA, Verjovski-Almeida S, Zago MA, et al. The contribution of 700,000 ORF sequence tags to the definition of the human transcriptome. Proc. Natl Acad. Sci. USA. 2001;98:12103–12108. doi: 10.1073/pnas.201182798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sironi M, Menozzi G, Comi GP, Cagliani R, Bresolin N, Pozzoli U. Analysis of intronic conserved elements indicates that functional complexity might represent a major source of negative selection on non-coding sequences. Hum. Mol. Genet. 2005;14:2533–2546. doi: 10.1093/hmg/ddi257. [DOI] [PubMed] [Google Scholar]
- 29.Ravasi T, Suzuki H, Pang KC, Katayama S, Furuno M, Okunishi R, Fukuda S, Ru KL, Frith MC, Gongora MM, et al. Experimental validation of the regulated expression of large numbers of non-coding RNAs from the mouse genome. Genome Res. 2006;16:11–19. doi: 10.1101/gr.4200206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.de Souza SJ, Camargo AA, Briones MR, Costa FF, Nagai MA, Verjovski-Almeida S, Zago MA, Andrade LE, Carrer H, El Dorry HF, et al. Identification of human chromosome 22 transcribed sequences with ORF expressed sequence tags. Proc. Natl Acad. Sci. USA. 2000;97:12690–12693. doi: 10.1073/pnas.97.23.12690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fonseca RD, Carraro DM, Brentani H. Mining ORESTES no-match database: can we still contribute to cancer transcriptome? Genet. Mol. Res. 2006;5:24–32. [PubMed] [Google Scholar]
- 32.Sorek R, Safer HM. A novel algorithm for computational identification of contaminated EST libraries. Nucleic Acids Res. 2003;31:1067–1074. doi: 10.1093/nar/gkg170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brentani R, Carraro D, Verjovski-Almeida S, Reis E, Neves E, de Souza S, Carvalho A, Brentani H, Reis L. Gene expression arrays in cancer research: methods and applications. Crit. Rev. Oncol. Hematol. 2005;54:95–105. doi: 10.1016/j.critrevonc.2004.12.006. [DOI] [PubMed] [Google Scholar]
- 34.Vangelder RN, Vonzastrow ME, Yool A, Dement WC, Barchas JD, Eberwine JH. Amplified RNA synthesized from limited quantities of heterogeneous cDNA. Proc. Natl Acad. Sci. USA. 1990;87:1663–1667. doi: 10.1073/pnas.87.5.1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gomes LI, Silva RLA, Stolf BS, Cristo EB, Hirata R, Soares FA, Reis LFL, Neves EJ, Carvalho AF. Comparative analysis of amplified and nonamplified RNA for hybridization in cDNA microarray. Anal. Biochem. 2003;321:244–251. doi: 10.1016/s0003-2697(03)00466-4. [DOI] [PubMed] [Google Scholar]
- 36.DeRisi J. In: DNA Microarrays: A Molecular Cloning Manual. Bowtell D, Sambrook J, editors. New York: Cold Spring Harbor Laboratory Press; 2003. pp. 187–193. [Google Scholar]
- 37.Yang YH, Speed T. Design issues for cDNA microarray experiments. Nat. Rev. Genet. 2002;3:579–588. doi: 10.1038/nrg863. [DOI] [PubMed] [Google Scholar]
- 38.Yang Y, Dudoit S, Luu P, Lin D, Peng V, Ngai J, Speed T. Normalization for cDNA microarray data: a robust composite method addressing single and multiple slide systematic variation. Nucleic Acids Res. 2002;30:e15. doi: 10.1093/nar/30.4.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kong L, Zhang Y, Ye ZQ, Liu XQ, Zhao SQ, Wei L, Gao G. CPC: assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007;35:W345–W349. doi: 10.1093/nar/gkm391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Washietl S, Hofacker IL, Stadler PF. Fast and reliable prediction of noncoding RNAs. Proc. Natl Acad. Sci. USA. 2005;102:2454–2459. doi: 10.1073/pnas.0409169102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.de Kok JB, Roelofs RW, Giesendorf BA, Pennings JL, Waas ET, Feuth T, Swinkels DW, Span PN. Normalization of gene expression measurements in tumor tissues: comparison of 13 endogenous control genes. Lab. Invest. 2005;85:154–159. doi: 10.1038/labinvest.3700208. [DOI] [PubMed] [Google Scholar]
- 42.Xu JC, Stolk JA, Zhang XQ, Silva SJ, Houghton RL, Matsumura M, Vedvick TS, Leslie KB, Badaro R, Reed SG. Identification of differentially expressed genes in human prostate cancer using subtraction and microarray. Cancer Res. 2000;60:1677–1682. [PubMed] [Google Scholar]
- 43.Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pedersen JS, Bejerano G, Siepel A, Rosenbloom K, Lindblad-Toh K, Lander ES, Kent J, Miller W, Haussler D. Identification and classification of conserved RNA secondary structures in the human genome. Plos Comput. Biol. 2006;2:251–262. doi: 10.1371/journal.pcbi.0020033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Griffiths-Jones S. Annotating noncoding RNA genes. Annu. Rev. Genomics Hum. Genet. 2007;8:279–298. doi: 10.1146/annurev.genom.8.080706.092419. [DOI] [PubMed] [Google Scholar]
- 46.Rymarquis LA, Kastenmayer JP, Huttenhofer AG, Green PJ. Diamonds in the rough: mRNA-like non-coding RNAs. Trends Plant Sci. 2008;13:329–334. doi: 10.1016/j.tplants.2008.02.009. [DOI] [PubMed] [Google Scholar]
- 47.Liu C, Bai B, Skogerbo G, Cai L, Deng W, Zhang Y, Bu D, Zhao Y, Chen R. NONCODE: an integrated knowledge database of non-coding RNAs. Nucleic Acids Res. 2005;33:D112–D115. doi: 10.1093/nar/gki041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Galante PAF, Vidal DO, de Souza JE, Camargo AA, de Souza SJ. Sense-antisense pairs in mammals: functional and evolutionary considerations. Genome Biol. 2007;8:R40. doi: 10.1186/gb-2007-8-3-r40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bussemakers MJ, van Bokhoven A, Verhaegh GW, Smit FP, Karthaus HF, Schalken JA, Debruyne FM, Ru N, Isaacs WB. DD3: a new prostate-specific gene, highly overexpressed in prostate cancer. Cancer Res. 1999;59:5975–5979. [PubMed] [Google Scholar]
- 50.Jiang Z, Woda BA, Wu CL, Yang XMJ. Discovery and clinical application of a novel prostate cancer marker - alpha-Methylacyl CoA racemase (P504S) Am. J. Clin. Pathol. 2004;122:275–289. doi: 10.1309/EJUY-UQPE-X1MG-68MK. [DOI] [PubMed] [Google Scholar]
- 51.Jiang Z, Woda BA, Rock KL, Xu YD, Savas L, Khan A, Pihan G, Cai F, Babcook JS, Rathanaswami P, et al. P504S - a new molecular marker for the detection of prostate carcinoma. Am. J. Surg. Pathol. 2001;25:1397–1404. doi: 10.1097/00000478-200111000-00007. [DOI] [PubMed] [Google Scholar]
- 52.Futreal P, Coin L, Marshall M, Down T, Hubbard T, Wooster R, Rahman N, Stratton M. A census of human cancer genes. Nat. Rev. Cancer. 2004;4:177–183. doi: 10.1038/nrc1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Griffiths-Jones S, Moxon S, Marshall M, Khanna A, Eddy SR, Bateman A. Rfam: annotating non-coding RNAs in complete genomes. Nucleic Acids Res. 2005;33:D121–D124. doi: 10.1093/nar/gki081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Machado-Lima A, del Portillo HA, Durham AM. Computational methods in noncoding RNA research. J. Math. Biol. 2008;56:15–49. doi: 10.1007/s00285-007-0122-6. [DOI] [PubMed] [Google Scholar]
- 55.Pollard KS, Salama SR, Lambert N, Lambot MA, Coppens S, Pedersen JS, Katzman S, King B, Onodera C, Siepel A, et al. An RNA gene expressed during cortical development evolved rapidly in humans. Nature. 2006;443:167–172. doi: 10.1038/nature05113. [DOI] [PubMed] [Google Scholar]
- 56.Cooper CS, Campbell C, Jhavar S. Mechanisms of Disease: biomarkers and molecular targets from microarray gene expression studies in prostate cancer. Nat. Clin. Pract. Urol. 2007;4:677–687. doi: 10.1038/ncpuro0946. [DOI] [PubMed] [Google Scholar]
- 57.LaTulippe E, Satagopan J, Smith A, Scher H, Scardino P, Reuter V, Gerald WL. Comprehensive gene expression analysis of prostate cancer reveals distinct transcriptional programs associated with metastatic disease. Cancer Res. 2002;62:4499–4506. [PubMed] [Google Scholar]
- 58.Varambally S, Dhanasekaran SM, Zhou M, Barrette TR, Kumar-Sinha C, Sanda MG, Ghosh D, Pienta KJ, Sewalt RGAB, Otte AP, et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature. 2002;419:624–629. doi: 10.1038/nature01075. [DOI] [PubMed] [Google Scholar]
- 59.Rhodes DR, Sanda MG, Otte AP, Chinnaiyan AM, Rubin MA. Multiplex biomarker approach for determining risk of prostate-specific antigen-defined recurrence of prostate cancer. J. Natl Cancer Inst. 2003;95:661–668. doi: 10.1093/jnci/95.9.661. [DOI] [PubMed] [Google Scholar]
- 60.Tomlins SA, Rhodes DR, Perner S, Dhanasekaran SM, Mehra R, Sun XW, Varambally S, Cao XH, Tchinda J, Kuefer R, et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science. 2005;310:644–648. doi: 10.1126/science.1117679. [DOI] [PubMed] [Google Scholar]
- 61.Reis EM, Ojopi EPB, Alberto FL, Rahal P, Tsukumo F, Mancini UM, Guimaraes GS, Thompson GMA, Camacho C, Miracca E, et al. Large-scale transcriptome analyses reveal new genetic marker candidates of head, neck, and thyroid cancer. Cancer Res. 2005;65:1693–1699. doi: 10.1158/0008-5472.CAN-04-3506. [DOI] [PubMed] [Google Scholar]
- 62.Panzitt K, Tschernatsch MMO, Guelly C, Moustafa T, Stradner M, Strohmaier HM, Buck CR, Denk H, Schroeder R, Trauner M, et al. Characterization of HULC, a novel gene with striking up-regulation in hepatocellular carcinoma, as noncoding RNA. Gastroenterology. 2007;132:330–342. doi: 10.1053/j.gastro.2006.08.026. [DOI] [PubMed] [Google Scholar]
- 63.Brito GC, Fachel AA, Vettore AL, Vignal GM, Gimba ER, Campos FS, Barcinski MA, Verjovski-Almeida S, Reis EM. Identification of protein-coding and intronic noncoding RNAs down-regulated in clear cell renal carcinoma. Mol. Carcinog. 2008;47:757–767. doi: 10.1002/mc.20433. [DOI] [PubMed] [Google Scholar]
- 64.Chen W, Bocker W, Brosius J, Tiedge H. Expression of neural BC200 RNA in human tumours. J. Pathol. 1997;183:345–351. doi: 10.1002/(SICI)1096-9896(199711)183:3<345::AID-PATH930>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 65.Iacoangeli A, Lin Y, Morley EJ, Muslimov IA, Bianchi R, Reilly J, Weedon J, Diallo R, Bocker W, Tiedge H. BC200 RNA in invasive and preinvasive breast cancer. Carcinogenesis. 2004;25:2125–2133. doi: 10.1093/carcin/bgh228. [DOI] [PubMed] [Google Scholar]
- 66.Ji P, Diederichs S, Wang WB, Boing S, Metzger R, Schneider PM, Tidow N, Brandt B, Buerger H, Bulk E, et al. MALAT-1, a novel noncoding RNA, and thymosin beta 4 predict metastasis and survival in early-stage non-small cell lung cancer. Oncogene. 2003;22:8031–8041. doi: 10.1038/sj.onc.1206928. [DOI] [PubMed] [Google Scholar]
- 67.Srikantan V, Zou ZQ, Petrovics G, Xu L, Augustus M, Davis L, Livezey JK, Connell T, Sesterhenn IA, Yoshino K, et al. PCGEM1, a prostate-specific gene, is overexpressed in prostate cancer. Proc. Natl Acad. Sci. USA. 2000;97:12216–12221. doi: 10.1073/pnas.97.22.12216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Petrovics G, Zhang W, Makarem M, Street JP, Connelly R, Sun L, Sesterhenn IA, Srikantan V, Moul JW, Srivastava S. Elevated expression of PCGEM1, a prostate-specific gene with cell growth-promoting function, is associated with high-risk prostate cancer patients. Oncogene. 2004;23:605–611. doi: 10.1038/sj.onc.1207069. [DOI] [PubMed] [Google Scholar]
- 69.Schalken JA, Hessels D, Verhaegh G. New targets for therapy in prostate cancer: Differential display code 3 (DD3(PCA3)) a highly prostate cancer-specific gene. Urology. 2003;62:34–43. doi: 10.1016/s0090-4295(03)00759-3. [DOI] [PubMed] [Google Scholar]
- 70.Hessels D, Gunnewiek JMTK, van Oort I, Karthaus HFM, van Leenders GJL, van Balken B, Kiemeney LA, Witjes JA, Schalken JA. DD3(PCA3)-based molecular urine analysis for the diagnosis of prostate cancer. Eur. Urol. 2003;44:8–15. doi: 10.1016/s0302-2838(03)00201-x. [DOI] [PubMed] [Google Scholar]
- 71.Tinzl M, Marberger M, Horvath S, Chypre C. DD3(PCA3) RNA analysis in urine - a new perspective for detecting prostate cancer. Eur. Urol. 2004;46:182–187. doi: 10.1016/j.eururo.2004.06.004. [DOI] [PubMed] [Google Scholar]
- 72.Lapointe J, Li C, Higgins JP, van de Rijn M, Bair E, Montgomery K, Ferrari M, Egevad L, Rayford W, Bergerheim U, et al. Gene expression profiling identifies clinically relevant subtypes of prostate cancer. Proc. Natl Acad. Sci. USA. 2004;101:811–816. doi: 10.1073/pnas.0304146101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Dhanasekaran SM, Barrette TR, Ghosh D, Shah R, Varambally S, Kurachi K, Pienta KJ, Rubin MA, Chinnaiyan AM. Delineation of prognostic biomarkers in prostate cancer. Nature. 2001;412:822–826. doi: 10.1038/35090585. [DOI] [PubMed] [Google Scholar]
- 74.Kobayashi T, Cohen P. Activation of serum- and glucocorticoid-regulated protein kinase by agonists that activate phosphatidylinositide 3-kinase is mediated by 3-phosphoinositide-dependent protein kinase-1 (PDK1) and PDK2. Biochem. J. 1999;339(Pt 2):319–328. [PMC free article] [PubMed] [Google Scholar]
- 75.Park J, Leong M, Buse P, Maiyar A, Firestone G, Hemmings B. Serum and glucocorticoid-inducible kinase (SGK) is a target of the PI 3-kinase-stimulated signaling pathway. EMBO J. 1999;18:3024–3033. doi: 10.1093/emboj/18.11.3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Shanmugam I, Cheng G, Terranova P, Thrasher J, Thomas C, Li B. Serum/glucocorticoid-induced protein kinase-1 facilitates androgen receptor-dependent cell survival. Cell Death Differ. 2007;14:2085–2094. doi: 10.1038/sj.cdd.4402227. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}