

Abstract
Current theories of neuropathic hypersensitivity include an imbalance of supraspinal inhibition and facilitation. Our overall hypothesis is that the locus coeruleus (LC), classically interpreted as a source of pain inhibition, may paradoxically result in facilitation after tibial and common peroneal nerve transection (spared sural nerve injury – SNI). We first tested the hypothesis that non-noxious tactile hindpaw stimulation of the spared sural innervation territory increases neuronal activity in the LC in male rats. We observed a bilateral increase in the stimulus-evoked expression of transcription factors Fos and phosphorylated CREB (pCREB) in LC after SNI but not sham surgery; these markers of neuronal activity correlated with the intensity of tactile allodynia. We next tested the hypothesis that noradrenergic neurons contribute to the development of neuropathic pain. To selectively destroy these neurons, we delivered anti-dopamine-β-hydroxylase saporin (anti-DβH-saporin) into the intracerebroventricular space two weeks before SNI. We found that anti-DβH-saporin, but not an IgG-saporin control, reduced behavioural signs of tactile allodynia, mechanical hyperalgesia, and cold allodynia from 3-28 d after SNI. Our final experiment tested the hypothesis that the LC contributes to the maintenance of neuropathic pain. We performed SNI, waited two weeks for maximal allodynia and hyperalgesia to develop, and then administered the local anaesthetic lidocaine (4%) directly into the LC parenchyma. Lidocaine reduced all behavioural signs of neuropathic pain in a reversible manner, suggesting that the LC contributes to pain facilitation. We conclude that, in addition to its well-known inhibition of acute and inflammatory pain, the LC facilitates the development and maintenance of neuropathic pain in the SNI model. Further studies are needed to determine the facilitatory pathways emanating from the LC.
Pontine noradrenergic A6 neurons (locus coeruleus, LC) provide the bulk of norepinephrine (NE) found in the CNS, with an elaborate network of ascending and descending projections (Grzanna and Molliver, 1980). As reviewed previously, the LC (as well as A5 and A7 regions) may contribute to the bidirectional modulation of pain (Millan, 2002, Holden and Pizzi, 2003). On one hand, numerous studies indicate that the LC is engaged by injurious noxious stimuli, inflammation, or nerve damage to promote feedback inhibition of pain. For example, descending noradrenergic projections to the spinal cord (Westlund and Coulter, 1980, Kwiat and Basbaum, 1992) were originally characterized as inhibitory to acute somatic pain (Jones and Gebhart, 1986, 1987), although extensive depletion of NE with electrolytic or noradrenergic lesions of the LC do not always increase transient nociception in uninjured rats (West et al., 1993, Martin et al., 1999, Taylor et al., 2000, Jasmin et al., 2003). Also, noradrenergic LC lesions increased inflammation-induced thermal hyperalgesia and dorsal horn neuronal responsiveness (Tsuruoka and Willis, 1996b, a, Wei et al., 1999, Tsuruoka et al., 2003b).
In contrast to pain inhibition, however, emerging evidence suggests a contribution of the LC to pain facilitation. For example, noradrenergic LC lesions significantly reduced tonic behavioural responses to intraplantar formalin injection (Martin et al., 1999, Taylor et al., 2000), and prevented autotomy in rats with peripheral nerve transection (Al-Adawi et al., 2002). Based on these findings and the extensive literature describing the rostral ventral medulla (RVM) as a pain facilitatory center (Ossipov et al., 2000, Dubner, 2004), we hypothesized that the LC may contribute to the induction and/or maintenance of allodynia and hyperalgesia in an established model of peripheral neuropathic pain (Decosterd and Woolf, 2000). Indeed, current theories of neuropathic hypersensitivity include an imbalance of inhibition and facilitation; we hypothesize that the LC, classically interpreted as a source of pain inhibition, may paradoxically result in facilitation after nerve injury.
We first determined whether an innocuous mechanical stimulus would increase markers of neuronal activity in the LC (Fos and phosphorylated cAMP response element-binding protein, or pCREB) that correlate with behavioural manifestations of neuropathic pain. Second, we determined whether destruction of LC neurons with the noradrenergic neurotoxin, anti-dopamine beta hydroxylase-saporin (anti-DβH-saporin), would prevent the development of injury-induced hypersensitivity. Finally, we disrupted synaptic activity in the LC with the microinjection of a local anaesthetic (lidocaine). If the LC tonically facilitates neuropathic pain, then this should decrease the tactile and cold hypersensitivity that develops after nerve injury.
EXPERIMENTAL PROCEDURES
Subjects
Male Sprague-Dawley rats (Harlan or Charles River, Houston, TX) weighing 250-310 g were housed individually in plastic cages with pine-chip bedding in a temperature-controlled room (25°C) under a 12-hr light/dark cycle (6 AM-6 PM) with ad libitum access to food and water. Rats were handled 5 min/day for 5 days prior to any experimental manipulation and all procedures were performed during the light cycle. All animal protocols were approved by the Institutional Animal Care and Use Committee of Tulane University. All procedures were conducted in accordance with the Ethical Guidelines of the International Association for the Study of Pain (Zimmermann, 1983).
Spared nerve injury surgery
Rats were anesthetized with a continuous supply of 1.5-5.0% isoflurane gas (Abbott laboratories, Chicago, IL). SNI surgery was performed as described previously (Decosterd and Woolf, 2000, Taylor et al., 2007). In brief, an incision was made in the skin at the level of the trifurcation of the left sciatic nerve. The overlying muscles were retracted, exposing the common peroneal, tibial, and sural nerves. The common peroneal and tibial nerves were ligated with 6.0 silk (Ethicon, Somerville, NJ) and the knot and adjacent nerve (2.0 mm) were transected. Care was taken to avoid touching the sural branch. Sham surgery was produced by skin incision at the level of the trifurcation, and by exposing but not touching the three sciatic branches. Following SNI or sham surgery, the muscle was sutured with absorbable 4.0 sutures (Ethicon) and the skin closed with wound clips.
Behavioural tests of allodynia and hyperalgesia
1) General procedures
To minimize stress-induced hypoalgesia (Aloisi et al., 1994) or stress-induced potentiation of drug-induced anti-nociception (Calcagnetti et al., 1990, Levesque and Holtzman, 1993), rats were acclimated to a stainless steel grid within individual Plexiglas boxes for 1 hr/day for 3 days prior to and on the day of testing. In addition, lidocaine was delivered by remote injection. To minimize observer subjectivity, the experimenter was blind to treatment group.
2) von Frey assessment of tactile threshold
In all rats, we first assessed tactile allodynia using von Frey hairs (Stoelting, Inc). The plantar region of each hindpaw was stimulated with an incremental series of 8 monofilaments of increasing logarithmic stiffness. We further localized the stimulus region to the sural innervation territory -- the lateral aspect of the plantar hind paw. The 50% withdrawal threshold was determined using the up-down method of Dixon, modified by Chaplan et al (Chaplan et al., 1994). First, an intermediate von Frey monofilament (number 4.31, 2.0 g) was applied perpendicular to the plantar skin, causing a slight bending. In case of a positive response (rapid withdrawal of the paw within 6 sec), a smaller filament was tested. In case of a negative response, a larger filament was tested. In the correlation studies, we calculated % change from baseline as (pre-SNI minus post-SNI) / preSNI.
3) Automated assessment of tactile threshold
Tactile allodynia was also assessed with a machine-mounted probe (MMP) (Dynamic Plantar Aesthesiometer™; Ugo Basile Inc, Comerio VA, Italy) (Gibbs et al., 2006, Grigg et al., 2007). This method provides a parametric alternative to the use of von Frey hairs for the assessment of mechanical allodynia. Under visual guidance with an attached angled mirror, the thin, blunt metal rod of this device was positioned under the lateral region of the plantar hindpaw. A trigger was depressed, initiating contact of the rod with the footpad. The applied force increased at a rate of 2.5 g/sec with a cut-off of 50 g at 20 sec. Upon paw withdrawal, the device automatically disengaged and displayed the applied force. Three measurements were averaged.
4) Test of mechanical hyperalgesia
Next, we gently applied the sharp edge of a diaper pin to the footpad, avoiding damage to the skin. The duration of paw withdrawal was recorded, with a 15 sec cut off. Three measurements were averaged.
5) Test of cold allodynia
Using a syringe connected to PE-90 tubing, flared at the tip to a diameter of 2 mm, we applied a drop of acetone to the plantar paw. Surface tension maintained the volume of the drop to 10-12 μl. The duration of paw withdrawal was recorded, with a 20 sec cut-off. Three observations were averaged.
We did not test heat hypersensitivity because it has not been reliably observed after SNI (Decosterd and Woolf, 2000).
Fos and pCREB immunohistochemisty
Experimental Protocol
Baseline behavioural testing revealed hypersensitivity to be maximal two weeks after SNI. At this time, rats with SNI or sham surgery were exposed to innocuous mechanical stimulation. With the rat wrapped in a towel for mild restraint, the experimenter’s thumb was used to stroke the hypersensitive (lateral) portion of the paw for 2 sec with 2 sec intervals of no stimulation for a total of 10 min (Ma and Woolf, 1996, Intondi et al., 2007, Zhang et al., 2007). Two hrs later, brains were removed for immunohistochemistry of an immediate-early gene product (Fos) and the phosphorylated form of CREB (pCREB). We also examined Fos expression in SNI rats under basal conditions (i.e. no stimulation) as additional controls. Rats were randomly assigned to one of four groups after baseline behavioural testing: Experimental – SNI-stimulated (N = 12); Controls – sham-stimulated (N = 8), SNI-unstimulated (N = 6), sham-unstimulated (N = 6).
Tissue Preparation
Two hr after somatosensory or sham stimulation, rats were deeply anesthetized with 1.0 ml/kg of a ketamine/xylazine mixture [ketamine (88.9 mg/ml)/xylazine (11.1 mg/ml)] (Butler, Columbus, OH) and perfused intracardially with 0.1 M PBS containing 0.05% heparin followed by 10% formalin. Brains were removed and placed in 10% formalin at 4°C overnight, after which they were transferred to a 30% sucrose solution in 0.1 M PB. Using a freezing microtome, 40 μm sections were taken from ~ -9.36 mm to -10.44 mm relative to bregma in order to map the entire rostro-caudal length of the LC (~ -9.48 mm to -10.32 mm) (Paxinos and Watson, 2005). Free-floating sections were washed 4×10 min in 0.1 M PBS followed by a 30 min pre-treatment in 1% H2O2 in 0.1 M PBS and then blocked for 1 hr in 0.2% triton-X and 5.0% normal goat serum in 0.1 M PBS (0.2% TXGPBS).
Immunohistochemistry
Fos and pCREB immunohistochemistry was performed using an HRP-DAB strategy as described previously (Abbadie et al., 1997, Colombo et al., 2003, Intondi et al., 2007). Adjacent tissue sections from each rat were incubated in either rabbit anti-Fos (1:20,000; Oncogene) or anti-pCREB (1:1000, 06-519; Upstate Biotechnology) for 48 hrs at 4°C on a slow rocker. After removal of the 1° antibody, the sections were washed 4×10 min in 0.1 M PBS, again blocked in 0.2% TXGPBS and incubated in biotinylated goat anti-rabbit 2° antibody (1:1000; Jackson ImmunoResearch Laboratories, West Grove, PA) for 2 hrs at room temperature (RT). Both 1° and 2° were prepared in 0.2% TXGPBS. Next, the sections were washed 4×10 min in 0.1 M PBS and incubated for 1 hr at RT with an avidin-biotin horseradish complex in 0.2% TX in 0.1 M PBS (Vectastain Elite Kit, Vector Laboratories, Burlingame, CA). The sections were washed 4×10 min in 0.1 M PBS and visualized with 0.05% diaminobenzidine/0.05 M Tris buffer (pH 7.6) activated by 0.003% H2O2. The reaction was terminated with dH2O, followed by 3 final 10 min washes in 0.01 M PBS. Sections were mounted onto slides in 0.01 M PBS, given 24 hrs to dry, and then coverslipped with Cytoseal™60 mounting media (Richard-Allan Scientific, Kalamazoo, MI).
Quantification and analysis
Sections were captured under bright-field illumination with a 10X or 15X objective using Meta Imaging Series software (Metamorph 4.500). Blinded to treatment, Fos- and pCREB-immunoreactive (Fos-ir and pCREB-ir)-positive profiles were counted using NIH Image J software (Rasband, 1997-2007, Abramoff et al., 2004). In brief, the images were first transformed into 16-bit greyscale and then the relative size (μm2) and circularity of a Fos-ir or pCREB-ir positive profile was determined from a random subset of images using the wand tool. Next, the region of interest (ROI) -- LC was outlined and each image was thresholded using the maximum entropy thresholding plugin (Fos-ir) or adjusted manually to the beginning of the upward slope of the density histogram using the image→adjust→threshold command. In both the left and right LC, highlighted profiles in the ROI were automatically counted using the analyze command. The high degree of pCREB staining resulted in numerous instances of overlapping profiles. These cases (approximately 10% of the total) were counted manually and added to the automated counts.
To evaluate the reliability of our NIH Image J-assisted quantification method, a different investigator (H.P.) randomly sampled 2 sections each from 7 SNI and 5 sham rats for Fos, and two investigators (H.P. and J.B.) randomly sampled 1 section from each of 8 SNI and 8 sham rats for pCREB. The number of labelled profiles was counted manually as in our previous reports (Intondi et al., 2007). There was a significant correlation of r = 0.95, p < 0.0001 between investigators. Remarkably, the total number of manually-counted Fos profiles (614) was only 1.63% greater than the number of computer-generated Fos profiles (604), and the average of H.P. and J.B. pCREB counts (1220) was only 0.25% greater than computer-generated counts (1217). Correlation analysis between the two methods yielded r values of 0.93 (p < 0.0001) and 0.95 (p < 0.000001) for Fos and pCREB, respectively.
We used 3 landmarks to examine the expression of Fos-ir and pCREB-ir across the rostro-caudal extent of the LC: contour of the fourth ventricle; abundance of Me5 cells; and shape and appearance of the facial nerve (7n). With these landmarks and with the stereotaxic atlas of Paxinos and Watson, we categorized sections according to their rostral-caudal level, relative to bregma (Paxinos and Watson, 2005). We then collated sections into four divisions: -9.48 to -9.65 mm; -9.65 to -9.9 mm; -9.9 to -10.15 mm; -10.15 to -10.32 mm (N = 3-10 sections/division). Fos and pCREB were not analyzed rostral to Paxinos -9.48 mm because the LC proper diminishes at this level, leaving a heterogeneous population of NE and non-NE containing subcoeruleus neurons connecting the A7 and A5 noradrenergic cell groups (Aston-Jones, 2004, Paxinos and Watson, 2005). Fos and pCREB were not analyzed caudal to -10.32 mm because the LC ends at the level of the genu of the facial nerve (Aston-Jones, 2004). This was confirmed by manual examination of far-rostral (-9.36 mm) and far-caudal (-10.44 mm) sections in which there was an absence of cellular staining for Fos or pCREB.
There were no significant differences in the number of Fos-ir profiles between left and right LC for either sham-stimulated (L = 7.7 ± 1.9, R = 8.1 ± 1.8; mean ± SEM) [t(52) = 1.13, p = 0.76] or SNI-stimulated (L = 34.5 ± 4.8, R = 34.1 ± 4.7) [t(56) = 1.04, p = 0.93] rats or within groups of unstimulated rats (p’s > .05) (data not shown). In addition, t-tests for individual samples revealed no significant differences in the number of pCREB-ir profiles between left and right LC for either sham-stimulated (L = 38.4 ± 8.7, R = 39.6 ± 6.7; mean ± SEM) [t(44) = 0.11, p = 0.92]) or SNI-stimulated (L = 80.2 ± 7.3, R = 94.3 ± 8.3) [t(72) = 1.29, p = 0.20] rats (data not shown). Therefore, left and right profile counts were summed for each tissue section. 2-3 sections/rat/coronal division were averaged to obtain a single value for each rat in each division. These averaged counts/section for an individual rat across each of the 4 divisions were averaged prior to between-groups analysis of total profile counts.
Stereotaxic lesions with anti-DβH-saporin
We used the noradrenergic-specific neurotoxin, anti-DβH-saporin, to selectively destroy DβH-containing neurons (Wrenn et al., 1996, Wiley and Kline, 2000). Anti-DβH-saporin exerts its neurotoxic effects by binding to the transitory extracellular exposure of the membrane-bound form of DβH during NE release. Subsequently anti-DβH containing vesicles undergo endocytosis and retrograde transport to the cell body (Studelska and Brimijoin, 1989, Picklo et al., 1994) where, saporin, a ribosome-inactivating protein, arrests protein synthesis (Barthelemy et al., 1993).
Surgery
Rats were administered anti-DβH-saporin 14 days prior to nerve injury to allow sufficient time for neurotoxic effects to take place (Wrenn et al., 1996, Wiley and Kline, 2000). A small hole was made in the skull at AP = -0.8 and ML = +1.5 mm according to bregma (Paxinos and Watson, 2005). Injections of 10 μl [5 μg anti-DβH-saporin or 5 μg mouse IgG-saporin (control)] (Advanced Targeting Systems, San Diego, CA), both in sterile saline, were infused over 5 min at a rate of 2 μl/min through a 28G cannula (Plastics One) lowered into the left ventricule to DV = -4.3 mm and then retracted to DV = -3.3 mm. The infusion cannula remained in place for an additional 10 min to facilitate diffusion away from the tip. The burr hole was sealed with bone wax and the incision closed with 3 wound clips. Mouse IgG-saporin, a conjugate of pre-immune mouse IgG and saporin, is an excellent control for anti-DβH-saporin. Manufactured according to the same protocol, the two agents differ in that mouse IgG-saporin is constructed from antibodies that have no specificity and no ability to target cells with the exception of cells that possess Fc receptors [(Advanced Targeting Systems; (Dinota et al., 1990)]. SNI surgery was performed 14 days following infusion. Rats were first assessed for baseline behaviour prior to SNI and then tested for allodynia and hyperalgesia on days 3, 7, 14, 21, and 28 after SNI.
Immunohistochemistry
Lesions were verified by DβH immunohistochemistry, as described previously (Taylor et al., 2000). Sections were incubated in mouse anti-DβH (1:1000; MAB308, Chemicon International, Inc., Temecula, CA) followed by incubation with biotinylated goat anti-mouse 2° antibody (1:1000; Jackson ImmunoResearch). All other procedures were identical to that used for Fos and pCREB immunohistochemistry. One rat was eliminated due to incomplete bilateral damage to the LC (substantial immunoreactivity remained).
Microinjection of lidocaine
Surgery
Three to five days after nerve injury, rats were secured in a stereotaxic frame (Stoelting, Wood Dale, IL) under isoflurane (1.5-5%) anaesthesia. After a 2 cm skin incision exposed the cranium, the skull was levelled in the DV plane at lambda and bregma. Two 26G guide cannulae, 10 mm in length and plugged with a 33G wire stylet, were implanted bilaterally at AP = -0.8, ML = ±1.3, DV = +4.0 mm with respect to the interaural line (Paxinos and Watson, 2005). Cannulae were secured with three machine screws and dental cement (Lang Dental, Wheeling, IL) and the skin closed with 6.0 silk sutures (Ethicon). Rats were allowed a recovery period of 6-8 days before testing.
Injection protocol
On the day of drug injection, each stylet was replaced with a 33G microinjector, lowered 1.0 mm below the guide cannulae, and connected to a 1.0 ul syringe via polyethylene tubing. Rats were habituated to the testing environment for 60 min to allow recovery from the restraint associated with cannulae insertion. After assessment of baseline behavior, 0.5 μl/side of saline (Baxter, Deerfield, IL) or 4% lidocaine (Henry Schein) was infused bilaterally via a multiple syringe infusion pump (Stoelting). Microinjectors were left in place an additional 5 min to minimize backflow along the cannulae tracts. Studies using similar procedures show that typical spread of fluid spans an area ~1.0 mm2 in diameter (Malpeli and Schiller, 1979, Sandkuhler et al., 1987, Martin, 1991). Behavioural hypersensitivity was measured at 15, 45 and 90 min after injection.
Histological examination of cannulae placement
After behavioural testing, rats received intra-LC infusions of India ink 25% v/v (0.5 μl/LC) and were perfused as described above. The brains were removed and 40 μm sections were taken throughout the LC and stained with cresyl violet. The following key landmarks were used for injection site mapping: the motor trigeminal nucleus, mesencephalic 5 nucleus, nucleus of the trapezoid body, middle cerebellar peduncle and sensory root of the 5th nerve. We only used results from rats where the guide cannulae were centered above the LC.
Data Analysis
Using Systat 11 and Statistica 6.0 software, differences between means of parametric data (machine automated tactile allodynia, mechanical hyperalgesia, cold allodynia) were analyzed by ANOVA while differences between nonparametric data (von Frey hairs) were analyzed by Kruskal-Wallis or Mann-Whitney U statistics. Total Fos or pCREB in the LC and at individual coronal divisions of the LC between SNI and sham groups in the simulated or unstimulated conditions were analyzed by one-way ANOVA. The Pearson product-moment correlation coefficient (PMCC) was calculated between behavior and Fos or pCREB. To analyze the effects of LC lesions on the development of behavioural hypersensitivity, an overall two-way repeated measures ANOVA was performed on data collapsed across days 3-28. To analyze the effects of lidocaine, a one-way-repeated measures ANOVA was performed, restricted to 15-45 min time-points. If significant, overall ANOVA’s were followed by post-hoc t-tests with Bonferroni correction to evaluate group differences at specific time-points. All data are presented as mean ± S.E.M. with significance set to p < 0.05.
RESULTS
As illustrated in Fig 1A, SNI significantly reduced the paw withdrawal threshold to a non-noxious mechanical stimulus (von Frey hairs), and increased paw withdrawal duration to application of either a noxious mechanical (pin) or cool (acetone) stimulus, indicating the presence of tactile allodynia, mechanical hyperalgesia, and cold allodynia, respectively (p’s < 0.001). Hypersensitivity did not develop in the contralateral paw (p’s > 0.05; data not shown).
Figure 1. Stimulus-induced Fos expression in the LC after SNI.
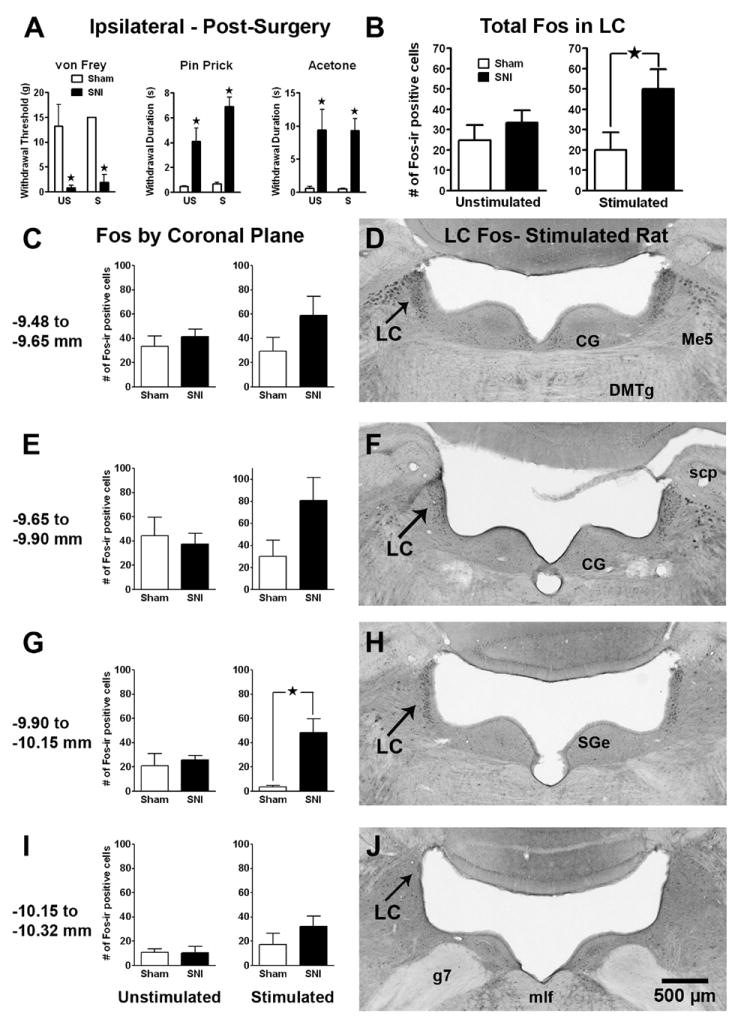
Panel A: Tactile allodynia (drop in von Frey threshold), mechanical hyperalgesia (increased response to pin prick), and cold allodynia (increased response to topical acetone) in rats with spared nerve injury (SNI) or sham surgery. There was no difference in hypersensitivity among SNI rats that were later assigned to the unstimulated (US) or stimulated with non-noxious stroking of the footpad (S) condition. Panel B: The total number of Fos-immunoreactive (Fos-ir) profiles in the LC (both sides) in sham and SNI rats 2 wk following SNI. Innocuous tactile footpad stimulation increased Fos in SNI rats. Panels C, E, G, I: The number of Fos-ir profiles in 4 distinct coronal divisions throughout the rostro-caudal extent of the LC. The effect of footpad stimulation in SNI rats reached statistical significance in the -9.90 to -10.15 mm division. Panels D, F, H, J: Representative photomicrographs of stimulated Fos-ir at each coronal division of the LC in an SNI rat. CG=central gray; DMTg=dorsomedial tegmental area; g7=facial nerve; Me5=mesencephalic 5th nucleus; mlf=medial longitudinal fasciculus; scp=superior cerebellar peduncle; SGe=supragenual nucleus. Magnification = 3X. Data is expressed as mean ± SEM. n=3-12. ★p < 0.05.
SNI increases stimulus-induced Fos expression in the LC
To test the hypothesis that SNI increases markers of neuronal activity in the LC, we compared the number of Fos-ir profiles in SNI and sham-operated rats in the presence or absence of sensory stimulation. We found that computer-assisted analysis of the number of Fos- or pCREB-ir profiles closely reflects manual counting methods. Fig 1B demonstrates that SNI + mechanical stimulation significantly increased Fos-ir across the entire rostro-caudal extent of the LC [F(1,18) = 4.93, p < 0.05, ANOVA]. Further analysis revealed that SNI + stimulation increased Fos expression within each coronal division (Figs 1C-J); this was significant from bregma -9.65 to -10.15 mm (p < 0.05).
There was a significant correlation between tactile allodynia (% change in VF threshold after SNI) and the number (across all coronal planes) of stimulus-induced Fos-ir (PMCC = 0.67, p = 0.025) positive profiles. Subsequent analysis revealed that this correlation extended to the -9.65 to -9.90 mm coronal division for Fos-ir (PMCC = 0.74, p = 0.035, n = 8, not shown).
As reported previously in the spinal cord dorsal horn (Catheline et al., 1999), Fig 2 illustrates that nerve injury alone did not change Fos-ir: there was no difference in profile number between sham and SNI rats in the unstimulated condition (p > 0.05). In addition, mechanical stimulation in the absence of nerve injury (in sham rats) did not change Fos-ir (p > 0.05).
Figure 2. Fos expression in the LC after Innocuous paw stimulation and/or SNI.
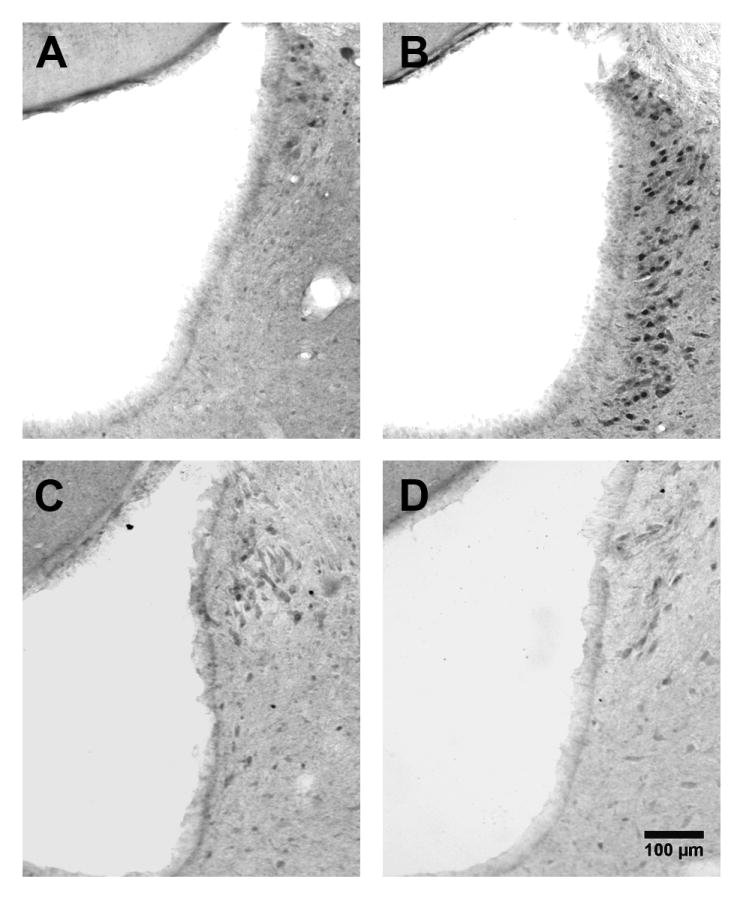
Representative photomicrographs of Fos expression in the LC (~ -9.90 mm from bregma) following footpad stimulation to a sham (A), or SNI rat (B), or following no stimulation of a sham (C) or SNI rat (D). Magnification = 10X.
SNI increases stimulus-induced pCREB expression in the LC
The phosphorylated form of the transcription factor CREB (pCREB) alters the expression of numerous genes (including Fos) and, like Fos, is a marker of cellular activity in the nervous system (Lonze and Ginty, 2002, Impey et al., 2004), including pain modulatory systems. Therefore, we extended our Fos results with quantification of pCREB expression in the LC after SNI and mechanical stimulation of the injured paw. Figs 3A-C show that this increased the overall number of pCREB-ir positive profiles as compared with sham-operated rats [F(1,18) = 6.98, p = 0.016]. When tissue sections were segregated into four coronal divisions (Fig 3D), we found that SNI increased pCREB-ir profiles within each division (p’s < 0.05).
Figure 3. Stimulus-induced pCREB expression in the LC after SNI.

Panel A: The total number of pCREB-immunoreactive (pCREB-ir) profiles in the LC (both sides) in sham and SNI rats 2 wk following SNI. The representative photomicrographs (-9.90 mm from bregma) from a sham (B) and an SNI rat (C) illustrate that innocuous tactile footpad stimulation in the setting of nerve injury increased pCREB-ir. Panel D: Quantification of pCREB-ir following footpad stimulation to sham and SNI rats within each of 4 coronal divisions of the LC. Magnification = 15X. n=5-12. ★p < 0.05.
There was a significant correlation between tactile allodynia (% change in VF threshold after SNI) and the number (across all coronal planes) of stimulus-induced pCREB-ir (PMCC = 0.75, p = 0.008) positive profiles. Subsequent analysis revealed that this correlation extended to the -10.15 to -10.32 mm coronal division for pCREB-ir (PMCC = 0.73, p = 0.016, n = 10, not shown).
Noradrenergic LC lesions retard the development of allodynia and hyperalgesia
Numerous studies indicate that LC lesions change inflammatory pain (Tsuruoka and Willis, 1996b, a, Wei et al., 1999, Taylor et al., 2000, Tsuruoka et al., 2003b). To extend the lesion approach to a model of neuropathic pain, we evaluated the effect of i.c.v. injection of 5 μg of anti-DβH-saporin, a noradrenergic-specific neurotoxin, on the density of noradrenergic neurons in A5, LC, and A7 and on the development of neuropathic pain. Fig 4 illustrates that, in contrast to mouse IgG-saporin controls, anti-DβH-saporin almost completely eliminated DβH-immunoreactivity (DβH-ir) in the LC, but neither A5 nor A7 nuclei. This confirms the relative specificity of the 5 μg i.c.v. dose for the LC (Wrenn et al., 1996, Rohde and Basbaum, 1998).
Figure 4. Pontine DβH immunoreactivity following anti-DβH-saporin.

Photomicrographs depicting low (2X, Panels A-B) and high (10X, Panels C-H) power images from a representative saporin IgG control (A, C, E, G) or an anti-DβH-saporin-treated rat (B, D, F, H). Anti-DβH-saporin almost completely eliminated DβH-immunoreactivity (DβH-ir) in the LC (C vs D) but neither A5 (E vs F) nor A7 nuclei (G vs H).
Fig 5 illustrates that anti-DβH-saporin, administered 2 weeks prior to SNI, reduced tactile allodynia, mechanical hyperalgesia, and cold allodynia compared with mouse IgG-treated controls. When collapsed across days 3 - 28, tactile allodynia was significantly lower in the anti-DβH-saporin-treated group as assessed by von Frey [Fig 5B, Mann-Whitney F = 5.0, p < 0.05] and automated von Frey (MMP) [Fig 5D, F(1,5) = 14.83, p < 0.05]. Both mechanical hyperalgesia (Fig 5F) [F(1,6) = 14.69, p < 0.01] and cold allodynia (Fig 5H) [F(1,6) = 27.10, p < 0.01] were also significantly lower in anti-DβH-saporin-treated rats. Further inspection of the time course data reveals that behavioural hypersensitivity never robustly developed in the anti-DβH-saporin group.
Figure 5. SNI-induced allodynia and hyperagesia after anti-DβH-saporin.

Line graphs in the left panels (A, C, E, G) illustrate time course data collected from the paw ipsilateral to SNI. Histograms at the right (B, D, F, H) summarize the change in behaviour (mean of day 3-28) at the ipsilateral and contralateral sides. Mechanical threshold was assessed with both von Frey hairs (A, B) and an automated machine-mounted probe (MMP, C, D). Mechanical hyperalgesia was assessed as paw withdrawal duration to a blunt pin (Pin Prick, E, F). Cold allodynia was assessed as paw withdrawal duration to topical plantar acetone (G, H). Anti-DβH-saporin reduced all behavioral signs of neuropathic pain [★p’s < 0.05, Mann-Whitney U test subsequent to Kruskal Wallis (A, B) or post-hoc Bonferroni subsequent to ANOVA (C-H)]. Values represent mean ± SEM. n=4.
Anti-DβH-saporin did not change behavioural responses elicited from the contralateral paw (p’s > 0.05, data not shown). Table 1 shows that 5 μg anti-DβH-saporin did not disrupt weight gain. In addition there were no overt signs of altered behavior (e.g. distress vocalization, loss of muscular coordination, muscular tremor, defensive aggression, freezing, papillary dilation, salivation, panting, reflex urination and defecation) in the experimental group, consistent with a previous report (Rohde and Basbaum, 1998). These data indicate that neurotoxin administration does not produce confounding non-specific effects.
Table 1.
Body weight (g) at various time points after vehicle or anti-DβH-saporin.
| Mouse IgG-saporin | i.c.v. infusion | SNI | Day 3 | Day 7 | Day 14 | Day 21 | Day 28 | |
|---|---|---|---|---|---|---|---|---|
| Day | 0 | 25 | 39 | 42 | 46 | 53 | 60 | 67 |
| Mean | 250.3 | 318.5 | 347.0 | 342.8 | 346.3 | 359.5 | 366.8 | 368.3 |
| ±SEM | 4.6 | 5.2 | 7.7 | 8.5 | 9.4 | 9.0 | 8.0 | 9.1 |
| anti-DβH-saporin | i.c.v. infusion | SNI | Day 3 | Day 7 | Day 14 | Day 21 | Day 28 | |
| Day | 0 | 25 | 38 | 42 | 45 | 52 | 59 | 66 |
| Mean | 249.8 | 313.5 | 340.3 | 333.5 | 336.8 | 351.5 | 355.5 | 352.0 |
| ±SEM | 7.7 | 13.4 | 17.1 | 18.4 | 19.2 | 18.5 | 16.0 | 14.8 |
Local anaesthesia of the LC reverses allodynia and hyperalgesia
We next evaluated whether local microinjection of lidocaine, a short acting anaesthetic, would reverse signs of established allodynia and hyperalgesia. As illustrated in Fig 6, saline did not change SNI-induced signs of allodynia and hyperalgesia. By contrast, lidocaine significantly decreased all 4 signs of hypersensitivity: tactile allodynia as assessed by von Frey hairs, at both 15 min and 45 min post-infusion [Kruskall-Wallis ANOVA by Ranks H(1,N=6) = 3.97, p < 0.05 and H(1,N=6) = 3.86, p < 0.05, respectively]; tactile allodynia as assessed with a machine-mounted probe [F(1,9) = 13.40, p = 0.005]; mechanical hyperalgesia [F(1,9) = 6.21, p = 0.034]; and cold allodynia [F(1,9) = 33.80, p < 0.001]. These effects peaked at 15 min and then subsided to baseline values by 90 min. Lidocaine did not change behavioural responses evoked from the contralateral paw (p’s > 0.05, data not shown). Fig 7 shows a representative example of a microinjection site within the LC, indicated by deposits of India ink.
Figure 6. SNI-induced allodynia and hyperalgesia after intra-LC lidocaine.
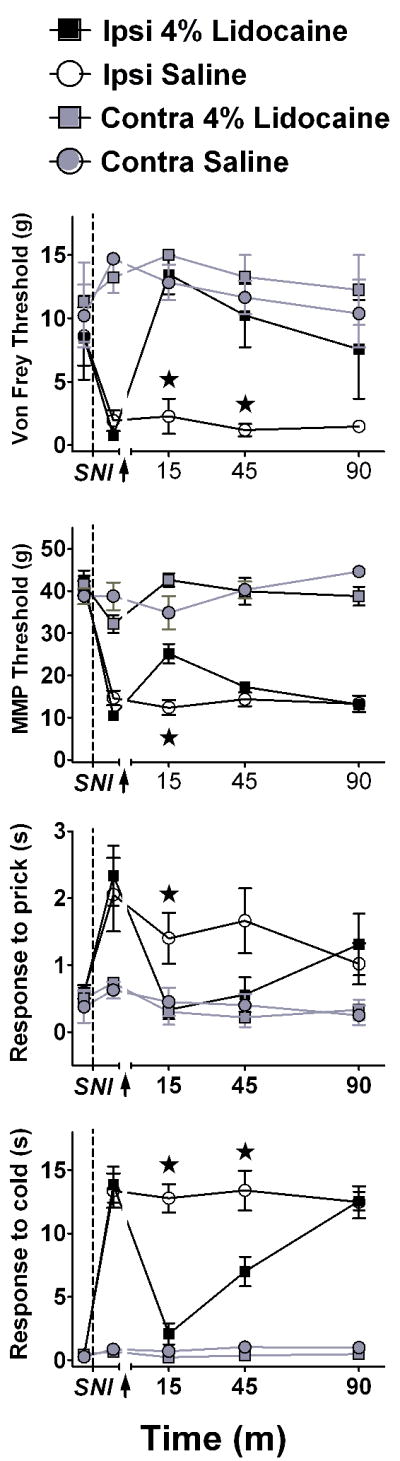
Line graphs illustrating time course data for mechanical threshold assessed with either von Frey hairs (A), or an automated machine-mounted probe (MMP, B), a noxious mechanical blunt pin (prick) stimulus (C), or a cold (topical acetone) stimulus (D). When delivered 2 wk after SNI, bilateral microinjection of lidocaine (4%), but not saline, reduced all signs of neuropathic pain at the paw ipsilateral to SNI. Data is expressed as mean ± SEM. n=3-6. ★p < 0.01.
Figure 7. Microinjection sites within the LC.
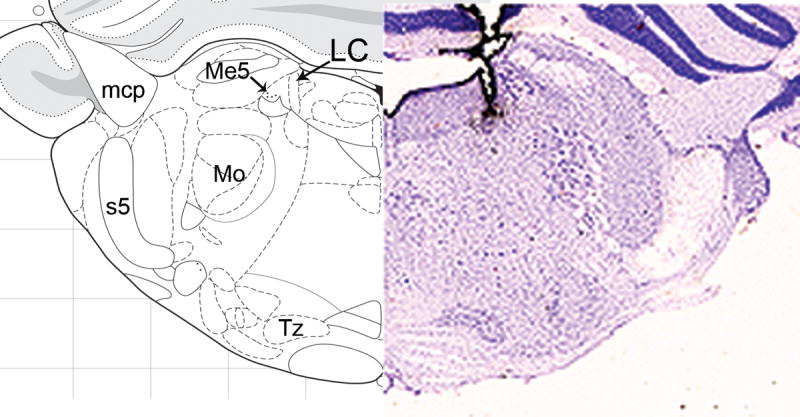
The Nissl section on the right illustrates a representative deposit of India ink within and above the LC (mapped to be approximately -0.8 mm from interaural). Landmarks are provided on the left. mcp=middle cerebellar peduncle; Me5=mesencephalic 5 nucleus; Mo=motor 5 nucleus; s5=sensory root of the 5th nucleus; Tz=nucleus of the trapezoid body.
DISCUSSION
Severe tissue injury or nerve damage triggers early neuroplastic changes in the brainstem that contribute to an endogenous feedback inhibition of pain. This serves as a beneficial, adaptive mechanism in times of stress and/or danger. With time, injury also recruits facilitatory mechanisms in the brain that ultimately sensitize pain transmission neurons in the spinal cord. Normally, such mechanisms aid in the prevention of further injury, thus promoting curative processes (Millan, 1999, 2002). Under certain conditions, however, CNS facilitation can persist long after the injury has healed, thereby contributing to central sensitization that drives the neural and behavioural manifestations of chronic pain (Urban and Gebhart, 1999, Porreca et al., 2002, Ren and Dubner, 2002). In addition to the traditional view of the LC as a pain inhibitory structure (Millan, 2002, Aston-Jones, 2004, Pertovaara, 2006), the present results indicate that the LC (whether directly or indirectly via other brain regions) might also participate in the facilitation of neuropathic pain.
Somatosensory stimulation increased Fos and pCREB in the LC after SNI
We found that somatosensory stimulation significantly increased Fos and pCREB in rats with SNI, and suggest that this results from the direct activation of dorsal horn neurons that project to noradrenergic neurons in the brainstem (Westlund and Craig, 1996). We could not elicit LC Fos expression with mechanical stimulation alone, e.g. in the absence of SNI. These results are consistent with Besson and colleagues, who found that somatosensory hindpaw stimulation induced Fos expression in the dorsal horn following CCI but not sham surgery (Catheline et al., 1999). Second, we found that stimulation-evoked induction of Fos was correlated with behavioral signs of allodynia and hyperalgesia. Although there is general (but not universal) agreement that increased Fos expression is a marker of increased neuronal activity (Harris, 1998, Coggeshall, 2005), it remains to be determined whether these neurons contribute to pain facilitation or inhibition. Interestingly, our results are consistent with the finding that spinal nerve ligation potentiated the responsiveness of LC neurons to noxious mechanical stimuli – this raises the intriguing idea that nerve injury sensitizes the LC, thus leading to pain facilitation (Viisanen and Pertovaara, 2007).
As with mechanical stimulation in the absence of SNI, we found that SNI in the absence of mechanical stimulation did not induce Fos in the LC. This is consistent with previous electrophysiological studies indicating that L5/L6 spinal nerve ligation does not change the spontaneous discharge rate of LC neurons (Viisanen and Pertovaara, 2007), but contrasts with the finding that chronic constriction injury of the sciatic nerve increased regional blood flow in the LC (Mao et al., 1993).
We found that stimulus-induced protein expression occurred to a similar degree within the LC of both hemispheres. This extends previous reports indicating that unilateral high intensity nerve stimulation (Voisin et al., 2005), paw inflammation with carrageenan (Tsuruoka et al., 2003a), intraplantar formalin (Palkovits et al., 1999), or CCI (Mao et al., 1993) increased LC Fos and/or neuronal activity in a bilateral pattern. This is not surprising since spinal projection neurons not only decussate and travel to the contralateral side of the brain, but also ascend bilaterally (Spike et al., 2003).
Noradrenergic LC neurons contribute to the development of neuropathic pain
We found that anti-DβH-saporin decreased the intensity of allodynia and hyperalgesia. In support of these findings, DSP4, another selective noradrenergic neurotoxin, prevented autotomy behavior (a possible sign of neuropathic pain) after sciatic nerve transection (Al-Adawi et al., 2002). These data suggest that targeted destruction of the LC noradrenergic system delays or even prevents the development of neuropathic pain.
Only two other studies have investigated the contribution of LC noradrenergic neurons to behavioral signs of neuropathic pain. In contrast to the i.c.v. route of administration of anti-DβH-saporin used in our studies, Li et. al. and Jasmin et. al. used the intrathecal route to more selectively destroy spinal noradrenergic terminals arising from the brainstem. They found that i.t. anti-DβH-saporin either did not change (Li et al., 2002) or decreased von Frey threshold (Jasmin et al., 2003). Short of performing additional pharmacological studies evaluating inhibitory and excitatory systems, several explanations can be proposed for the difference in outcome between the i.c.v and i.t. studies. First, compared to the i.c.v. route, intrathecal anti-DβH-saporin yields a less-complete destruction of noradrenergic neurons in the LC and their corresponding ascending forebrain and descending spinal projections (Martin et al., 1999, Jasmin et al., 2003).
Second, the anatomical distribution of neuron loss differs after intrathecal and i.c.v. administration. While both preferentially destroy cell bodies in the LC, with a lesser destruction of A5 and A7 (Wrenn et al., 1996), only i.c.v. administration reduced noradrenergic terminal staining in the periaqueductal gray, parabrachial area, hypothalamus, and amygdala (Li et al., 2002, Jasmin et al., 2003). Since our studies used the i.c.v. route, pain facilitation by the LC may be mediated by ascending noradrenergic pathways rather than descending coeruleospinal pathways. Further studies are needed to distinguish these pathways, perhaps involving pharmacologic definition with specific noradrenergic antagonists delivered to discreet CNS sites.
A third possibility concerns differences in experimental pain model; our studies employed SNI, while Jasmin et al and Li et al used SNL (Kim and Chung, 1992). These models share much in common, but they do differ in terms of neuropathic symptoms (e.g. allodynia and/or hyperalgesia), mode of expression (e.g. tactile and/or thermal), duration, and degree (Ossipov et al., 2006).
As in previous studies, anti-DβH-saporin destroyed a few A5 and A7 noradrenergic neurons. Like the LC, the A5 and A7 cell groups modulate acute nociception (Sagen and Proudfit, 1986, Burnett and Gebhart, 1991, Holden et al., 1999, Nuseir and Proudfit, 2000). In contrast to the LC, however, few if any reports discuss the contribution of A5 and A7 neurons to chronic pain (Holden and Pizzi, 2003). Based on our immunohistochemical, neurotoxin, and lidocaine microinjection data presented here, we believe that the massive destruction of LC neurons decreased allodynia and hyperalgesia, although we cannot unequivocally rule out a relatively small impact of the neurotoxin on A5 and A7 neurons.
The LC contributes to the maintenance of allodynia and hyperalgesia
We found that microinjection of lidocaine dramatically reversed all behavioural signs of established allodynia and hyperalgesia. The duration of action was in accordance with the pharmacokinetics of lidocaine, and its reversibility argues against non-specific (e.g. toxic) actions.
Since lidocaine blocks Na+ channel function, it prevents the conduction of nerve impulses not only in cells but also in axons; therefore, we cannot rule out an effect on fibers of passage (Sandkuhler et al., 1987, Martin, 1991). Furthermore, our lidocaine injection experiments were not designed to rule out a contribution of surrounding areas known to be important in nociceptive transmission and modulation, such as the parabrachial and A7 nuclei. However, when considered together with our anti-DβH-saporin studies, which bypass issues of fibers of passage and drug diffusion, we believe that lidocaine acted at least in part through direct inhibition of LC cells. We speculate that in the setting of peripheral nerve injury, noradrenergic neurons in the LC exert a tonic facilitatory influence on behavioural signs of neuropathic pain. As the present studies did not evaluate spinal norepinephrine content or study descending noradrenergic innervations, we are unable to determine if the LC contributes to allodynia or hyperalgesia via ascending or descending projections.
Reference to analogous studies in the RVM provides a framework for understanding the role of the LC in neuropathic pain. Porreca and colleagues demonstrated that selective lesions of opioid receptor-expressing neurons in the RVM, or local administration of lidocaine, reversed tactile allodynia. These data indicate that the RVM maintains neuropathic pain (Porreca et al., 2001, Burgess et al., 2002, Gardell et al., 2003). However, while we found that anti-DβH-saporin reduced early signs of allodynia, neither dermorphin-saporin RVM lesions (Porreca et al., 2001), nor lidocaine inactivation of the RVM (Burgess et al., 2002) reduced tactile allodynia at early time points, suggesting that the RVM contributes to the maintenance but not the initiation of neuropathic pain. By contrast, the results of our neurotoxin and microinjection studies suggest that the LC contributes to both the maintenance and initiation of neuropathic pain.
Broad spectrum modulation of sensory modality by the LC
Our model of neuropathic pain (SNI) and experimental design allowed us to assess withdrawal threshold to a non-noxious mechanical stimulus and withdrawal duration to noxious mechanical or cool stimuli. Thus, our results allow us to conclude that NE-containing neurons in the LC contribute not only to nerve injury-induced tactile allodynia, but also to cold allodynia and mechanical hyperalgesia. This suggests that the LC acts globally to modulate multiple sensory modalities. Similarly, descending facilitation from the RVM participates in the expression of multiple pain modalities (tactile allodynia, heat hyperalgesia) associated with SNL (Pertovaara et al., 1996, Burgess et al., 2002, Vera-Portocarrero et al., 2006).
Summary
We found that SNI increased Fos and pCREB in the LC. Second, noradrenergic LC lesions inhibited the development of allodynia and hyperalgesia. And third, reversible inactivation of the LC reduced established neuropathic pain. We conclude that NE neurons in the LC participate in the development and/or maintenance of allodynia and hyperalgesia in the setting of peripheral nerve injury, and propose that the traditional view of the LC as a pain inhibitory structure be modified to account for its capacity as a pain facilitator. While noradrenergic reuptake inhibitors such as duloxetine decrease neuropathic pain (Brecht et al., 2007), it remains important to pursue the current findings with future studies to determine the pharmacology and the contribution of ascending or descending pathways underlying LC-mediated pain facilitation. Indeed, Rahman et al recently found that intrathecal administration of the alpha(2)-adrenoceptor antagonist, atipamezole, increased the evoked responses of dorsal horn neurons to low-intensity mechanical stimuli in control animals but was without effect in animals with peripheral nerve injury. This shift in the balance of noradrenergic controls from inhibition to facilitation may contribute to neuropathic pain (Rahman et al., 2008).
Acknowledgments
The study was supported by NIH grants R01DA018732 and K02DA019656 to BKT and a 2007 post-doctoral fellowship from the PhRMA foundation and NRSA FNS056889A to JJB. We would also like to thank Christopher L. Kreidt for his technical contributions to the lidocaine study and Heather A. Scuderi Porter (H.P.) for her contribution to the Fos analysis.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abbadie C, Taylor BK, Peterson MA, Basbaum AI. Differential contribution of the two phases of the formalin test to the patter of c-fos expression in the rat spinal cord: studies with remifentanil and lidocaine. Pain. 1997;69:101–110. doi: 10.1016/s0304-3959(96)03285-x. [DOI] [PubMed] [Google Scholar]
- Abramoff MD, Magelhaes PJ, Ram SJ. Image Processing with ImageJ. Biophotonics International. 2004;11:36–42. [Google Scholar]
- Al-Adawi S, Dawe GS, Bonner A, Stephenson JD, Zarei M. Central noradrenergic blockade prevents autotomy in rat: implication for pharmacological prevention of postdenervation pain syndrome. Brain Res Bull. 2002;57:581–586. doi: 10.1016/s0361-9230(01)00747-x. [DOI] [PubMed] [Google Scholar]
- Aloisi AM, Steenbergen HL, van de Poll NE, Farabollini F. Sex-dependent effects of restraint on nociception and pituitary-adrenal hormones in the rat. Physiology and Behavior. 1994;55:789–793. doi: 10.1016/0031-9384(94)90061-2. [DOI] [PubMed] [Google Scholar]
- Aston-Jones G. Locus Coeruleus, A5 and A7 Noradrenergic Cell Groups. In: Paxinos G, editor. The Rat Nervous System. China: Elsevier; 2004. [Google Scholar]
- Barthelemy I, Martineau D, Ong M, Matsunami R, Ling N, Benatti L, Cavallaro U, Soria M, Lappi DA. The expression of saporin, a ribosome-inactivating protein from the plant Saponaria officinalis, in Escherichia coli. J Biol Chem. 1993;268:6541–6548. [PubMed] [Google Scholar]
- Brecht S, Courtecuisse C, Debieuvre C, Croenlein J, Desaiah D, Raskin J, Petit C, Demyttenaere K. Efficacy and safety of duloxetine 60 mg once daily in the treatment of pain in patients with major depressive disorder and at least moderate pain of unknown etiology: a randomized controlled trial. J Clin Psychiatry. 2007;68:1707–1716. doi: 10.4088/jcp.v68n1110. [DOI] [PubMed] [Google Scholar]
- Burgess SE, Gardell LR, Ossipov MH, Malan TP, Jr, Vanderah TW, Lai J, Porreca F. Time-dependent descending facilitation from the rostral ventromedial medulla maintains, but does not initiate, neuropathic pain. J Neurosci. 2002;22:5129–5136. doi: 10.1523/JNEUROSCI.22-12-05129.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnett A, Gebhart GF. Characterization of descending modulation of nociception from the A5 cell group. Brain Res. 1991;546:271–281. doi: 10.1016/0006-8993(91)91491-i. [DOI] [PubMed] [Google Scholar]
- Calcagnetti DJ, Fleetwood SW, Holtzman SG. Pharmacological profile of the potentiation of opioid analgesia by restraint stress. Pharmacol Biochem Behav. 1990;37:193–199. doi: 10.1016/0091-3057(90)90061-l. [DOI] [PubMed] [Google Scholar]
- Catheline G, Le Guen S, Honore P, Besson JM. Are there long-term changes in the basal or evoked Fos expression in the dorsal horn of the spinal cord of the mononeuropathic rat? Pain. 1999;80:347–357. doi: 10.1016/s0304-3959(98)00234-6. [DOI] [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. Journal of Neuroscience Methods. 1994;53:55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- Coggeshall RE. Fos, nociception and the dorsal horn. Prog Neurobiol. 2005;77:299–352. doi: 10.1016/j.pneurobio.2005.11.002. [DOI] [PubMed] [Google Scholar]
- Colombo PJ, Brightwell JJ, Countryman RA. Cognitive strategy-specific increases in phosphorylated cAMP response element-binding protein and c-Fos in the hippocampus and dorsal striatum. J Neurosci. 2003;23:3547–3554. doi: 10.1523/JNEUROSCI.23-08-03547.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decosterd I, Woolf CJ. Spared nerve injury: an animal model of persistent peripheral neuropathic pain. Pain. 2000;87:149–158. doi: 10.1016/S0304-3959(00)00276-1. [DOI] [PubMed] [Google Scholar]
- Dinota A, Tazzari PL, Michieli M, Visani G, Gobbi M, Bontadini A, Tassi C, Fanin R, Damiani D, Grandi M, et al. In vitro bone marrow purging of multidrug-resistant cells with a mouse monoclonal antibody directed against Mr 170,000 glycoprotein and a saporin-conjugated anti-mouse antibody. Cancer Res. 1990;50:4291–4294. [PubMed] [Google Scholar]
- Dubner R. The neurobiology of persistent pain and its clinical implications. Suppl Clin Neurophysiol. 2004;57:3–7. doi: 10.1016/s1567-424x(09)70337-x. [DOI] [PubMed] [Google Scholar]
- Gardell LR, Vanderah TW, Gardell SE, Wang R, Ossipov MH, Lai J, Porreca F. Enhanced evoked excitatory transmitter release in experimental neuropathy requires descending facilitation. J Neurosci. 2003;23:8370–8379. doi: 10.1523/JNEUROSCI.23-23-08370.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs JL, Flores CM, Hargreaves KM. Attenuation of capsaicin-evoked mechanical allodynia by peripheral neuropeptide Y Y1 receptors. Pain. 2006;124:167–174. doi: 10.1016/j.pain.2006.04.013. [DOI] [PubMed] [Google Scholar]
- Grigg P, Robichaud DR, 2nd, Bove GM. A feedback-controlled dynamic linear actuator to test foot withdrawal thresholds in rat. J Neurosci Methods. 2007;163:44–51. doi: 10.1016/j.jneumeth.2007.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grzanna R, Molliver ME. The locus coeruleus in the rat: an immunohistochemical delineation. Neuroscience. 1980;5:21–40. doi: 10.1016/0306-4522(80)90068-8. [DOI] [PubMed] [Google Scholar]
- Harris JA. Using c-fos as a neural marker of pain. Brain Res Bull. 1998;45:1–8. doi: 10.1016/s0361-9230(97)00277-3. [DOI] [PubMed] [Google Scholar]
- Holden JE, Pizzi JA. The challenge of chronic pain. Adv Drug Deliv Rev. 2003;55:935–948. doi: 10.1016/s0169-409x(03)00097-8. [DOI] [PubMed] [Google Scholar]
- Holden JE, Schwartz EJ, Proudfit HK. Microinjection of morphine in the A7 catecholamine cell group produces opposing effects on nociception that are mediated by alpha1-and alpha2-adrenoceptors. Neuroscience. 1999;91:979–990. doi: 10.1016/s0306-4522(98)00673-3. [DOI] [PubMed] [Google Scholar]
- Impey S, McCorkle SR, Cha-Molstad H, Dwyer JM, Yochum GS, Boss JM, McWeeney S, Dunn JJ, Mandel G, Goodman RH. Defining the CREB regulon: a genome-wide analysis of transcription factor regulatory regions. Cell. 2004;119:1041–1054. doi: 10.1016/j.cell.2004.10.032. [DOI] [PubMed] [Google Scholar]
- Intondi AB, Dahlgren MN, Eilers MA, Taylor BK. Intrathecal neuropeptide Y reduces behavioral and molecular markers of inflammatory or neuropathic pain. Pain. 2007 doi: 10.1016/j.pain.2007.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jasmin L, Boudah A, Ohara PT. Long-term effects of decreased noradrenergic central nervous system innervation on pain behavior and opioid antinociception. J Comp Neurol. 2003;460:38–55. doi: 10.1002/cne.10633. [DOI] [PubMed] [Google Scholar]
- Jones SL, Gebhart GF. Quantitative characterization of coeruleospinal inhibition of nociceptive transmission in the rat. J Neurophysiol. 1986;56:1397–1410. doi: 10.1152/jn.1986.56.5.1397. [DOI] [PubMed] [Google Scholar]
- Jones SL, Gebhart GF. Spinal pathways mediating tonic, coeruleospinal, and raphe-spinal descending inhibition in the rat. J Neurophysiol. 1987;58:138–159. doi: 10.1152/jn.1987.58.1.138. [DOI] [PubMed] [Google Scholar]
- Kim SH, Chung JM. An experimental model for peripheral neuropathy produced by segmental spinal nerve ligation in the rat. Pain. 1992;50:355–363. doi: 10.1016/0304-3959(92)90041-9. [DOI] [PubMed] [Google Scholar]
- Kwiat GC, Basbaum AI. The origin of brainstem noradrenergic and serotonergic projections to the spinal cord dorsal horn in the rat. Somatosens Mot Res. 1992;9:157–173. doi: 10.3109/08990229209144768. [DOI] [PubMed] [Google Scholar]
- Levesque D, Holtzman SG. The potentiating effects of restraint stress and continuous naloxone infusion on the analgesic potency of morphine are additive. Brain Research. 1993;617:176–180. doi: 10.1016/0006-8993(93)90633-x. [DOI] [PubMed] [Google Scholar]
- Li X, Conklin D, Ma W, Zhu X, Eisenach JC. Spinal noradrenergic activation mediates allodynia reduction from an allosteric adenosine modulator in a rat model of neuropathic pain. Pain. 2002;97:117–125. doi: 10.1016/s0304-3959(02)00011-8. [DOI] [PubMed] [Google Scholar]
- Lonze BE, Ginty DD. Function and regulation of CREB family transcription factors in the nervous system. Neuron. 2002;35:605–623. doi: 10.1016/s0896-6273(02)00828-0. [DOI] [PubMed] [Google Scholar]
- Ma QP, Woolf CJ. Basal and touch-evoked fos-like immunoreactivity during experimental inflammation in the rat. Pain. 1996;67:307–316. doi: 10.1016/0304-3959(96)03132-6. [DOI] [PubMed] [Google Scholar]
- Malpeli JG, Schiller PH. A method of reversible inactivation of small regions of brain tissue. J Neurosci Methods. 1979;1:143–151. doi: 10.1016/0165-0270(79)90011-6. [DOI] [PubMed] [Google Scholar]
- Mao J, Mayer DJ, Price DD. Patterns of increased brain activity indicative of pain in a rat model of peripheral mononeuropathy. J Neurosci. 1993;13:2689–2702. doi: 10.1523/JNEUROSCI.13-06-02689.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin JH. Autoradiographic estimation of the extent of reversible inactivation produced by microinjection of lidocaine and muscimol in the rat. Neurosci Lett. 1991;127:160–164. doi: 10.1016/0304-3940(91)90784-q. [DOI] [PubMed] [Google Scholar]
- Martin WJ, Gupta NK, Loo CM, Rohde DS, Basbaum AI. Differential effects of neurotoxic destruction of descending noradrenergic pathways on acute and persistent nociceptive processing. Pain. 1999;80:57–65. doi: 10.1016/s0304-3959(98)00194-8. [DOI] [PubMed] [Google Scholar]
- Millan MJ. The induction of pain: an integrative review. Prog Neurobiol. 1999;57:1–164. doi: 10.1016/s0301-0082(98)00048-3. [DOI] [PubMed] [Google Scholar]
- Millan MJ. Descending control of pain. Prog Neurobiol. 2002;66:355–474. doi: 10.1016/s0301-0082(02)00009-6. [DOI] [PubMed] [Google Scholar]
- Nuseir K, Proudfit HK. Bidirectional modulation of nociception by GABA neurons in the dorsolateral pontine tegmentum that tonically inhibit spinally projecting noradrenergic A7 neurons. Neuroscience. 2000;96:773–783. doi: 10.1016/s0306-4522(99)00603-x. [DOI] [PubMed] [Google Scholar]
- Ossipov MH, Lai J, Malan TP, Jr, Porreca F. Spinal and supraspinal mechanisms of neuropathic pain. Ann N Y Acad Sci. 2000;909:12–24. doi: 10.1111/j.1749-6632.2000.tb06673.x. [DOI] [PubMed] [Google Scholar]
- Ossipov MH, Lai J, Porreca F. Mechanisms of experimental neuropathic pain: integration from animal models. In: McMahon SB, Koltzenburg M, editors. Wall and Melzack’s Textbook of Pain. China: Elsevier; 2006. pp. 929–946. [Google Scholar]
- Palkovits M, Baffi JS, Pacak K. The role of ascending neuronal pathways in stress-induced release of noradrenaline in the hypothalamic paraventricular nucleus of rats. J Neuroendocrinol. 1999;11:529–539. doi: 10.1046/j.1365-2826.1999.00365.x. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates: The new coronal set. Burlington, MA: Elsevier; 2005. [Google Scholar]
- Pertovaara A. Noradrenergic pain modulation. Prog Neurobiol. 2006;80:53–83. doi: 10.1016/j.pneurobio.2006.08.001. [DOI] [PubMed] [Google Scholar]
- Pertovaara A, Wei H, Hamalainen MM. Lidocaine in the rostroventromedial medulla and the periaqueductal gray attenuates allodynia in neuropathic rats. Neurosci Lett. 1996;218:127–130. doi: 10.1016/s0304-3940(96)13136-0. [DOI] [PubMed] [Google Scholar]
- Picklo MJ, Wiley RG, Lappi DA, Robertson D. Noradrenergic lesioning with an anti-dopamine beta-hydroxylase immunotoxin. Brain Res. 1994;666:195–200. doi: 10.1016/0006-8993(94)90772-2. [DOI] [PubMed] [Google Scholar]
- Porreca F, Burgess SE, Gardell LR, Vanderah TW, Malan TP, Jr, Ossipov MH, Lappi DA, Lai J. Inhibition of neuropathic pain by selective ablation of brainstem medullary cells expressing the mu-opioid receptor. J Neurosci. 2001;21:5281–5288. doi: 10.1523/JNEUROSCI.21-14-05281.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porreca F, Ossipov MH, Gebhart GF. Chronic pain and medullary descending facilitation. Trends Neurosci. 2002;25:319–325. doi: 10.1016/s0166-2236(02)02157-4. [DOI] [PubMed] [Google Scholar]
- Rahman W, D’Mello R, Dickenson AH. Peripheral nerve injury-induced changes in spinal alpha(2)-adrenoceptor-mediated modulation of mechanically evoked dorsal horn neuronal responses. J Pain. 2008;9:350–359. doi: 10.1016/j.jpain.2007.11.010. [DOI] [PubMed] [Google Scholar]
- Rasband WS. ImageJ. U. S. National Institutes of Health; Bethesda, Maryland, USA: 19972007. http://rsb.info.nih.gov/ij. [Google Scholar]
- Ren K, Dubner R. Descending modulation in persistent pain: an update. Pain. 2002;100:1–6. doi: 10.1016/s0304-3959(02)00368-8. [DOI] [PubMed] [Google Scholar]
- Rohde DS, Basbaum AI. Activation of coeruleospinal noradrenergic inhibitory controls during withdrawal from morphine in the rat. J Neurosci. 1998;18:4393–4402. doi: 10.1523/JNEUROSCI.18-11-04393.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagen J, Proudfit HK. Alterations in nociception following lesions of the A5 catecholamine nucleus. Brain Res. 1986;370:93–101. doi: 10.1016/0006-8993(86)91108-x. [DOI] [PubMed] [Google Scholar]
- Sandkuhler J, Maisch B, Zimmermann M. The use of local anaesthetic microinjections to identify central pathways: a quantitative evaluation of the time course and extent of the neuronal block. Experimental brain research Experimentelle Hirnforschung. 1987;68:168–178. doi: 10.1007/BF00255242. [DOI] [PubMed] [Google Scholar]
- Spike RC, Puskar Z, Andrew D, Todd AJ. A quantitative and morphological study of projection neurons in lamina I of the rat lumbar spinal cord. Eur J Neurosci. 2003;18:2433–2448. doi: 10.1046/j.1460-9568.2003.02981.x. [DOI] [PubMed] [Google Scholar]
- Studelska DR, Brimijoin S. Partial isolation of two classes of dopamine beta-hydroxylase-containing particles undergoing rapid axonal transport in rat sciatic nerve. J Neurochem. 1989;53:622–631. doi: 10.1111/j.1471-4159.1989.tb07379.x. [DOI] [PubMed] [Google Scholar]
- Taylor BK, Abhyankar SS, Vo NT, Kriedt CL, Churi SB, Urban JH. Neuropeptide Y acts at Y1 receptors in the rostral ventral medulla to inhibit neuropathic pain. Pain. 2007;131:83–95. doi: 10.1016/j.pain.2006.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor BK, Roderick RE, Basbaum AI. Brainstem noradrenergic control of nociception is abnormal in the spontaneously hypertensive rat. Neurosci Lett. 2000;291:139–142. doi: 10.1016/s0304-3940(00)01389-6. [DOI] [PubMed] [Google Scholar]
- Tsuruoka M, Arai YC, Nomura H, Matsutani K, Willis WD. Unilateral hindpaw inflammation induces bilateral activation of the locus coeruleus and the nucleus subcoeruleus in the rat. Brain Res Bull. 2003a;61:117–123. doi: 10.1016/s0361-9230(03)00099-6. [DOI] [PubMed] [Google Scholar]
- Tsuruoka M, Matsutani K, Inoue T. Coeruleospinal inhibition of nociceptive processing in the dorsal horn during unilateral hindpaw inflammation in the rat. Pain. 2003b;104:353–361. doi: 10.1016/s0304-3959(03)00042-3. [DOI] [PubMed] [Google Scholar]
- Tsuruoka M, Willis WD. Descending modulation from the region of the locus coeruleus on nociceptive sensitivity in a rat model of inflammatory hyperalgesia. Brain Research. 1996a;743:86–92. doi: 10.1016/s0006-8993(96)01025-6. [DOI] [PubMed] [Google Scholar]
- Tsuruoka M, Willis WD., Jr Bilateral lesions in the area of the nucleus locus coeruleus affect the development of hyperalgesia during carrageenan-induced inflammation. Brain Res. 1996b;726:233–236. [PubMed] [Google Scholar]
- Urban MO, Gebhart GF. Supraspinal contributions to hyperalgesia. Proc Natl Acad Sci U S A. 1999;96:7687–7692. doi: 10.1073/pnas.96.14.7687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vera-Portocarrero LP, Zhang ET, Ossipov MH, Xie JY, King T, Lai J, Porreca F. Descending facilitation from the rostral ventromedial medulla maintains nerve injury-induced central sensitization. Neuroscience. 2006;140:1311–1320. doi: 10.1016/j.neuroscience.2006.03.016. [DOI] [PubMed] [Google Scholar]
- Viisanen H, Pertovaara A. Influence of peripheral nerve injury on response properties of locus coeruleus neurons and coeruleospinal antinociception in the rat. Neuroscience. 2007;146:1785–1794. doi: 10.1016/j.neuroscience.2007.03.016. [DOI] [PubMed] [Google Scholar]
- Voisin DL, Guy N, Chalus M, Dallel R. Nociceptive stimulation activates locus coeruleus neurones projecting to the somatosensory thalamus in the rat. J Physiol. 2005;566:929–937. doi: 10.1113/jphysiol.2005.086520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei F, Dubner R, Ren K. Nucleus reticularis gigantocellularis and nucleus raphe magnus in the brain stem exert opposite effects on behavioral hyperalgesia and spinal Fos protein expression after peripheral inflammation. Pain. 1999;80:127–141. doi: 10.1016/s0304-3959(98)00212-7. [DOI] [PubMed] [Google Scholar]
- West WL, Yeomans DC, Proudfit HK. The function of noradrenergic neurons in mediating antinociception induced by electrical stimulation of the locus coeruleus in two different sources of Sprague-Dawley rats. Brain Res. 1993;626:127–135. doi: 10.1016/0006-8993(93)90571-4. [DOI] [PubMed] [Google Scholar]
- Westlund KN, Coulter JD. Descending projections of the locus coeruleus and subcoeruleus/medial parabrachial nuclei in monkey: axonal transport studies and dopamine-beta-hydroxylase immunocytochemistry. Brain Res. 1980;2:235–264. doi: 10.1016/0165-0173(80)90009-0. [DOI] [PubMed] [Google Scholar]
- Westlund KN, Craig AD. Association of spinal lamina I projections with brainstem catecholamine neurons in the monkey. Experimental brain research Experimentelle Hirnforschung. 1996;110:151–162. doi: 10.1007/BF00228547. [DOI] [PubMed] [Google Scholar]
- Wiley RG, Kline IR. Neuronal lesioning with axonally transported toxins. J Neurosci Methods. 2000;103:73–82. doi: 10.1016/s0165-0270(00)00297-1. [DOI] [PubMed] [Google Scholar]
- Wrenn CC, Picklo MJ, Lappi DA, Robertson D, Wiley RG. Central noradrenergic lesioning using anti-DBH-saporin: anatomical findings. Brain Res. 1996;740:175–184. doi: 10.1016/s0006-8993(96)00855-4. [DOI] [PubMed] [Google Scholar]
- Zhang ET, Ossipov MH, Zhang DQ, Lai J, Porreca F. Nerve injury-induced tactile allodynia is present in the absence of FOS labeling in retrogradely labeled post-synaptic dorsal column neurons. Pain. 2007;129:143–154. doi: 10.1016/j.pain.2006.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann M. Ethical guidelines for investigations of experimental pain in conscious animals. Pain. 1983;16:109–110. doi: 10.1016/0304-3959(83)90201-4. [DOI] [PubMed] [Google Scholar]