

Abstract
Modern biomedical research is evolving with the rapid growth of diverse data types, biophysical characterization methods, computational tools and extensive collaboration among researchers spanning various communities and having complementary backgrounds and expertise. Collaborating researchers are increasingly dependent on shared data and tools made available by other investigators with common interests, thus forming communities that transcend the traditional boundaries of the single research lab or institution. Barriers, however, remain to the formation of these virtual communities, usually due to the steep learning curve associated with becoming familiar with new tools, or with the difficulties associated with transferring data between tools. Recognizing the need for shared reference data and analysis tools, we are developing an integrated knowledge environment that supports productive interactions among researchers. Here we report on our current collaborative environment, which focuses on bringing together structural biologists working in the area of mass spectrometric based methods for the analysis of tertiary and quaternary macromolecular structures (MS3D) called the Collaboratory for MS3D (C-MS3D). C-MS3D is a web-portal designed to provide collaborators with a shared work environment that integrates data storage and management with data analysis tools. Files are stored and archived along with pertinent meta data in such a way as to allow file handling to be tracked (data provenance) and data files to be searched using keywords and modification dates. While at this time the portal is designed around a specific application, the shared work environment is a general approach to building collaborative work groups. The goal of which is to not only provide a common data sharing and archiving system but also to assist in the building of new collaborations and to spur the development of new tools and technologies.
Keywords: Collaboratory, data analysis tools, structural biology, mass spectrometry
Introduction
High throughput biological research has become increasingly complex and data intensive. Scientists are facing tremendous challenges in optimizing experimental designs, archiving and mining data, maintaining data provenance and accessing common data analysis tools, all of which limit the ability to efficiently collaborate with colleagues. To address these issues, cyber infrastructure programs have been created to meet data sharing and collaboration challenges across scientific disciplines by developing individual tools or focusing on larger scientific areas.1-7 For example, the Biomedical Informatics Research Network (BIRN)8 has been established to address critical issues in neuroimaging research by creating a collaborative environment that is taking advantage of distributed computing, database federation, and other e-science capabilities to revolutionize the way biomedical research and clinical information management are carried out. The BioCore (Biological Collaborative Research Environment) web-based collaboratory provides a suite of molecular dynamics and visualization tools, as well as access to high performance computers to perform MD simulations and calculations.2 In a similar fashion, the support of cutting-edge information technologies will put many emerging communities in the position of making crucial contributions to their respective fields. The Collaboratory for Multi-Scale Chemical Science (CMCS)9-11 incorporates advances in informatics, semantic web, collaboratory, and grid research to facilitate collaboration among and within interdisciplinary combustion science communities. This project, a first generation combustion knowledge grid, was very successful at both enhancing the state of chemical sciences and enhancing the state of collaborative science. A result of this collaboratory is an open-source science-independent knowledge environment, the Knowledge Environment for Collaborative Science (KnECS),12 that can be used to facilitate the development of new knowledge environments for specific science applications.
A growing community of scientists dedicated to developing and applying approaches that combine chemical cross-linking and covalent labeling techniques with mass spectrometry and molecular modeling for investigating tertiary and quaternary structures of macromolecules (MS3D) has been coming together according to the familiar path of other emerging communities of investigators with diverse backgrounds but common interests. Scientists involved in this type of collaborative research work together to generate and share experimental data, develop and test new informatics tools, and design novel approaches to tackle the next challenge. Although the interactions necessary to complete a specific project may be typically limited to a small number of collaborators, there are infrastructural barriers to sharing data and analysis tools, which can severely hamper the progress towards the common goal. For this reason, a scientific problem-solving environment is critical to integrate tools and data in the context of an interdisciplinary collaboration. However, a successful knowledge environment is expected to provide scientific value, and not simply technical value, in order to maximize the impact of the new technology.
In support of the MS3D community and to foster collaborations among MS3D researchers and the wider structural biology community, a new initiative called the Collaboratory for MS3D (C-MS3D) has been established as a knowledge environment to support the efforts of investigators engaged in the development of enabling technologies for MS3D.13 In this context, new software tools, reference databases, and data schema are being implemented to facilitate mass spectrometry data analysis and the integration of spatial constraints from different sources in the construction of 3D models, with the overarching goal of enabling the structural elucidation of otherwise inaccessible biomolecules. For this reason, C-MS3D was designed to facilitate collaborations among biochemists, biophysicists, structural biologists and mass spectrometrists utilizing chemical cross-linking and covalent labeling techniques to investigate the structure and function of macromolecules and macromolecular complexes. Over time, C-MS3D is expected to expand to attract participation from investigators who are developing and utilizing other structural elucidation methods such as FRET and Dipolar EPR that, like MS3D, typically generate ‘sparse’ or ‘minimal’ constraint sets, and who will greatly benefit from sharing knowledge and tools to extend the reach of these approaches for structural analysis.
In this report, we describe the first implementation of the C-MS3D infrastructure substantiated by a web portal located at ms3d.org. An overview is provided of the general features designed to facilitate the work of investigators collaborating on a common project, including tools for data/metadata management, notification, application integration, and security. A general MS3D workflow is proposed to guide users through the wide range of possible steps involved in a typical structural elucidation project. Over time, each step will be populated with tools intended to aid the completion of each specific task in a highly integrated fashion. As an example, a brief description is included of a set of research tools dedicated to enable the MS-based characterization of products obtained by probing target biomolecules with mono- and bi-functional alkylating agents.
Methods
C-MS3D is an integrated knowledge environment with a portal interface that provides access to a data repository and set of tools to support the MS3D workflow. The portal at the heart of C-MS3D is based on the Knowledge Environment for Collaborative Science (KnECS),12 an open source informatics toolkit designed to enable knowledge grids that interconnect science communities, unique facilities, data, and tools. The primary user environment of KnECS is a web portal, built upon the CompreHensive collaborativE Framework (CHEF).14 KnECS leverages the Scientific Annotation Middleware (SAM)15 software for data and metadata services. KnECS was originally developed in support of the Collaboratory for Multi-Scale Chemical Sciences,9-11 a first generation combustion knowledge grid supporting collaboration among and within interdisciplinary combustion science communities.
C-MS3D is deployed on a dual Intel Xeon 3.4GHz CPU machine (Super Micro Computer, Inc., San Jose, CA) with 16GB of memory and a 250GB hard drive, running a Linux operating system (Version 2.4.21-20.EL, http://www.linux.org/). We are taking advantage of a number of open source tools to deploy the C-MS3D portal within the KnECS infrastructure. The underlying open source software platform includes the Apache Tomcat servlet container (Version 5.0.28, http://tomcat.apache.org/), the OpenJMS implementation of the Java Message Service API (Application Programming Interface) (Version 0.7.5, http://openjms.sourceforge.net/), and a MySQL database (Version 4.1.10, http://www.mysql.com/). Tomcat servers support both the portal and SAM (Scientific Annotation Middleware, http://collaboratory.emsl.pnl.gov/docs/collab/sam/) while MySQL provides the metadata storage mechanism for SAM. The Apache Axis implementation of SOAP (Simple Object Access Protocol) (Version 1.1, http://ws.apache.org/axis/) is used to deploy web services. Web services provide access to software such as the OpenBabel (Version 1.100.0, http://openbabel.sourceforge.net/) chemical toolbox and ImageMagick (Version 5.5.6, http://www.imagemagick.org/) to translate from one format to another within the portal, as well as access to portlets that have been developed for this project. These MS3D tools, which have been integrated into the portal as web services, are described in the following sections.
Results and Discussion
The C-MS3D initiative has taken an iterative, phased approach to the development and integration of collaborative tools, MS3D applications, enabling data schema, and data management tools. According to this approach, a web portal (ms3d.org) has been deployed with direct input from scientists leading the development of MS3D, while feedback is constantly solicited from users to refine the requirements and enable the evolution of the initial implementation to fulfill the emerging needs of this community. An open source model has been adopted for the development of the portal and related tools to facilitate the contributions of investigators interested in the diverse facets of biomolecular structure determinations.
The Knowledge Environment for Collaborative Science (KnECS)
The Knowledge Environment for Collaborative Science (KnECS),12 an open-source toolkit for deploying science portals, provides a multi-layer architecture on which science communities can deploy a web portal, customize interfaces and KnECS services, add new data manipulation and analysis tools, and integrate science-specific applications (Figure 1). To deploy C-MS3D, we branded the KnECS web portal to provide a unique look and feel for the MS3D portal. The availability of KnECS allowed a fully functional C-MS3D portal and data repository to be customized and deployed with ease, allowing us to concentrate on integrating specific tools that support the MS3D workflow, including data translators and MS3D-related data analysis tools. KnECS has a number of tools and features that are used by scientists and science applications; we will not list all of these here, but rather mention some that illustrate the utility of the C-MS3D portal as a tool for collaborative biomedical research. The utility of these features will be shown in greater detail throughout this section.
Figure 1.
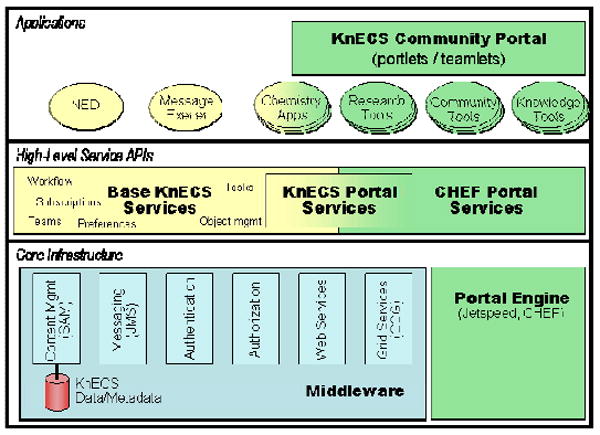
C-MS3D Informatics Architecture. A schematic view of the KnECS multi-tier architecture for integrating data and tools.
KnECS features include team and data collaboration tools, lightweight federation of data, provenance tracking, and multi-level support for application integration. KnECS leverages the Scientific Annotation Middleware (SAM)15 software for data and metadata services, such as storing and retrieving data and metadata, searching, providing access control, as well as extensible mechanisms for automatically extracting metadata from files, performing translations, and managing data relationships. KnECS has a basic service that facilitates asynchronous collaboration, a subscription service. The KnECS Notification Email Daemon (NED) monitors events matching user or team notification subscriptions and sends immediate, daily or weekly digest email messages summarizing the events.
With respect to security, KnECS includes middleware components that implement authentication and authorization. All users have accounts with login ids and passwords; the accounts give users access to the portal and the data repository. The portal service is running over an encrypted Secure Sockets Layer (SSL). Authorization is managed via the Java Authentication and Authorization Service (JAAS) interfaces to support pluggable security. Each individual user has a private workspace in which he or she can store data and may have access to team workspaces, where access controls are provided by SAM.
KnECS has a number of features that support application integration. We use a number of these in the development of MS3D data interpretation portlets. Requests to the MS3D web services are submitted to an asynchronous queue-based service and the service then notifies the user when results are available. Furthermore, inputs to the tools can be uploaded directly from the data repository and outputs from the computations can be downloaded using a file chooser that directly interfaces with the data repository.
Enabling team collaboration and data sharing
The C-MS3D portal contains general-purpose collaboration tools, such as chat, shared calendars, threaded discussions, shared task lists, and announcements that support teams and community interactions. Users can form groups to focus on a specific project and create communities to explore a specific aspect of MS3D. These collaboration tools facilitate the communication between subscribing members and are therefore vital to the group’s continued progress. The notification subscription service, allows both individuals and teams, to be recipients of email notifications for specified actions within the portal. This service can alert users to the availability of new data, users can monitor when their collaborators access their shared workspace, and users can be notified when a specific workflow tool has completed its computations.
In the shared data repository, both public and private workspaces are automatically set up for each team on the data server upon team creation. Private workspaces are used to share and perhaps develop data within a team while the public area can be used to publish verified reference data. Users can control access to workspaces. Team members can share web links through Bookmarks, providing a customizable library of web site links that project members can control by adding, removing, and editing web resources.
The portal contains a Data Browser (Figure 2), which constitutes an immediate and self-explanatory way of interacting with the data repository through the web. The Data Browser allows users to perform standard file and directory operations, control permissions, view and edit metadata, perform data translations, and visualize data in various formats. The Data Browser also integrates a provenance graph tool that displays data relationships as a 2D graph, a search tool which supports data discovery, and an annotation tool that allows users to annotate data with text, sound, images, equations, or whiteboard drawings.
Figure 2.
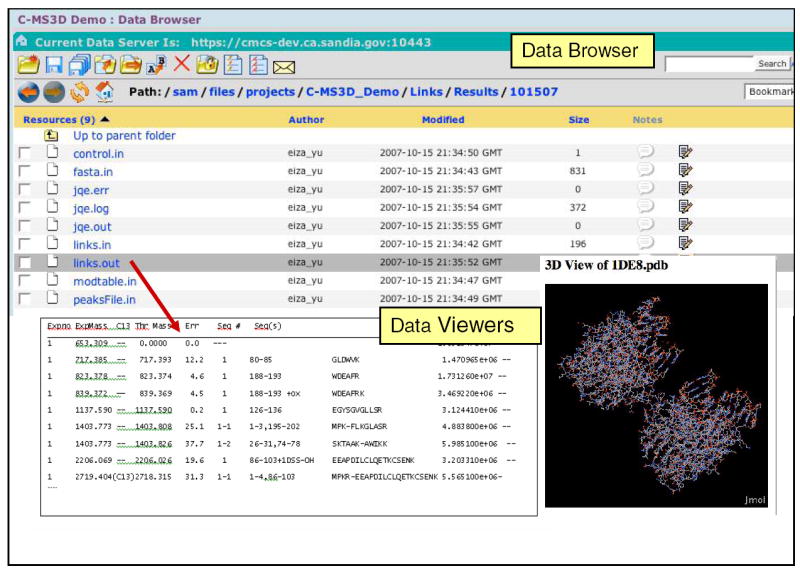
The Data Browser allows users to browse and view data and metadata. Users can manage files in teamspace by adding, removing, editing, and securing file access by permissions. Annotations and file translations/ viewers are also available for some file types (in this case, a PDB viewer).
Data can be stored in a specific format, defined by a MIME type, and then data translators can be registered in SAM to translate the data into a number of other formats (vide infra). This is a real-time translation, and the results are not stored. However, users have the option of saving the results of any data translation. Similar features are employed in the portal, so that the data can be visualized in other formats, as well.
MS3D workflow
The first portal implementation includes a graphic user interface (GUI) replicating a putative MS3D workflow, which introduces the key elements of a typical project and provides the entry points to the different groups of tasks and tools (Figure 3). The various tasks are loosely grouped in three general categories corresponding to typical operations performed during the structural elucidation of target biomolecules by virtually any approach, such as the acquisition of structural information, the translation of spatial constraints into molecular models, and the validation of the resulting structures. More specifically, the workflow includes discrete steps performed to secure pre-existing biochemical information, select appropriate probes and experimental procedures, map the position of chemical modifications and crosslinks, extract actual constraints from the experimental data, utilize this information to generate an initial model, refine and validate the final 3D structure.
Figure 3.
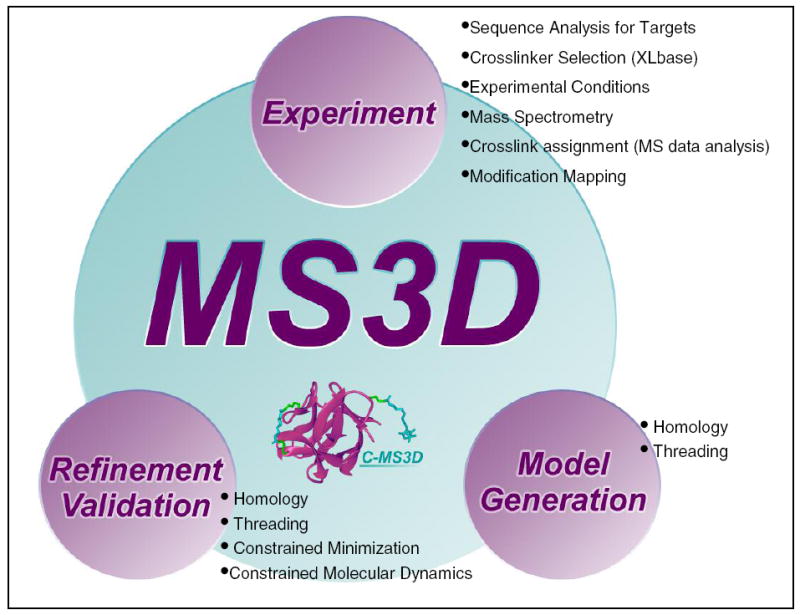
Screenshot of the initial implementation of the MS3D workflow graphic user interface (GUI) accessible through ms3d.org. A putative workflow comprises three key processes performed during a typical MS3D project, such as the acquisition of structural information (EXPERIMENT), the translation of spatial constraints into molecular models (MODEL GENERATION), and the validation of the resulting structures (REFINEMENT/VALIDATION). Each category is populated with hyperlinks to databases and existing 3rd party applications, websites, and tools that will assist in experimental design and execution. This workflow is provided as a guide for users through the wide range of possible steps involved in a typical MS3D project.
One of the objectives of the C-MS3D initiative consists of populating each category in the GUI with actual tools and instructions to assist the experimentalist throughout the entire project in a seamless integrated environment. The integration between the various tasks is ensured by the use of appropriate data formats that make the output of a certain tool acceptable as the input for any other tool included in the workflow, directly or through appropriate translators. Considering the success of MS-based techniques in supporting the characterization of products obtained by probing a variety of biomolecular substrates, many of the current tasks deal specifically with the acquisition and interpretation of data generated by this analytical platform. However, the intrinsic flexibility afforded by the workflow architecture is expected to enable the integration of information provided by other platforms and associated tools. Although the tasks are listed in a somewhat sequential fashion in this initial GUI implementation (Figure 3), the corresponding tools are accessible from anywhere in the workflow to allow for possible reiterations and accommodate any variations of experimental design.
The possibly large volumes of information obtained from different sources, the sheer number of operations necessary to complete a typical project, and the highly iterative nature of this type of work make effective data management an imperative. In order to assist in bookkeeping and annotation, an electronic lab notebook provided by SAM was implemented to enable interfacing with the data repository. Available hyperlinks to database and existing 3rd party applications, websites, and tools that perform the computations needed in the MS3D workflow are also offered through the portal. Outputs from such applets/tools can be saved in the data repository in the appropriate format for interoperability with downstream workflow tools.
Data interoperability
With the goal of facilitating data exchange and tool interoperability, the C-MS3D project has adopted both the mzXML format developed at the Seattle Proteome Center6 and the mzData format implemented by the Proteomic Standards Initiative (PSI),16, 17 which share an open representation of mass spectrometric data. Based on generic extensible markup language (XML), these standards have been introduced by the proteomics community to provide a universal interface between the native binary formats employed by the various MS platforms and the downstream data analysis pipelines. In their current forms, they encode different and somewhat complementary scopes of information, with mzXML including more intrinsic details about the raw spectra and mzData comprising additional information resulting from spectra processing (i.e, peaklists). There are plans to merge the current formats into a new standard, mzML,18 to eliminate any confusion among users and minimize the burden on software developers. The ability to work with both mzXML and mzData will allow for a quick adaptation of the current MS3D tools to the new standard, once its implementation is finalized.
The tools included in the portal rely on translators that are being developed to convert vendor-specific formats into mzXML/mzData, thus dispensing with the need to directly support multiple input formats. This approach is expected to simplify the integration of different instruments within the same analytical framework and enable the direct exchange and validation of MS data.6, 17 Therefore, the MS3D tools will be able to accept mzData as input, perform the desired task, and provide an output in compatible format, which will be directly readable by downstream tools. At the time of submission of this report, the current translators could process data generated in mzData (v. 1.05), mascot generic format (mgf), and from free MS peak reduction software such as PNNL’s Decon2Ls.19 The translators will be updated to handle mzML as soon as this format is implemented.
Annotating data with pedigree metadata is critical for verification and archiving purposes, as well as for tracking the operations of any of the tools employed in the MS3D workflow. Knowing the provenance of certain items helps the investigator make connections to other useful data, which is especially important in a diffuse collaborative environment where data is being shared. The open nature of the mzXML, mzData, and mzML formats is particularly well suited for accommodating the development of automatic methods for capturing references, managing data annotations, and enabling effective data searching. In this direction, the KnECS architecture directly supports the automatic extraction of metadata when a file is deposited in the data repository. These metadata become searchable and browsable through the Data Browser described earlier.
Analysis tools for footprinting and crosslinking data
A series of programs for the analysis of MS data provided by substrates treated with selected chemical probes has been included in the initial version of the portal and are discussed in this report to exemplify the type and scope of featured applications. This software consists of the Links20 and MS2Links21, 22 programs, which were originally introduced as stand-alone packages to support the processing of data obtained respectively from the bottom-up and top-down analysis of crosslinked/covalently-labeled proteins and nucleic acids. While the underlying algorithms remained essentially unchanged, the new web-based implementations contain additional features aimed at expanding their applicability and increasing their user-friendliness, which are expected to make them more accessible to a wider range of MS3D users (Figure 4).
Figure 4.
Links tools integrated into C-MS3D portal. Figure shows program’s main input interface. A unique feature of Links/MS2Links is the ability to handle both nucleic acid and protein sequences in data analysis. Highly customizable modification table editor (see Figure 5 for more details) allows greater flexibility and more comprehensive assignment of peaks generated from crosslinking reactions. Multiple peaklists formats (mgf, mzData and csv from Decon2Ls) are supported in Links and MS2Links by providing data translators for common peak deconvolution/reduction output file formats. Links also has flexible search parameters, matching monoisotopic or average masses, as well as providing a theoretical library of masses based on sequence and modification entries.
Links is a tool for assigning MS peak lists generated from chemical crosslinking and/or modification experiments followed by enzymatic digestion and MS analysis. MS2Links performs the analogous task on peak lists provided by tandem mass spectrometry of chemically modified products. Initially, both programs require the sequence of the target biopolymer (i.e., single or multiple proteins, RNA, DNA, and combinations thereof) and specific information about the chemical modifier, the expected residue specificity, and the protease/nuclease employed for digestion. Links calculates the theoretical masses of all possible crosslinked/alkylated and digested products, whereas MS2Links generates a list of all possible fragment ions produced by submitting any precursor ion to gas-phase activation. Finally, each program matches the theoretical libraries with the actual peak lists obtained from the pertinent mass mapping or sequencing experiment. The output returns putative assignments within a defined mass error threshold for the given experiment. Together, the position and type of modification (e. g., monoversus bi-functional) obtained from a given reagent provide the spatial information that is the objective of this section of the MS3D workflow.
The web implementations provide greater flexibility in analyzing MS-data generated from progressively larger biomolecular complexes, including multiprotein (> 2 subunits) and nucleoprotein assemblies, which are expected to constitute the targets of choice for alternative approaches of structural elucidation. The portal implementation is capable of accepting experimental data in the form of mzData input files, which was not allowed by the original stand-alone package. This is accomplished through translators that extract peak lists and other necessary information from mzData files generated by a variety of vendor-specific platforms that support these formats. In addition, Links and MS2Links can accept ASCII files containing data in mgf format (e. g., m/z vs. intensity) to allow for the processing of data provided by instrumentation that is not mzData compliant. In the current implementation, the utilization of vendor-specific or 3rd party software is recommended to complete an initial round of data reduction and processing before using Links/MS2Links. The development of a data reduction algorithm to be integrated in the MS3D workflow is underway.
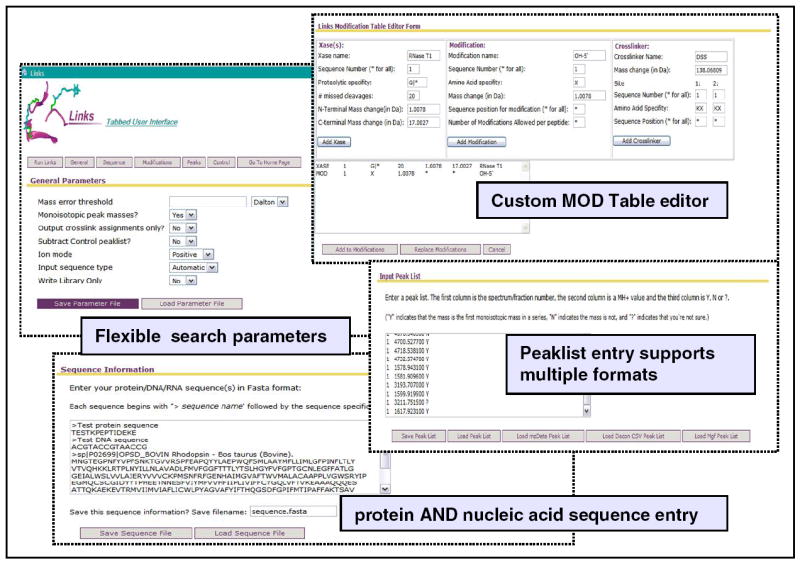
An important new feature of the web-based version is a robust user interface for user-input definitions, called the MOD Table Editor (Figure 5). A drop-down menu aids users in creating a custom modification list, which should contain the definitions of all of the pertinent crosslinkers and modifications to be used by Links/MS2Links. Indeed, users can take advantage of predefined modifications, or create new definitions for searchable items. At any given time, the list may include common modifications (e. g., phosphorylation, oxidation, disulfide bonds), probe-dependent modifications (e. g., crosslinks, dead-ends, alkylation adducts, etc.), and products induced by the specific experimental conditions (e. g., breakdown products, known contaminants, etc.). Each definition should include information about residue-or position-specificity of the reagent, together with the expected mass shift. In this direction, links to 3rd party calculators for generating accurate monoisotopic and average masses has been included in Bookmarks to enable proper calculations of the mass shifts associated with a specific modification.
Figure 5.

The Links Modification Table Editor. Options for cleavage (Xase), modifications, and crosslinkers used in MS3D are defined using this interface. Some commonly used proteases, nucleases, crosslinkers and modifications arising from typical reactions are included and can be selected using the drop-down menus. Using the editor interface, users can add custom modifications (user-defined). Links analyses are highly flexible and exhaustive as multiple configurations can be defined and interrogated in a single run.
The MOD Table editor does not present intrinsic restrictions in terms of number of modifications that can be considered at any given time. Therefore, a subset of the available definitions, or all of them at once, can be readily used in the same Links session. The only caveat is that increasing the number of definitions per session is expected to result in the generation of a sizeable combinatorial library of theoretical products, which will increase the time necessary to perform an exhaustive search. In general, a session involving a typical 300 residues protein substrate and 5 modifications is completed in a couple of minutes. However, in specific cases in which the expected duration may extend beyond a reasonable limit, users can logout and take advantage of an e-mail notification scheme that will alert them when the job is finished.
It should be noted that the theoretical libraries generated by Links/MS2Links can be saved and retrieved as stand-alone files. This feature enables users to inspect the library at their convenience and look for desirable products. By allowing for MS3D experiments to be completed in silico before they are actually performed in vitro, in analogy to what is done in VIRTUALMSLAB,23 these packages present additional value as powerful predicting tools, which will guide the selection of the best reagent/analytical approach to tackle a given substrate. Outputs are written and stored with all associated input information in the MS3D data repository, as shown in Figure 2. In the current implementation, the format of the output files is still ASCII, but future versions will switch to XML. Links/MS2Links output data is tagged with identification and workflow metadata to enable archiving, easy retrieval and comparison of replicate experiments.
A knowledge-based approach to crosslinking experiments
The outcome of a MS3D experiment depends largely in the selection of suitable crosslinking reagents for the particular biomolecule or biomolecular complex under study. The C-MS3D portal includes XLBase, an annotated crosslinkers database to support the experimental design and interpretation of crosslinking data. The database includes critical information about known reagents that have been already tested and discussed in the general literature. More specifically, each entry includes the following fields:
names, chemical formula, images of 2D and 3D structures;
reaction chemistry schemes and conditions;
observed breakdown products and/or contaminants;
crosslinking distance statistics (reported as average, mode, and distance range);
full mass assignments and associated MS;
notes on experience with such crosslinker (from users, and published material);
reference list;
link to manufacturers/suppliers.
Knowledge of a crosslinker’s specificity and length range is essential during the cross-linker selection and experimental design processes, as well as during model building. As shown in molecular dynamics (MD) simulations, distance constraints imposed by crosslinks do not correspond to strictly defined lengths, but rather fall within a range of values that are vastly different from the reported maximum calculated linker lengths.24, 25 Range values provide a more reasonable approximation/sampling of the distance constrained by the crosslinkers and in turn generate a more plausible/realistic structural model.
In general, the interpretation of MS3D data is greatly facilitated by direct knowledge of the products generated by the probing reaction, as well by that of unwanted species arising from side reactions (i. e., with buffer components, cross-reactivity with other residues, reagent contaminants/impurities, degradation). Including such details in the database of each reagent would enable to account for most, if not all, the species observed in a given mass spectrum and would thus increases the confidence with which the crosslinked products are identified. In similar fashion, knowledge of the gas-phase dissociation behavior of crosslinked and modified peptides/oligonucleotides is crucial in the interpretation of tandem mass spectra and is therefore essential in assigning the correct position of putative crosslinks.
The current version of XLBase includes entries from which MS3D-type data have been obtained and are thus available for querying, adding, and updating information. The database can be queried on any field/s in the database. Query outputs can be viewed as a list of crosslinker hits as well as a single page layout, where all the annotations associated with the single crosslinker hit is displayed (Figure 6). Finally, to accomplish the full potential and utility of the database, user input is necessary, and a mechanism is put in place (through XLBase Support) for users to be able to share experiences and disseminate valuable practical information on performing crosslinking experiments and data analysis within the community.
Figure 6.
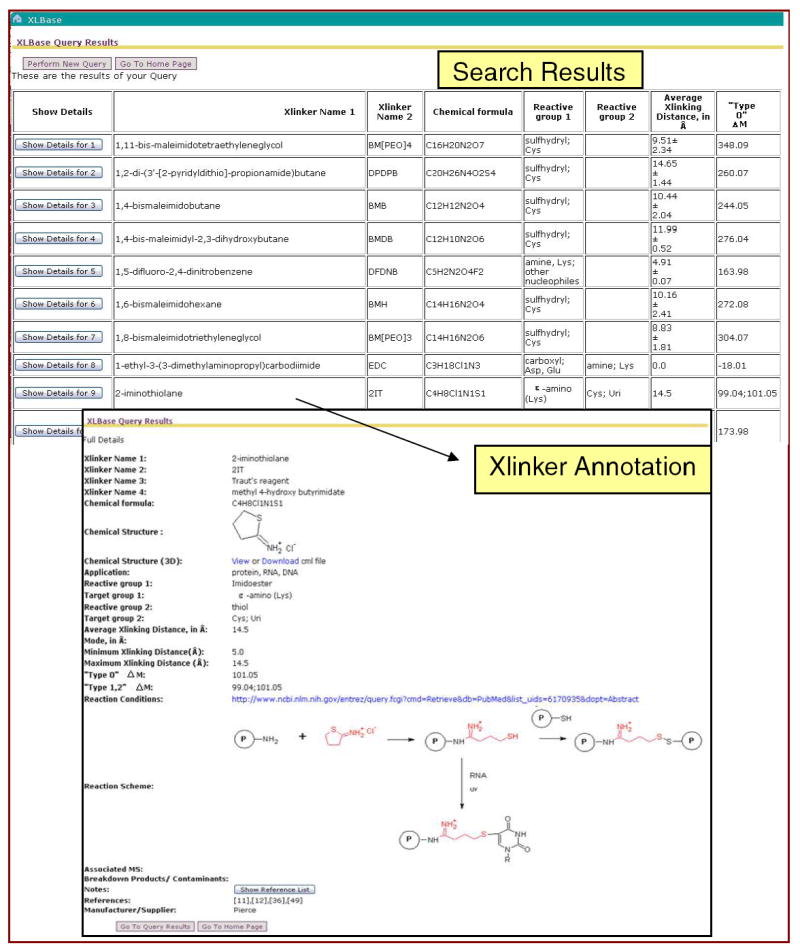
XLBase Interface. Query results can be viewed in tabular format for immediate comparison of crosslinking reagents (upper screen dump) or as single page to show full annotation for a specific crosslinker (lower screen dump).
We also envision that XLBase will expand and include analogous annotation for footprinting monofunctional reagents. Future work includes the integration of XLBase with Links and MS2Links, automatically extracting the currently available list of incremental masses for automated assignment of MS data. XLBase will also be used as a source of structure and/or crosslinking distance range information for structure calculations and structure prediction algorithms in the MS3D workflow.
Conclusions
A new initiative called the Collaboratory for MS3D (C-MS3D) has been established as a knowledge environment to support the efforts of investigators engaged in the development of alternative and enabling structural biology technologies, such as MS3D. The features provided by the first implementation of the C-MS3D portal have already proven successful by supporting the development and testing of recently deployed MS3D tools, such as Links, MS2Links, and XLBase. A measure of the significance of this collaborative initiative for the MS3D community is provided by the fact that the available tools (i.e., Links, MS2Links) are already being used by a beta test community in their MS3D investigations. Following our phased/iterative approach for the C-MS3D implementation, the current tools will be more closely integrated with each other according to users feedback. New tools are expected to be added by current participants and new contributors. Through the C-MS3D portal, we hope to foster collaborations among MS3D researchers and the wider structural biology community, particularly in adapting these types of distance constraints in current molecular modeling suites as well as development of dedicated software tools. The next development phase will involve populating the MS3D workflow, completing the collection of tools necessary to guide neophytes in each step to complete their MS3D projects.
Acknowledgments
Funding for the Collaboratory for MS3D (C-MS3D) is provided jointly by NIH (RR019864-01 to C.P.) and NSF (Chem 0439067 to D.F.). Sandia is a multiprogram laboratory operated by Sandia Corporation, a Lockheed Martin Company, for the United States Department of Energy’s National Nuclear Security Administration under contract DE-AC04-94AL85000.
Footnotes
Supporting information. N/A
References
- 1.Adams MW, Dailey HA, DeLucas LJ, Luo M, Prestegard JH, Rose JP, Wang BC. The Southeast Collaboratory for Structural Genomics: a high-throughput gene to structure factory. Acc Chem Res. 2003;36(3):191–8. doi: 10.1021/ar0101382. [DOI] [PubMed] [Google Scholar]
- 2.Bhandarkar M, Budescu G, Humphrey WF, Izaguirre JA, Izrailev S, Kale LV, Kosztin D, Molnar F, Phillips JC, Schulten K. In: Bruzzone AG, Uchrmacher A, Page EH, editors. BioCoRE: A collaboratory for structural biology; SCS International Conference on Web-Based Modeling and Simulation; San Francisco,CA. 1999; San Francisco,CA: 1999. pp. 242–251. [Google Scholar]
- 3.Burkhardt K, Schneider B, Ory J. A biocurator perspective: annotation at the Research Collaboratory for Structural Bioinformatics Protein Data Bank. PLoS Comput Biol. 2006;2(10):e99. doi: 10.1371/journal.pcbi.0020099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gantenbein RE. Designing an Internet-based collaboratory for biomedical research. Biomed Sci Instrum. 2002;38:399–404. [PubMed] [Google Scholar]
- 5.Myers D, Allison TC, Bittner S, Didier B, Frenklach M, Green WHJ, Ho Y-L, Hewson J, Koegler W, Lansing C, Leahy D, Lee M, McCoy R, Minkoff M, Nijsure S, von Laszewski G, Montoya D, Pancerella C, Pinzon R, Pitz W, Rahn LA, Ruscic B, Schuchardt K, Stephan E, Wagner A, Windus T, Yang C. A Collaborative Informatics Infrastructure for Multi-Scale Science. Cluster Computing. 2005;8:243–253. [Google Scholar]
- 6.Pedrioli PG, Eng JK, Hubley R, Vogelzang M, Deutsch EW, Raught B, Pratt B, Nilsson E, Angeletti RH, Apweiler R, Cheung K, Costello CE, Hermjakob H, Huang S, Julian RK, Kapp E, McComb ME, Oliver SG, Omenn G, Paton NW, Simpson R, Smith R, Taylor CF, Zhu W, Aebersold R. A common open representation of mass spectrometry data and its application to proteomics research. Nat Biotechnol. 2004;22(11):1459–66. doi: 10.1038/nbt1031. [DOI] [PubMed] [Google Scholar]
- 7.Schwenk TL, Green LA. The Michigan Clinical Research Collaboratory: following the NIH Roadmap to the community. Ann Fam Med. 2006;4 Suppl 1:S49–54. doi: 10.1370/afm.538. discussion S58-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Grethe JS, Baru C, Gupta A, James M, Ludaescher B, Martone ME, Papadopoulos PM, Peltier ST, Rajasekar A, Santini S, Zaslavsky IN, Ellisman MH. Biomedical informatics research network: building a national collaboratory to hasten the derivation of new understanding and treatment of disease. Stud Health Technol Inform. 2005;112:100–9. [PubMed] [Google Scholar]
- 9.Pancerella C, Myers JD, Allison TC, Amin K, Bittner S, Didier B, Frenklach M, Green WHJ, Ho Y-L, Hewson J, Koegler W, Lansing C, Leahy D, Lee M, McCoy R, Minkoff M, Nijsure S, von Laszewski G, Montoya D, Pinzon R, Pitz W, Rahn L, Ruscic B, Schuchardt K, Stephan E, Wagner A, Wang B, Windus T, Xu L, Yang C. Metadata in the Collaboratory for Multi-Scale Chemical Science. Proceedings of the 2003 Dublin Core Conference: Supporting Communities of Discourse and Practice Metadata Research and Applications (DC 2003); Seattle, Washington. 2003. [Google Scholar]
- 10.Myers JD, Allison TC, Bittner S, Didier D, Frenklach M, Green WHJ, Ho Y-L, Hewson J, Koegler W, Lansing C, Leahy D, Lee M, McCoy R, Minkoff M, Nijsure S, von Laszewski G, Montoya D, Pancerella C, Pinzon R, Pitz W, Rahn L, Ruscic B, Schuchardt K, Stephan E, Wagner A, Windus T, Yang C. A Collaborative Informatics Infrastructure for Multi-Scale Science. Cluster Computing Special Issue: Challenges of Large Applications in Distributed Environments. 2005;8(4):243–253. [Google Scholar]
- 11.Myers JD, Pancerella C, Lansing C, Schuchardt KL, Didier B. Multi-scale Science: Supporting Emerging Practice with Semantically-Derived Provenance. Proceedings of the Workshop on Semantic Web Technologies for Searching and Retrieving Scientific Data held at the 2nd International Semantic Web Conference; Sanibel Island, Florida. 2003. [Google Scholar]
- 12.KnECS open source software. http://sourceforge.net/projects/knecs/
- 13.Pancerella C, Leahy D, Yang C, Young MM, Sale K, Rahn L, Kuntz ID, Fabris D. Data portal enabling new protein structure collaboration. 19th Protein Society Meeting; Boston, MA. 2005. [Google Scholar]
- 14.CHEF Collaborative Portal Framework website. http://www.chefproject.org.
- 15.Myers JD, Chappell A, E M, G A, Schwidder J. Re-Integrating the Research Record. IEEE Computing in Science and Engineering. 2003:44–50. [Google Scholar]
- 16.Orchard S, Hermjakob H, Apweiler R. The proteomics standards initiative. Proteomics. 2003;3(7):1374–6. doi: 10.1002/pmic.200300496. [DOI] [PubMed] [Google Scholar]
- 17.Orchard S, Hermjakob H, Julian RK, Jr, Runte K, Sherman D, Wojcik J, Zhu W, Apweiler R. Common interchange standards for proteomics data: Public availability of tools and schema. Proteomics. 2004;4(2):490–1. doi: 10.1002/pmic.200300694. [DOI] [PubMed] [Google Scholar]
- 18.Proteomics Standards Initiative. mzML Development. http://www.psidev.info/index.php?q=node/257.
- 19.Jaitly Navdeep, M ME, Tolić Nikola, Littlefield Kyle, Daly Don S, Adkins Joshua N, Anderson Gordon A, Smith Richard D. Open Source Tools for the Accurate Mass and Time (AMT) Tag Proteomics Pipeline; 54th ASMS Conference on Mass Spectrometry; Seattle, Washington. 2006; Seattle, Washington: 2006. [Google Scholar]
- 20.Young MM, Tang N, Hempel JC, Oshiro CM, Taylor EW, Kuntz ID, Gibson BW, Dollinger G. High throughput protein fold identification by using experimental constraints derived from intramolecular cross-links and mass spectrometry. Proc Natl Acad Sci U S A. 2000;97(11):5802–6. doi: 10.1073/pnas.090099097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kellersberger KA, Yu E, Kruppa GH, Young MM, Fabris D. Top-down characterization of nucleic acids modified by structural probes using high-resolution tandem mass spectrometry and automated data interpretation. Anal Chem. 2004;76(9):2438–45. doi: 10.1021/ac0355045. [DOI] [PubMed] [Google Scholar]
- 22.Schilling B, Row RH, Gibson BW, Guo X, Young MM. MS2Assign, automated assignment and nomenclature of tandem mass spectra of chemically crosslinked peptides. J Am Soc Mass Spectrom. 2003;14(8):834–50. doi: 10.1016/S1044-0305(03)00327-1. [DOI] [PubMed] [Google Scholar]
- 23.de Koning LJ, Kasper PT, Back JW, Nessen MA, Vanrobaeys F, Van Beeumen J, Gherardi E, de Koster CG, de Jong L. Computer-assisted mass spectrometric analysis of naturally occurring and artificially introduced cross-links in proteins and protein complexes. Febs J. 2006;273(2):281–91. doi: 10.1111/j.1742-4658.2005.05053.x. [DOI] [PubMed] [Google Scholar]
- 24.Green NS, Reisler E, Houk KN. Quantitative evaluation of the lengths of homobifunctional protein cross-linking reagents used as molecular rulers. Protein Sci. 2001;10(7):1293–304. doi: 10.1110/ps.51201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang Q, Crosland E, Fabris D. Nested Arg-specific bifunctional crosslinkers for MS-based structural analysis of proteins and protein assemblies. Anal Chim Acta. 2008 doi: 10.1016/j.aca.2008.05.074. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]