

Abstract
Although melanoma ultimately progresses to a highly aggressive and metastatic disease that is typically resistant to currently available therapy, it often begins as a benign nevus consisting of a clonal population of hyperplastic melanocytes that cannot progress because they are locked in a state of cellular senescence. Once senescence is overcome, the nevus can exhibit dysplastic features and readily progress to more lethal stages. Recent advances have convincingly demonstrated that senescence represents a true barrier to the progression of many types of cancer, including melanoma. Thus, understanding the mechanism(s) by which melanoma evades senescence has become a priority in the melanoma research community. Senescence in most cells is regulated through some combination of activities within the RB and p53 pathways. However, differences discovered among various tumor types, some subtle and others quite profound, have revealed that senescence frequently operates in a context-dependent manner. Here we review what is known about melanocyte senescence, and how such knowledge may provide a much-needed edge in our struggles to contain or perhaps vanquish this often-fatal malignancy.
The fundamental process of cellular senescence was originally characterized by Hayflick and Moorhead.1 Cellular senescence was first recognized as the phenomenon whereby normal diploid somatic cells lose the ability to divide after a finite number of divisions. This irreversible cell-cycle arrest following extensive proliferation is now known as "replicative senescence" or the "Hayflick phenomenon". Cells can also undergo a more rapid senescence in response to various physiological stresses, a process called “stress-induced senescence”.2–4 This includes oncogene-induced senescence seen following oncogene activation in normal cells.5–7 Other triggers of senescence include telomere shortening (in replicative senescence), DNA damage, and cellular and oxidative stresses. Senescence represents a cellular program executed in response to cell damage.2;8 Senescence is now understood to be a crucial barrier to tumorigenesis.5;6 It is accompanied by a set of morphological and functional changes: the cell becomes bigger and flatter, and undergoes changes in chromatin structure and gene expression,4;9 and displays markers such as senescence-associated-β-galactosidase (SA-β-gal) and heterochromatin protein 1-γ (Fig. 1).
Fig. 1.

Growing and senescent melanocytes. Growing Arf-deficient (A) and senescent wild-type (B) melanocytes by bright field microscopy after staining for SA-β–gal. Blue indicates senescent cells. C, D. Heterochromatin protein 1-γ-positive senescence-associated heterochromatin foci. Tp53-deficient melanocytes were infected with pBabe retrovirus containing Arf cDNA (D) or an empty vector control (C). Cells were fixed 48 h after infection and incubated with anti-HP1γ antibody, followed by a secondary antibody conjugated with FITC, and then viewed with a fluorescent microscope. Shown are green nuclei with more intensely stained green foci, marking senescence. All images are at the same magnification. E. p16-deficient melanocytes at the same passage level as in A and B by bright field microscopy (no SA-β–gal staining). Portions of this figure are reproduced from Ref. 43, with permission.
p53 and the retinoblastoma protein (RB) are considered classic central players in the induction of cellular senescence, and are both activated upon entry into senescence2;10–12 (Fig. 2). During senescence the p53 protein is stabilized by its negative regulator protein, mouse double minute 2 (Mdm2), which acts as an E3 ubiquitin ligase to target p53 for proteasomal degradation. p53 proceeds to activate its transcriptional targets, such as p21CIP1/WAF1.13 Activated (hypophosphorylated) RB binds to E2F-family transcription factors to repress their transcriptional targets and thus inhibit cell-cycle progression.14 However, it is now appreciated that senescence is much more complex, and cell context dependent.
Fig. 2.
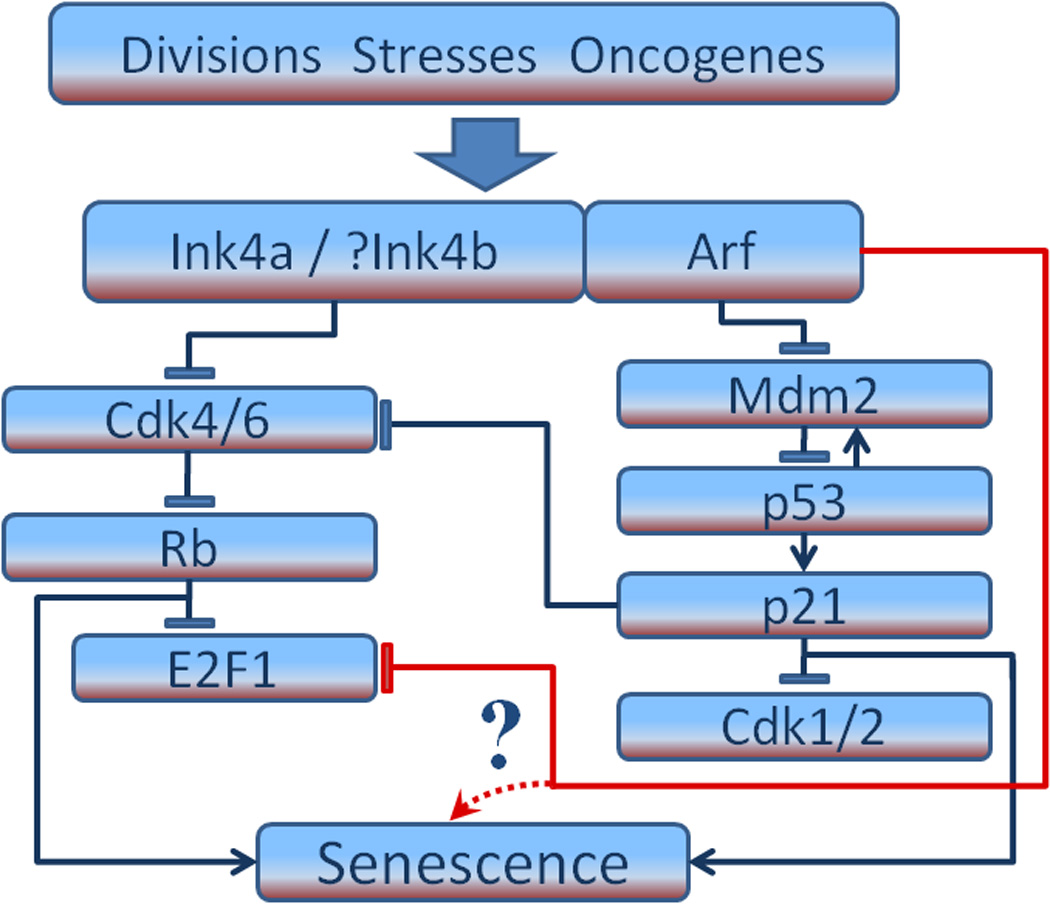
p53-independent action of Arf on melanocyte senescence. Shown are Ink4a/Rb and Arf/p53 pathways implicated in senescence, mostly derived from work with fibroblasts. In mouse melanocytes Arf can promote the p53-independent degradation of E2F1. This, and perhaps other mechanisms (indicated by question mark), could trigger senescence.
The currently accepted model for regulation of senescence in mouse embryo fibroblasts (MEFs) is a linear one where p53 plays a main role: its transcriptional activation of p21, a broad-spectrum cyclin/cyclin-dependent kinases (Cdks) inhibitor, leads to cell-cycle arrest. Inactivation of p53 in MEFs is sufficient to prevent senescence.15 Rb-null MEFs senesce normally but inactivation of an additional member of the Rb family, p107 and p130, prevents senescence.16 Activation of both the p53 and the Rb pathways is needed to maintain senescence in MEFs.15;16 However, p21-null MEFs senesce normally17, implying additional mechanisms. In human fibroblasts the activation of both p53 and RB is required to initiate and maintain senescence.2;12 Senescence of human fibroblasts in culture occurs in two phases.9 The first phase (normal replicative senescence) can be overridden by addition of viral oncogenes such as simian virus 40 large T antigen or human papilloma virus 16 oncogenes E6 and E7, whose protein products abrogate both the p53 and RB pathways.12 It can also be prevented by expression of human telomerase reverse transcriptase (hTERT) which elongates telomeres.10;12;18 After more divisions fibroblast cultures stop proliferating again, in a phase called crisis. Crisis is caused by extreme telomere shortening, involves telomere fusions and chromosomal breakage, and can be overcome by the expression of hTERT, leading to immortalization.9;10;18;19
A central locus in the control of cell senescence that engages the p53 and the RB pathways is the CDKN2 (Ink4-Arf in mouse) locus on chromosome 9p21. This locus is frequently implicated in various types of cancer20;21 and is the best-known familial melanoma locus, commonly altered in both heritable and sporadic melanoma.22;23 The Ink4-Arf locus encodes three tumour suppressors, Ink4a (CDKN2A) and Ink4b (CDKN2B), and Arf (also called p19Arf in mouse and p14ARF in human).24;25 Ink4b appears to be derived from a tandemly duplicated Ink4a gene. Whereas Ink4b has its own open reading frame, Ink4a and Arf have different first exons that are spliced to a common second and third exon. Although these two genes share exons 2 and 3, Arf is read from an alternative reading frame relative to Ink4a. Ink4a and Ink4b are inhibitors of cyclin D/Cdk 4 and 6 and are thus activators of Rb. They block the phosphorylation of Rb and its family members, p130 and p107.26 Arf activates p53 by binding to and inactivating Mdm2.24;25 This has been the prevailing view on how Arf functions as a cell-cycle inhibitor (Fig. 2), although Arf pathways now appear to be more intricate (see below).
While the tumour suppressor functions for INK4A and ARF have been under intensive examination, the role of INK4B has been studied much less. In 2007 however, Krimpenfort et al. showed that mice deficient for all three open reading frames (Ink4b, Ink4a and Arf) are more tumour-prone and develop a wider spectrum of tumours than Ink4a/Arf-null mice, with a preponderance of skin tumours and soft tissue sarcomas.27 Ink4a/Ink4b/Arf-null MEFs were substantially more sensitive to oncogenic transformation than Ink4a/Arf-null MEFs, and under conditions of stress, levels of Ink4b protein were significantly elevated in Ink4a-null MEFs. The authors proposed that Ink4b acts to fulfil a critical backup function for Ink4a.27 Moreover, INK4B has been proposed to be a marker of oncogene-induced senescence.28 To date, the majority of reported mutations in the CDKN2 locus associated with familial melanoma affect INK4A function, some affect ARF and a few affect INK4B.22;23;29 In contrast with other tumour types, p53 is mutated relatively infrequently in melanomas (9%).22
The relative importance of the p53 and RB pathways in the control of senescence varies between different cell types. The Arf/p53 pathway seems to be more important than the Ink4a/Ink4b/Rb pathway in senescence of mouse fibroblasts. Ink4a-null and Ink4b-null fibroblasts with functional Arf senesce normally,30–32 while Arf-null fibroblasts with normal Ink4a33 and Ink4a-Arf-null fibroblasts do not senesce.33;34 At the same time, loss of either INK4A/ARF product can cooperate with H-RAS activation to produce clinically indistinguishable melanomas.35 In 2002, Sviderskaya et al. showed that Ink4a-Arf-null mouse melanocytes with intact p53 function do not senesce, in other words are immortal, whereas wild-type melanocytes senesce within 4–5 weeks of culture.36 Ink4a-Arf hemizygous melanocytes show defective senescence unlike hemizygous mouse fibroblasts which senesce normally.34
A study of human melanocytes from melanoma patients with two inactive INK4A alleles but functional ARF showed that these have high rates of apoptosis, which are reduced when the cells are grown in the presence of keratinocytes or keratinocyte-derived factors, human stem cell factor and endothelin 1.37 With these growth factors the INK4A-deficient melanocytes exhibit impaired senescence (unlike INK4A-deficient human fibroblasts, which senesce normally).38 Levels of p53 and p21 are elevated in senescent INK4A-null melanocytes but not in senescent normal melanocytes. hTERT is sufficient to immortalise these cells but not normal human melanocytes. Gray-Schopfer et al. (2006) used gene transfer to confirm that in cultured human melanocytes disruption of the INK4A/RB pathway and telomerase activation are required for escape from senescence leading to immortalization.39 Together, these data show that INK4A plays an important role in normal human melanocyte senescence, and raise the possibility that in INK4A-deficient melanocytes p53 may contribute to a delayed form of senescence that requires telomere shortening.37 However, it is worth noting that in relation to melanoma progression, senescence in benign melanocytic nevi involves Ink4a but not p53 or p21 upregulation. At the same time, expression of both Ink4a and p21 is lost in advanced melanomas.39 Thus, a general impression has emerged that the INK4A/RB-dependent form of senescence is especially important in human melanocytes. In comparison, fibroblasts (and some other cell types) undergo senescence involving the ARF/p53 as well as the INK4A/RB pathways. This difference may help to explain the special importance of INK4A mutations in human melanoma.
Based on the senescence studies, a genetic model of primary melanoma progression has been proposed.10;39 Benign nevi (moles) are clones of melanocytes which proliferate for some time following an oncogenic mutation, and then stop proliferating in Ink4a-dependent senescence. They are considered to be potential precursors of melanoma.40;41 The next stage of melanoma progression is dysplastic nevi which represent escape from INK4A/RB senescence, or crisis with chromosomal abnormalities.10 Activation of telomerase in dysplastic nevi could lead to radial growth phase (RGP) melanoma, proposed to consist of immortal but keratinocyte-dependent cells. The final stage of melanoma progression is called vertical growth phase (VGP). It is suggested to be keratinocyte-independent, invasive and to require mutation(s) in a pathway(s) suppressing apoptosis.10;42
In support of a portion of this genetic model, Michaloglou et al. recently showed that oncogenic BRAF signalling could induce senescence, accompanied by expression of INK4A. 43 However, a further comparison between the pattern of expression of INK4A and the senescence marker SA-β-gal revealed a marked mosaic induction of INK4A in human nevi, suggesting that factors other than INK4A also contribute to the prevention of oncogenic BRAF-driven proliferation, and therefore the induction of senescence. What then are the other factors that could be involved in oncogene-induced senescence in nevi? Peeper and colleagues have very recently demonstrated that oncogene-induced senescence is linked to inflammation, specifically with secretion of IL-6, as well as to INK4B upregulation (Cell, in press).44 A study on senescence of mouse melanocytes showed that Arf-null melanocytes (with retention of Ink4a and Tp53) have a very high rate of cell proliferation and a complete lack of senescence, raising the possibility that ARF is a key melanocyte senescence factor.45 Interestingly, mouse Ink4a-null melanocyte growth is impaired in the absence of keratinocytes or keratinocyte-derived factors, similarly to that of human INK4A-null melanocytes,37 while mouse Arf-null melanocytes grow robustly in the absence of these factors, and completely fail to senesce.45
Until recently, the common view was that ARF, MDM2 and p53 act in a linear pathway (Fig. 2). However, it has been demonstrated that (i) ARF interacts with many proteins other then MDM2, including MYC, E2F1, NFκB and CtBP (reviewed by Sherr),46 (ii) Arf-null and Tp53-null mice are not identical in the types of tumors they develop,47;47;48;48 and (iii) ARF has p53-independent functions including ones in development, ribosome biosynthesis, DNA-damage response, autophagy and apoptosis.31 These recent findings suggest that ARF might function in a p53-independent manner. This notion was supported in a recent study by Ha et al., which dissected the role of p53 and its positive regulator ARF in the process of melanocyte senescence.45 A genetically engineered mouse model was used to demonstrate that deficiency in Arf, but not p53, facilitated rapid development of UV-inducible melanoma. This phenotypic difference was accounted for, at least in part, by the surprising fact that unlike fibroblasts, melanocyte senescence is strongly regulated by Arf, but less so by p53. Notably, Tyner et al. found that transgenic mice that broadly express a hyperactive form of p53 exhibit accelerated cellular aging in a variety of tissues but normal pigmentation in hair and skin, supporting the notion that p53 has a subdued role in melanocyte senescence.49 Ha et al. also showed that activated N-RAS, a relatively common mutation in melanoma patients, collaborated with deficiency in Arf in a p53-independent manner to fully transform melanocytes.45 It is worth noting that an effect of p53 loss on melanocyte transformation was observed in these studies, but only in melanocytes already deficient in Arf. Thus, it was concluded that Arf can act as a melanoma tumor suppressor by inducing p53-independent senescence, and may in fact facilitate oncogene-induced senescence; a comparison between the pattern of ARF expression and senescence markers in nevi will be needed to further substantiate a role for ARF in human melanocyte senescence. It is also worth noting that senescence is a highly complex process, and a role for p53 in senescence in melanocytic cells cannot be excluded. In fact, recent evidence from Maria Soengas shows that in human melanoma cell lines, blocking MDM2 or the p53-MDM2 interaction, genetically or pharmacologically, can induce a senescent response (personal communication).
These many studies suggest that ARF, INK4a and p53 have critical, non-overlapping functions in melanocyte growth control and melanoma suppression. The relevance of the loss of ARF function as a distinct event in melanoma is supported by several reports of ARF-specific, INK4a-independent mutations in human tumors, as well as examples of ARF-specific promoter methylation (reviewed by Chin et al.).23 Polsky and colleagues have recently determined that inactivation of ARF in melanoma, either through genetic or epigenetic mechanisms, is in fact a relatively common event (JNCI, in press).50 In contrast, although TP53 is the most frequently mutated gene in human cancer, it is a relatively infrequent target in melanoma.22;51;52 And when mutant p53 factors are found, they often retain transactivation function.53 Taken together, recent human and mouse data support a significant, p53-independent role for ARF in melanomagenesis. How then, does ARF regulate melanocyte senescence and suppress melanomagenesis in a p53-independent fashion?
Ha et al.45 showed that E2F1 is regulated in melanocytes by functional ARF through proteasomal degradation, as reported for other mouse and human cell types.54–56 ARF has also been reported to inhibit the formation of active E2F1-DP1 transcriptional complexes.57 Members of the E2F family are best known for their role in promoting cell cycle transition from G1 to S phase, but they also regulate apoptosis, DNA repair and differentiation (reviewed by Johnson and Degregori).58 Clearly, the E2F family is quite complex and their functions context dependent; in fact, members can function as both oncogenes and tumor suppressors. Interestingly, ARF itself has been reported to be both positively and negatively regulated by different members of the E2F family.59–61 A recent study by Chang et al. examined the p53-independent regulation of E2F1 by ARF from a different perspective.62 ARF was shown to regulate transit through the cell cycle by directly disrupting MDM2-Rb interaction, blocking proteasome-dependent Rb degradation, thereby preventing activation of E2F1. Accordingly, ablation of Rb was found to impair the ability of ARF to suppress proliferation, demonstrating crosstalk between these two pathways. Although these data indicate a role for E2F family members in ARF-mediated, p53-independent regulation of melanocyte senescence (Fig. 2), the complexity of this process predicts that additional intriguing factors will emerge over time.
In closing it is important to emphasize that senescence appears to be regulated in melanocytes in a manner that is, at least partially, distinct from other studied cell types. While differences in mouse and human cell senescence are well documented,63 even when considering cells derived from the same species p53 appears to have a prominent role in the senescence of fibroblasts and a more limited role in melanocytes. Although the reason for this difference is still unclear, it helps explain key observations concerning the relative infrequency of p53 mutations in human melanoma, and should be kept in mind when designing future anti-melanoma therapy. p53 is currently being considered as a therapeutic target for a variety of cancer patients.64 Matheu et al. have shown that genetically engineered mice with an extra gene dose of both Arf and Tp53 exhibit increased, cooperative protection against several tumor types.65 Based on discussions in this review, therapy designed to restore p53 function alone in melanoma cannot be expected to assuage all the oncogenic consequences of deficiency in ARF, which may provide an effective therapeutic alternative, or one that could be used successfully in combination with other agents against melanoma.
AKNOWLEDGEMENTS
We thank Professor Dorothy C. Bennett for critical reading of the manuscript and useful discussions. The work in the authors’ laboratories was supported in part by the intramural research program of the NIH, National Cancer Institute, Center for Cancer Research and Wellcome Trust Grant 078327.
Reference List
- 1.Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res. 1961;25:585–621. doi: 10.1016/0014-4827(61)90192-6. [DOI] [PubMed] [Google Scholar]
- 2.Ben Porath I, Weinberg RA. The signals and pathways activating cellular senescence. Int J Biochem Cell Biol. 2005;37:961–976. doi: 10.1016/j.biocel.2004.10.013. [DOI] [PubMed] [Google Scholar]
- 3.Collado M, Blasco MA, Serrano M. Cellular senescence in cancer and aging. Cell. 2007;130:223–233. doi: 10.1016/j.cell.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 4.Lloyd AC. Limits to lifespan. Nat Cell Biol. 2002;4:E25–E27. doi: 10.1038/ncb0202-e25. [DOI] [PubMed] [Google Scholar]
- 5.Bartek J, Bartkova J, Lukas J. DNA damage signalling guards against activated oncogenes and tumour progression. Oncogene. 2007;26:7773–7779. doi: 10.1038/sj.onc.1210881. [DOI] [PubMed] [Google Scholar]
- 6.Mooi WJ, Peeper DS. Oncogene-induced cell senescence--halting on the road to cancer. N Engl J Med. 2006;355:1037–1046. doi: 10.1056/NEJMra062285. [DOI] [PubMed] [Google Scholar]
- 7.Yaswen P, Campisi J. Oncogene-induced senescence pathways weave an intricate tapestry. Cell. 2007;128:233–234. doi: 10.1016/j.cell.2007.01.005. [DOI] [PubMed] [Google Scholar]
- 8.Sharpless NE, Depinho RA. How stem cells age and why this makes us grow old. Nat Rev Mol Cell Biol. 2007;8:703–713. doi: 10.1038/nrm2241. [DOI] [PubMed] [Google Scholar]
- 9.Shay JW, Wright WE. Senescence and immortalization: role of telomeres and telomerase. Carcinogenesis. 2005;26:867–874. doi: 10.1093/carcin/bgh296. [DOI] [PubMed] [Google Scholar]
- 10.Bennett DC. Human melanocyte senescence and melanoma susceptibility genes. Oncogene. 2003;22:3063–3069. doi: 10.1038/sj.onc.1206446. [DOI] [PubMed] [Google Scholar]
- 11.Sherr CJ. The INK4a/ARF network in tumour suppression. Nat Rev Mol Cell Biol. 2001;2:731–737. doi: 10.1038/35096061. [DOI] [PubMed] [Google Scholar]
- 12.Wynford-Thomas D. Cellular senescence and cancer. J Pathol. 1999;187:100–111. doi: 10.1002/(SICI)1096-9896(199901)187:1<100::AID-PATH236>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 13.Kulju KS, Lehman JM. Increased p53 protein associated with aging in human diploid fibroblasts. Exp Cell Res. 1995;217:336–345. doi: 10.1006/excr.1995.1095. [DOI] [PubMed] [Google Scholar]
- 14.Narita M, Lowe SW. Executing cell senescence. Cell Cycle. 2004;3:244–246. [PubMed] [Google Scholar]
- 15.Dirac AM, Bernards R. Reversal of senescence in mouse fibroblasts through lentiviral suppression of p53. J Biol Chem. 2003;278:11731–11734. doi: 10.1074/jbc.C300023200. [DOI] [PubMed] [Google Scholar]
- 16.Sage J, Mulligan GJ, Attardi LD, Miller A, Chen S, Williams B, Theodorou E, Jacks T. Targeted disruption of the three Rb-related genes leads to loss of G(1) control and immortalization. Genes Dev. 2000;14:3037–3050. doi: 10.1101/gad.843200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pantoja C, Serrano M. Murine fibroblasts lacking p21 undergo senescence and are resistant to transformation by oncogenic Ras. Oncogene. 1999;18:4974–4982. doi: 10.1038/sj.onc.1202880. [DOI] [PubMed] [Google Scholar]
- 18.Bodnar AG, Ouellette M, Frolkis M, Holt SE, Chiu CP, Morin GB, Harley CB, Shay JW, Lichtsteiner S, Wright WE. Extension of life-span by introduction of telomerase into normal human cells. Science. 1998;279:349–352. doi: 10.1126/science.279.5349.349. [DOI] [PubMed] [Google Scholar]
- 19.Artandi SE, Depinho RA. Mice without telomerase: what can they teach us about human cancer? Nat Med. 2000;6:852–855. doi: 10.1038/78595. [DOI] [PubMed] [Google Scholar]
- 20.Gil J, Peters G. Regulation of the INK4b-ARF-INK4a tumour suppressor locus: all for one or one for all. Nat Rev Mol Cell Biol. 2006;7:667–677. doi: 10.1038/nrm1987. [DOI] [PubMed] [Google Scholar]
- 21.Sharpless NE. INK4a/ARF: a multifunctional tumor suppressor locus. Mutat Res. 2005;576:22–38. doi: 10.1016/j.mrfmmm.2004.08.021. [DOI] [PubMed] [Google Scholar]
- 22.Bennett DC. How to make a melanoma: what do we know of the primary clonal events? Pigment Cell Melanoma Res. 2008;21:27–38. doi: 10.1111/j.1755-148X.2007.00433.x. [DOI] [PubMed] [Google Scholar]
- 23.Chin L, Garraway LA, Fisher DE. Malignant melanoma: genetics and therapeutics in the genomic era. Genes Dev. 2006;20:2149–2182. doi: 10.1101/gad.1437206. [DOI] [PubMed] [Google Scholar]
- 24.Kim WY, Sharpless NE. The regulation of INK4/ARF in cancer and aging. Cell. 2006;127:265–275. doi: 10.1016/j.cell.2006.10.003. [DOI] [PubMed] [Google Scholar]
- 25.Sherr CJ. Principles of tumor suppression. Cell. 2004;116:235–246. doi: 10.1016/s0092-8674(03)01075-4. [DOI] [PubMed] [Google Scholar]
- 26.Helmbold H, Deppert W, Bohn W. Regulation of cellular senescence by Rb2/p130. Oncogene. 2006;25:5257–5262. doi: 10.1038/sj.onc.1209613. [DOI] [PubMed] [Google Scholar]
- 27.Krimpenfort P, Ijpenberg A, Song JY, van d V, Nawijn M, Zevenhoven J, Berns A. p15Ink4b is a critical tumour suppressor in the absence of p16Ink4a. Nature. 2007;448:943–946. doi: 10.1038/nature06084. [DOI] [PubMed] [Google Scholar]
- 28.Collado M, Gil J, Efeyan A, Guerra C, Schuhmacher AJ, Barradas M, Benguria A, Zaballos A, Flores JM, Barbacid M, Beach D, Serrano M. Tumour biology: senescence in premalignant tumours. Nature. 2005;436:642. doi: 10.1038/436642a. [DOI] [PubMed] [Google Scholar]
- 29.Halaban R. Rb/E2F: a two-edged sword in the melanocytic system. Cancer Metastasis Rev. 2005;24:339–356. doi: 10.1007/s10555-005-1582-z. [DOI] [PubMed] [Google Scholar]
- 30.Krimpenfort P, Quon KC, Mooi WJ, Loonstra A, Berns A. Loss of p16Ink4a confers susceptibility to metastatic melanoma in mice. Nature. 2001;413:83–86. doi: 10.1038/35092584. [DOI] [PubMed] [Google Scholar]
- 31.Latres E, Malumbres M, Sotillo R, Martin J, Ortega S, Martin-Caballero J, Flores JM, Cordon-Cardo C, Barbacid M. Limited overlapping roles of P15(INK4b) and P18(INK4c) cell cycle inhibitors in proliferation and tumorigenesis. EMBO J. 2000;19:3496–3506. doi: 10.1093/emboj/19.13.3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sharpless NE, Bardeesy N, Lee KH, Carrasco D, Castrillon DH, Aguirre AJ, Wu EA, Horner JW, Depinho RA. Loss of p16Ink4a with retention of p19Arf predisposes mice to tumorigenesis. Nature. 2001;413:86–91. doi: 10.1038/35092592. [DOI] [PubMed] [Google Scholar]
- 33.Kamijo T, Zindy F, Roussel MF, Quelle DE, Downing JR, Ashmun RA, Grosveld G, Sherr CJ. Tumor suppression at the mouse INK4a locus mediated by the alternative reading frame product p19ARF. Cell. 1997;91:649–659. doi: 10.1016/s0092-8674(00)80452-3. [DOI] [PubMed] [Google Scholar]
- 34.Serrano M, Lee H, Chin L, Cordon-Cardo C, Beach D, Depinho RA. Role of the INK4a locus in tumor suppression and cell mortality. Cell. 1996;85:27–37. doi: 10.1016/s0092-8674(00)81079-x. [DOI] [PubMed] [Google Scholar]
- 35.Sharpless NE, Kannan K, Xu J, Bosenberg MW, Chin L. Both products of the mouse Ink4a/Arf locus suppress melanoma formation in vivo. Oncogene. 2003;22:5055–5059. doi: 10.1038/sj.onc.1206809. [DOI] [PubMed] [Google Scholar]
- 36.Sviderskaya EV, Hill SP, Evans-Whipp TJ, Chin L, Orlow SJ, Easty DJ, Cheong SC, Beach D, Depinho RA, Bennett DC. p16(Ink4a) in melanocyte senescence and differentiation. J Natl Cancer Inst. 2002;94:446–454. doi: 10.1093/jnci/94.6.446. [DOI] [PubMed] [Google Scholar]
- 37.Sviderskaya EV, Gray-Schopfer VC, Hill SP, Smit NP, Evans-Whipp TJ, Bond J, Hill L, Bataille V, Peters G, Kipling D, Wynford-Thomas D, Bennett DC. p16/cyclin-dependent kinase inhibitor 2A deficiency in human melanocyte senescence, apoptosis, and immortalization: possible implications for melanoma progression. J Natl Cancer Inst. 2003;95:723–732. doi: 10.1093/jnci/95.10.723. [DOI] [PubMed] [Google Scholar]
- 38.Brookes S, Rowe J, Gutierrez DA, Bond J, Peters G. Contribution of p16(INK4a) to replicative senescence of human fibroblasts. Exp Cell Res. 2004;298:549–559. doi: 10.1016/j.yexcr.2004.04.035. [DOI] [PubMed] [Google Scholar]
- 39.Gray-Schopfer VC, Cheong SC, Chong H, Chow J, Moss T, Abdel-Malek ZA, Marais R, Wynford-Thomas D, Bennett DC. Cellular senescence in naevi and immortalisation in melanoma: a role for p16? Br J Cancer. 2006;95:496–505. doi: 10.1038/sj.bjc.6603283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bennett DC, Medrano EE. Molecular regulation of melanocyte senescence. Pigment Cell Res. 2002;15:242–250. doi: 10.1034/j.1600-0749.2002.02036.x. [DOI] [PubMed] [Google Scholar]
- 41.Hayward N. New developments in melanoma genetics. Curr Oncol Rep. 2000;2:300–306. doi: 10.1007/s11912-000-0022-z. [DOI] [PubMed] [Google Scholar]
- 42.Bedogni B, Powell MB. Skin hypoxia: a promoting environmental factor in melanomagenesis. Cell Cycle. 2006;5:1258–1261. doi: 10.4161/cc.5.12.2810. [DOI] [PubMed] [Google Scholar]
- 43.Michaloglou C, Vredeveld LC, Soengas MS, Denoyelle C, Kuilman T, van der Horst CM, Majoor DM, Shay JW, Mooi WJ, Peeper DS. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature. 2005;436:720–724. doi: 10.1038/nature03890. [DOI] [PubMed] [Google Scholar]
- 44.Kuilman T, Michaloglou C, Vredeveld LCW, Douma S, Van Doorn R, Desmet CJ, Aarden LA, Mooi WJ, Peeper DS. Oncogene-induced cellular senescence relayed by a cell-autonomous interleukin-dependent network. Cell. 2008 doi: 10.1016/j.cell.2008.03.039. In press. [DOI] [PubMed] [Google Scholar]
- 45.Ha L, Ichikawa T, Anver M, Dickins R, Lowe S, Sharpless NE, Krimpenfort P, Depinho RA, Bennett DC, Sviderskaya EV, Merlino G. ARF functions as a melanoma tumor suppressor by inducing p53-independent senescence. Proc Natl Acad Sci U S A. 2007;104:10968–10973. doi: 10.1073/pnas.0611638104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sherr CJ. Divorcing ARF and p53: an unsettled case. Nat Rev Cancer. 2006;6:663–673. doi: 10.1038/nrc1954. [DOI] [PubMed] [Google Scholar]
- 47.Moore L, Venkatachalam S, Vogel H, Watt JC, Wu CL, Steinman H, Jones SN, Donehower LA. Cooperativity of p19ARF, Mdm2, and p53 in murine tumorigenesis. Oncogene. 2003;22:7831–7837. doi: 10.1038/sj.onc.1206985. [DOI] [PubMed] [Google Scholar]
- 48.Weber JD, Jeffers JR, Rehg JE, Randle DH, Lozano G, Roussel MF, Sherr CJ, Zambetti GP. p53-independent functions of the p19(ARF) tumor suppressor. Genes Dev. 2000;14:2358–2365. doi: 10.1101/gad.827300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tyner SD, Venkatachalam S, Choi J, Jones S, Ghebranious N, Igelmann H, Lu X, Soron G, Cooper B, Brayton C, Hee PS, Thompson T, Karsenty G, Bradley A, Donehower LA. p53 mutant mice that display early ageing-associated phenotypes. Nature. 2002;415:45–53. doi: 10.1038/415045a. [DOI] [PubMed] [Google Scholar]
- 50.Freedberg DE, Rigas SH, Russak J, Gai W, Kaplow M, Osman I, Turner F, Randerson-Moor JA, Houghton A, Busam K, Bishop T, Bastian BC, Newton-Bishop JA, Polsky D. Frequent p16-independent inactivation of p14ARF in human melanoma. J Natl Cancer Inst. 2008 doi: 10.1093/jnci/djn157. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hussein MR, Haemel AK, Wood GS. p53-related pathways and the molecular pathogenesis of melanoma. Eur J Cancer Prev. 2003;12:93–100. doi: 10.1097/00008469-200304000-00002. [DOI] [PubMed] [Google Scholar]
- 52.Petitjean A, Mathe E, Kato S, Ishioka C, Tavtigian SV, Hainaut P, Olivier M. Impact of mutant p53 functional properties on TP53 mutation patterns and tumor phenotype: lessons from recent developments in the IARC TP53 database. Hum Mutat. 2007;28:622–629. doi: 10.1002/humu.20495. [DOI] [PubMed] [Google Scholar]
- 53.Soussi T, Kato S, Levy PP, Ishioka C. Reassessment of the TP53 mutation database in human disease by data mining with a library of TP53 missense mutations. Hum Mutat. 2005;25:6–17. doi: 10.1002/humu.20114. [DOI] [PubMed] [Google Scholar]
- 54.Eymin B, Karayan L, Seite P, Brambilla C, Brambilla E, Larsen CJ, Gazzeri S. Human ARF binds E2F1 and inhibits its transcriptional activity. Oncogene. 2001;20:1033–1041. doi: 10.1038/sj.onc.1204220. [DOI] [PubMed] [Google Scholar]
- 55.Martelli F, Hamilton T, Silver DP, Sharpless NE, Bardeesy N, Rokas M, Depinho RA, Livingston DM, Grossman SR. p19ARF targets certain E2F species for degradation. Proc Natl Acad Sci U S A. 2001;98:4455–4460. doi: 10.1073/pnas.081061398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mason SL, Loughran O, La Thangue NB. p14(ARF) regulates E2F activity. Oncogene. 2002;21:4220–4230. doi: 10.1038/sj.onc.1205524. [DOI] [PubMed] [Google Scholar]
- 57.Datta A, Sen J, Hagen J, Korgaonkar CK, Caffrey M, Quelle DE, Hughes DE, Ackerson TJ, Costa RH, Raychaudhuri P. ARF directly binds DP1: interaction with DP1 coincides with the G1 arrest function of ARF. Mol Cell Biol. 2005;25:8024–8036. doi: 10.1128/MCB.25.18.8024-8036.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Johnson DG, Degregori J. Putting the Oncogenic and Tumor Suppressive Activities of E2F into Context. Curr Mol Med. 2006;6:731–738. doi: 10.2174/1566524010606070731. [DOI] [PubMed] [Google Scholar]
- 59.Bates S, Phillips AC, Clark PA, Stott F, Peters G, Ludwig RL, Vousden KH. p14ARF links the tumour suppressors RB and p53. Nature. 1998;395:124–125. doi: 10.1038/25867. [DOI] [PubMed] [Google Scholar]
- 60.Lomazzi M, Moroni MC, Jensen MR, Frittoli E, Helin K. Suppression of the p53- or pRB-mediated G1 checkpoint is required for E2F-induced S-phase entry. Nat Genet. 2002;31:190–194. doi: 10.1038/ng891. [DOI] [PubMed] [Google Scholar]
- 61.Rowland BD, Denissov SG, Douma S, Stunnenberg HG, Bernards R, Peeper DS. E2F transcriptional repressor complexes are critical downstream targets of p19(ARF)/p53-induced proliferative arrest. Cancer Cell. 2002;2:55–65. doi: 10.1016/s1535-6108(02)00085-5. [DOI] [PubMed] [Google Scholar]
- 62.Chang DL, Qiu W, Ying H, Zhang Y, Chen CY, Xiao ZX. ARF promotes accumulation of retinoblastoma protein through inhibition of MDM2. Oncogene. 2007;26:4627–4634. doi: 10.1038/sj.onc.1210254. [DOI] [PubMed] [Google Scholar]
- 63.Rangarajan A, Weinberg RA. Opinion: Comparative biology of mouse versus human cells: modelling human cancer in mice. Nat Rev Cancer. 2003;3:952–959. doi: 10.1038/nrc1235. [DOI] [PubMed] [Google Scholar]
- 64.Wang W, El Deiry WS. Restoration of p53 to limit tumor growth. Curr Opin Oncol. 2008;20:90–96. doi: 10.1097/CCO.0b013e3282f31d6f. [DOI] [PubMed] [Google Scholar]
- 65.Matheu A, Maraver A, Klatt P, Flores I, Garcia-Cao I, Borras C, Flores JM, Vina J, Blasco MA, Serrano M. Delayed ageing through damage protection by the Arf/p53 pathway. Nature. 2007;448:375–379. doi: 10.1038/nature05949. [DOI] [PubMed] [Google Scholar]