

Abstract
Two double-cysteine mutants of a small protein judiciously modified so that the cysteines appear at axially opposite sides of the native fold were prepared such that different axes were defined in the two mutants. Upon reduction, the disulfide bonds are broken, and the proteins act as bifunctional ligands toward Ag nanoparticles, encouraging their assembly into nanoparticle dimers and small aggregates such that, when excited with laser light, the proteins are automatically located at electromagnetic hot spots within the aggregates. Because the protein molecules are small (~2.3 nm) and because the electromagnetic energy at a hot spot tends to increase as the size of the interparticle gap decreases, this nanoparticle–protein–nanoparticle geometry significantly enhances the Raman emission at the metallic surface. Exploiting this effect, we have recorded surface-enhanced Raman spectra (SERS) of the proteins at near-single-molecule level. The observed SERS spectra were dominated by the vibrations of molecular groups near the anchor points of the proteins.
Introduction
Raman spectroscopy is extensively and routinely used as a sensitive vibrational spectroscopic method for analysis and sensing. With the advent of new instrumentation and intense light sources, Raman spectroscopy is no less sensitive nor more difficult than infrared, with the added advantage that Raman is compatible with aqueous systems, an almost sine qua non for biological applications.1-3 An embodiment of Raman spectroscopy called surface-enhanced Raman spectroscopy (SERS) renders Raman potentially far more sensitive than infrared.4,5 With the enhancement offered by SERS, the cross sections normally measured for Raman (10−30–10−25 cm2 per molecule)1-3 can, under favorable circumstances, be increased by as much as 14 orders of magnitude (single-molecule SERS experiments),2,6,7 that is, to a point where the Raman cross sections rival those of intense fluorescence. The SERS effect occurs when the targeted molecules reside at appropriate sites at or near nanostructured systems composed of certain metals such as silver or gold, and on average, the enhancement factor ranges between 3 and 6 orders of magnitude. Theory8,9 predicts and experiment confirms that exceptionally large increases in Raman cross sections are associated with molecules located in the nanosized interstitial sites of aggregates of interacting silver nanoparticles (Ag-NPs).10-12 These favorable locations for exciting intense SERS are often referred to as “hot spots”. The greatly enhanced signal observed at hot spots arises largely as a result of the increase in the magnitude of both the incident and the scattered electromagnetic (EM) fields resulting from the excitation of coupled, localized surface plasmons.
Almost since its discovery, SERS has been employed as a means of characterizing the interaction between metal surfaces and biological molecules (such as proteins or DNA) or detecting the formation of specific antigen–antibody complexes at such a surface.14-17 To produce the level of enhancement required to report the presence of the Raman-nonresonant target species at very low concentrations (such as those employed in this study), a large Raman cross-section “label” (such as resonant dye) has been used. The Raman-resonant labels are covalently attached to the target species, and the resultant conjugate is used to indirectly probe the presence of the species of interest.15-17 That is, the surface-enhanced resonance Raman spectra (SERRS) of the label are detected rather than those of the biomolecules themselves. Label-free detection of biomolecules, in contrast, offers the advantage that it reports direct spectroscopic evidence associated with the target molecule itself rather than the label. Accordingly, a great deal of effort is currently being applied to label-free biosensing and detection. Whereas great strides have been made in the label-free sensing of DNA, the ability to selectively detect proteins at very low concentrations in the absence of labels remains fairly limited.18-20
In this article, we report the label-free SERS spectra of two mutants of a small protein at very low concentration. The two mutants belonging to the single-domain protein FynSH3 were well characterized21 and modified so that they can act as bifunctional linkers for Ag-NPs, but no Raman-resonant labels were covalently attached to the proteins. The NPs were then assembled by the protein “linkers” into small clusters with the proteins located in the hot spots. It is well-known that the electromagnetic field enhancement depends critically on the distance between the silver particles and (at appropriate wavelengths) falls off rapidly as the interparticle gap increases.8-12 Because the protein molecules are small (~2.3 nm), the nanoparticle–protein–nanoparticle geometry significantly enhances the Raman signal collected from the nanogap, enabling direct detection with minimal sample volume (i.e., in the absence of any Raman-resonant labels as previously reported in the literature15-17).
Experimental Section
Chemicals and Procedures
All starting materials involved in the preparation of the Ag colloid and proteins were purchased from commercial sources as analytical pure reagents.
Ag Colloid Preparation
A slightly modified Creighton sol was produced by borohydride reduction of AgNO3. Briefly, a AgNO3 solution (3.4 mg in 20 mL of water) cooled to approximately 10 °C was added dropwise, with constant stirring, to a NaBH4 solution (4.53 mg in 60 mL of water) precooled to 2 °C. Stirring was continued for another 50 min.
Protein Preparation
Double-cysteine mutants of the FynSH3 domain were generated and confirmed by sequencing or by mass spectroscopy. The proteins were expressed (pET vector in Escherichia coli) and purified by affinity chromatography (Qiagen) and reverse-phase HPLC (C4, HP 1100, Waters).21
The proteins were conjugated to Ag-NPs at the cysteine sites. The protein was dissolved in a 50 mM NaPO4 buffer solution (pH 7.2), and the disulfide bonds were reduced with a 20-fold excess of tris(2-carboxyethyl)phosphine HCl (Fluka). The reducing agent was removed using a Sephadex G-25 gel filtration column (BioRad), after which protein solutions of different concentrations were immediately pipetted into the aqueous suspension of Ag-NPs and allowed to incubate for several days to allow for efficient adsorption. However, several peaks could be detected within the first hour of incubation (e.g., the C–S stretching mode at 677 cm−1). If NaCl was added as an aggregation agent (0.25 mM final concentration), the aggregation followed almost immediately (Figure S1 in Supporting Information). The final concentrations of the protein in the Ag colloid were 5 × 10−5 and 5 × 10−9 M.
Instrumentation
Micro-Raman Spectroscopy and SERS
Micro-Raman and SERS spectra were recorded with a LabRam system (Horiba Jobin Yvon) equipped with 1200 groove/mm holographic gratings. The 514.5-nm output of a Spectra Physics argon-ion laser (model 164) was used to excite the spectra. The output at the laser head was 70 mW. A laser power sensor was used to determine the laser power at the sample and the optical losses introduced by different optical components (such as optical filters, two Notch filters for the rejection of the Rayleigh scattering, external mirrors, etc.). The laser power and the acquisition time were carefully adjusted to increase the signal intensity in all spectra and minimize the background arising from the decomposition of the sample under the laser beam or water. The background significantly decreased or disappeared when the colloidal aggregates were deposited on a glass cover slip at very low concentrations (5 × 10−9 M) and measured using acquisition times of 1 s (Figure 3 below). A small background was also noticed for the aggregated Ag sol in buffer, in the absence of protein (for the same acquisition parameters). The spectra were collected in the back-scattering geometry with a confocal Raman microscope (high-stability BX40) equipped with Olympus objectives (LMPlanFl 100×, 50×, MPlan 10×). A Peltier-cooled CCD camera served as the detector. The spectral resolution was 1 cm−1.
Figure 3.
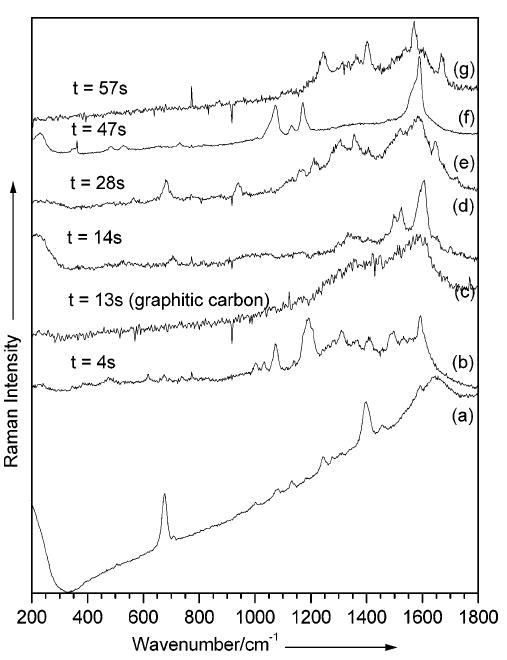
Time-resolved SERS spectra collected at 1-s intervals (b–g) from the metal–(K105C/S115C protein)–metal nanogap of an aggregate deposited on a glass cover slip (5 × 10−9 M). Six spectra were selected to highlight sudden spectral changes in intensity and position. Excitation line, 514.5 nm; laser power at the sample, 0.05 mW; acquisition time, 1 s. (a) Average SERS spectrum obtained in a buffer solution (from Figure 2b), included for comparison reasons.
Electron Microscopy
A drop of the final solution was placed on a marked 400-mesh lacey carbon-coated copper TEM grid (SPI Supplies) or a silicon wafer and allowed to dry. The samples were imaged by TEM (JEOL JEM-1230 transmission EM) and high-resolution SEM (JEOL 6340F). The beam intensity was kept at a minimum to reduce damage to the sample.
Results and Discussion
The 55-residue, predominantly β-sheet protein that was used in this study has been the subject of exhaustive kinetic and thermodynamic studies.22 In both of the employed mutants, the cysteine residues were judiciously located at opposite sites of the protein (K105C/S115C and A95C/V141C), so that the introduced thiolate groups could act as linkers to the metallic NPs (Figure 1a). The native-state distance between the two disulfide groups in the two mutants is less than 2.3 nm.
Figure 1.

(a) Two small FynSH3 proteins used as a linker between Ag-NPs (denoted K105C/S115C and A95C/V141C). The protein is 70 amino acids long, but only 56 are structured, as shown. The remaining unstructured portion of the protein is represented by a methyl group at the beginning and 13 amino acids His-tag on the end. The cysteine sites bearing sulfur atoms are indicated and connected by lines that denote pairs present in individual constructs. The native-state distance between the two disulfide groups and the sequence separation of each construct are also shown. (b) TEM image of a small NPs-dimer and trimer resulting from cross-linking with the K105C/S115C mutant. The Ag-NPs can be identified as black, round particles that adhere to the inner edge of the holes in the lacey carbon film of the grid. (c) SEM images of a small NPs dimer (marked by a white oval) resulting from cross-linking with K105C/S115C. The NPs dimer appears in this case as two very close white dots. At such low concentrations (i.e., 5 × 10−9 M), most of the examined spots on the silicon wafer contain monomers, and only a small percentage of the observed particles is represented by dimers or other small aggregates.
The mutant proteins were dissolved in a sodium phosphate buffer (pH 7.2) and reduced with a 20-fold excess of tris(2-carboxyethyl)phosphine HCl. Afterward, the reducing agent was removed using a gel filtration column, and protein solutions of different concentrations were immediately reacted with Ag-NPs. Ag-NPs with diameters of ~20 nm (Figure 1b and c) were prepared by the borohydride reduction of silver nitrate. The final concentrations of the protein in the Ag colloid were 5 × 10−5 and 5 × 10−9 M.
Breaking of the disulfide bonds during the reduction process makes the ensuing thiolate groups accessible to the metal surface. As the new Ag–S bonds are formed, the protein draws the metal particles together (Figure 1b and c), generating a SERS-active nanogap with the protein molecule ideally positioned within it.
That such metal–protein–metal aggregates (primarily dimers) can produce intense and detailed SERS spectra even at concentrations in the low nanomolar range is illustrated in Figure 2b and c. Similar SERS signatures were obtained at higher, micromolar concentrations (Figures S2 and S3 in the Supporting Information). No SERS spectra could be obtained under similar conditions at very low concentrations and without a specific reduction of the disulfides bonds.
Figure 2.
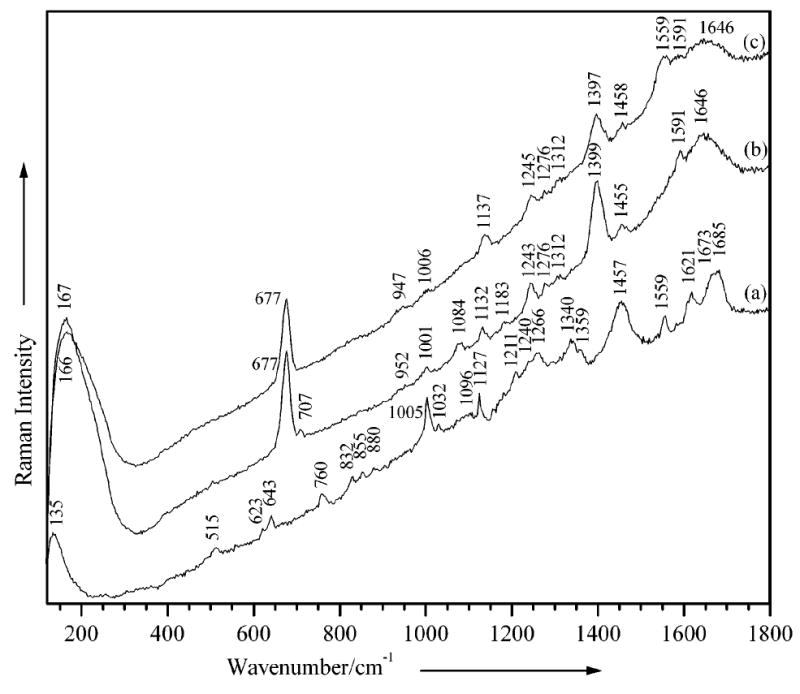
(a) Micro-Raman spectrum of the K105C/S115C-modified protein measured in the solid state and (b,c) SERS spectra of the buffer solution of the two mutant proteins in Ag colloid (5 × 10−9 M), (b) K105C/S115C and (c) A95C/V141C in Ag colloid (5 × 10−9 M). Excitation line, 514.5 nm; laser power at the sample, 1–10 mW; acquisition time, (a) 20 and (b,c) 200 s.
The SERS signal in Figure 2 was collected from a small focal volume (3.08 × 10−18 m3) within a cell, illuminated with 514.5-nm laser light. Straightforward calculations (see the Supporting Information) indicate that, on average, the focal volume contains approximately two NPs, with several protein molecules attached. At the lowest final concentration studied here (5 × 10−9 M), we estimate that the average number of protein molecules in the focal volume is approximately nine.
Ag–NP aggregates have high SERS activity toward the red portion of the visible spectrum, where the plasmon resonances of aggregates normally lie. However, these resonances are broad, and thus, the intensity of the SERS spectra that we observe at 514.5 nm are only approximately an order of magnitude lower than those of the ideal case for Ag–NP dimers of the diameters used in this study and for gap sizes of ~2 nm.11 In passing, we note that, despite the fact that few NP clusters exist in solution, such clusters have a propensity to be drawn into the tightly focused laser beam by the optical field gradient operating on the dipole induced in the linked NP clusters.23
Upon close examination, it is apparent that the SERS and ordinary Raman spectra of the K105C/S115C-modified protein (Figure 2) exhibit significant differences, suggesting that the SERS spectrum depends on the relative disposition of the protein with respect to the metal surface. It should be noted that ordinary solution-phase Raman spectra of these proteins could not be recorded at the concentrations used for our SERS studies. While new peaks emerged or changed their profile (e.g., position, relative intensity, full width at half-maximum) in the SERS spectrum, others were absent. In SERS, such significant changes are often observed for chemisorbed molecules, when the molecular and metal orbitals overlap resulting in new metal–complex species.3,4 Moreover, vibrational fingerprint modes involving portions of the molecule in the vicinity of the metal nanostructure should emerge as the most prominent bands in the spectrum.
The S–S stretching mode24 observed at 515 cm−1 in the ordinary Raman spectrum of the neat solid was absent in the SERS spectrum of the K105C/S115C mutant (Figure 2a and b), consistent with the rupture of this disulfide bond upon reduction and protein-induced aggregation of the NPs in solution. The C–S stretching mode associated with the cysteine groups,24,25 in contrast, emerges as a very strong peak at 677 cm−1. Moreover, the strong and broad band at approximately 167 cm−1 is due to the stretching vibration of newly formed Ag–S bonds.25 This means that the cysteine groups are in the immediate vicinity of the nanoparticle, with the C–S bonds most probably vertically oriented and strongly anchored to the metal surface through the S atoms. The conclusion that metal–molecule–metal bridging occurs after the disulfide bond cleavage is further supported by the increase in intensity or the appearance of vibrational modes characteristic of aromatic amino acid groups (Table S1 in the Supporting Information) located close to the cysteine thiolate and therefore in the immediate vicinity of the two NPs. These mostly involve histidine (H, at 1399 and 1591 cm−1), tryptophan (W, at 707, 1084, 1132, and 1243 cm−1), and phenylalanine (F, at 1183 and 1591 cm−1).24,26-28 The amino acid sequence in the two mutants is listed at the bottom of Figure 1a, where the cysteine sites bearing sulfur atoms in the two mutants are marked in red (i.e., K and S for K105C/S115C) and blue (i.e., A and V for A95C/V141C).
SERS spectra of the K105C/S115C mutant were also obtained at 1-s intervals from a colloidal solution (final protein concentration of 5 × 10−9 M) deposited on a glass cover slip. Some of the spots investigated along the sample showed temporal signal fluctuations (“blinking“). In a series of spectra recorded over a time period of 200 s, new peaks appeared or disappeared, changed their profile or intensity, or significantly shifted. It is generally agreed that such sudden spectral changes reflect the dynamics of one or very few molecules. A close examination of the time evolution of the spectra in Figure 3 shows that the SERS signal of the K105C/S115C protein alternates with that of graphitic carbon. This is represented by two distinct broad bands at about 1360 and 1570 cm−1. In most cases, the signal disappeared completely after a certain period of continuous exposure to the laser light, when water dried out completely. A possible scenario that can explain the blinking pattern observed here is decomposition of the bridging protein molecule into graphitic carbon and its replacement by another molecule that has migrated into the hot spot and resumed the role of a molecular bridge. Such a process would continue until all protein molecules were “consumed” and the signal disappeared completely.
The SERS spectrum of the A95C/V141C protein is similar to that of K105C/S115C. However, several Raman bands due to tryptophan (W18 and W14 at 707 and 1084 cm−1, respectively)24 and phenylalanine (F9a at 1183 cm−1)24,27 amino acids could not be detected for this system (Figure 2c). Such a result could be expected, given that, for this mutant the cysteine groups located close to the metallic surface are not surrounded by the mentioned residues (see the amino acid sequence in Figure 1a). Hence, it appears that the modes involving vibrational motions in the immediate vicinity of the metal–molecule interface are preferentially enhanced in the hot spot. This is a common observation in SERS. In this case, however, it illustrates how, by encouraging what is essentially the same protein to bind along different axes of its tertiary structure, significantly different SERS spectra are observed.
Conclusions
In conclusion, label-free SERS spectra were obtained with visible excitation lasers from two mutants of a small Raman-nonresonant protein illustrating several key features: (1) The double-cysteine proteins bearing disulfide groups at opposite sites “self-assemble” with Ag-NPs in such a way that the target molecules help create the electromagnetic hot spot in which they reside. (2) The metal–molecule–metal geometry enables label-free and direct detection of different presentations of the same protein with minimal sample volume (i.e., in the absence of any Raman-resonant labels covalently attached to the biomolecule of interest). Although visible excitation was used for this study, many of the Raman bands of the main and side chains of the small proteins, including disulfide, aromatic amino acids, histidine, and carboxyl groups, were observed with sufficient intensity to allow the identity of the molecule to be surmised for biosensing applications.
Acknowledgments
Funding from the Institute for Collaborative Biotechnologies through Grant DAAD19-03-D-0004 from the U.S. Army Research Office and from Lawrence Livermore National Laboratories through a UCDRD grant is gratefully acknowledged.
Footnotes
Supporting Information Available: Estimate of the number of Ag colloidal particles and analyte molecules in the focal volume, SERS spectra, and observed wavenumbers of the experimental Raman and SERS spectra. This material is available free of charge via the Internet at http://pubs.acs.org.
References and Notes
- 1.Schrader B, editor. Infrared and Raman Spectroscopy: Methods and Applications. John Wiley & Sons; Chichester, U.K.: 1995. [Google Scholar]
- 2.Kneipp K, Kneipp H, Itzkan I, Dasari RR, Feld MS. Chem Rev. 1999;99:2957–2975. doi: 10.1021/cr980133r. [DOI] [PubMed] [Google Scholar]
- 3.Chalmers JM, Griffiths PR. Handbook of Vibrational Spectroscopy. John Wiley & Sons; Chichester, U.K.: 2002. [Google Scholar]
- 4.Chang RK, Furtak TE, editors. Surface Enhanced Raman Scattering. Plenum Press; New York: 1982. [Google Scholar]
- 5.Moskovits M. J Raman Spectrosc. 2005;36:485–496. [Google Scholar]
- 6.Kneipp K, Wang Y, Kneipp H, Perelman LT, Itzkan I, Dasari RR, Feld MS. Phys Rev Lett. 1997;78:1667–1670. [Google Scholar]
- 7.Nie SM, Emery SR. Science. 1997;275:1102–1106. doi: 10.1126/science.275.5303.1102. [DOI] [PubMed] [Google Scholar]
- 8.Aravind PK, Nitzan A, Metiu H. Surf Sci. 1981;110:189–204. [Google Scholar]
- 9.Liver N, Nitzan A, Gersten JI. Chem Phys Lett. 1984;111:449–454. [Google Scholar]
- 10.Tsai DP, Kovacs J, Wang Z, Moskovits M, Shalaev VM, Suh JS, Botet R. Phys Rev Lett. 1994;72:4149–4152. doi: 10.1103/PhysRevLett.72.4149. [DOI] [PubMed] [Google Scholar]
- 11.Xu H, Aizpurua J, Käll M, Apell P. Phys Rev E. 2000;62:4318–4324. doi: 10.1103/physreve.62.4318. [DOI] [PubMed] [Google Scholar]
- 12.Imura K, Okamoto H, Hossain MK, Kitajima M. Nano Lett. 2006;6:2173–2176. doi: 10.1021/nl061650p. [DOI] [PubMed] [Google Scholar]
- 13.Ahern A, Garrell RL. Langmuir. 1991;7:254–261. [Google Scholar]
- 14.Grabbe ES, Buck RP. J Electroanal Chem. 1991;308:227–237. [Google Scholar]
- 15.Cao YWC, Jin R, Mirkin CA. Science. 2002;297:1536–1540. doi: 10.1126/science.297.5586.1536. [DOI] [PubMed] [Google Scholar]
- 16.Rosi NL, Mirkin CA. Chem Rev. 2005:1547–1562. doi: 10.1021/cr030067f. [DOI] [PubMed] [Google Scholar]
- 17.Xu S, Ji X, Xu W, Zhao B, Dou X, Bai Y, Ozaki Y. J Biomed Opt. 2005;10 doi: 10.1117/1.1915487. 031112(1–12) [DOI] [PubMed] [Google Scholar]
- 18.Wang WU, Chen C, Lin K-H, Fang Y, Lieber M. Proc Natl Acad Sci U S A. 2005;102:3208–3212. doi: 10.1073/pnas.0406368102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kyo M, Usui-Aoki K, Koga H. Anal Chem. 2005;77:7115–7121. doi: 10.1021/ac050884a. [DOI] [PubMed] [Google Scholar]
- 20.Drachev VP, Nashine VC, Thoreson MD, Ben-Amotz D, Jo Davisson V, Shalaev VM. Langmuir. 2005;21:8368–8373. doi: 10.1021/la0502490. [DOI] [PubMed] [Google Scholar]
- 21.McCarney E, Werner JH, Bernstein SL, Ruczinski I, Makarov DE, Goodwin PM, Plaxco KW. J Mol Biol. 2005;352:672–682. doi: 10.1016/j.jmb.2005.07.015. [DOI] [PubMed] [Google Scholar]
- 22.Plaxco KW, Guijarro JI, Morton CJ, Pitkeathly M, Campbell ID, Dobson CM. Biochemistry. 1998;37:2529–2537. doi: 10.1021/bi972075u. [DOI] [PubMed] [Google Scholar]
- 23.Calander N, Willander M. Phys Rev Lett. 2002;89 doi: 10.1103/PhysRevLett.89.143603. 143603(1–4) [DOI] [PubMed] [Google Scholar]
- 24.Harada I, Takeuchi H. In: Spectroscopy of Biological Systems. Chapter 3 Clark RJH, Hester RE, editors. Vol. 13. John Wiley & Sons; New York: 1986. [Google Scholar]
- 25.Compagnini G, Galati C, Pignataro S. Phys Chem Chem Phys. 1999;1:2351–2353. [Google Scholar]
- 26.Kitagawa T. Prog Biophys Mol Biol. 1992;58:1–18. doi: 10.1016/0079-6107(92)90009-u. [DOI] [PubMed] [Google Scholar]
- 27.Okishio N, Tanaka T, Nagai M, Fukuda R, Nagatomo S, Kitagawa T. Biochemistry. 2001;40:15797–15804. doi: 10.1021/bi011339g. [DOI] [PubMed] [Google Scholar]
- 28.Okishio N, Tanaka T, Fuduka R, Nagai M. Biochemistry. 2003;42:208–216. doi: 10.1021/bi020475y. [DOI] [PubMed] [Google Scholar]