

Abstract
The extracellular superoxide dismutase (SOD3), a secretory copper-containing enzyme, regulates angiotensin II (Ang II)–induced hypertension by modulating levels of extracellular superoxide anion. The present study was designed to determine the role of the copper transporter Menkes ATPase (MNK) in Ang II–induced SOD3 activity and hypertension in vivo. Here we show that chronic Ang II infusion enhanced systolic blood pressure and vascular superoxide anion production in MNK mutant (MNKmut) mice as compared with those in wild-type mice, which are associated with impaired acetylcholine-induced endothelium-dependent vasorelaxation in MNKmut mice. These effects in MNKmut mice are rescued by infusion of the SOD mimetic Tempol. By contrast, norepinephrine-induced hypertension, which is not associated with an increase in vascular superoxide anion production, is not affected in MNKmut mice. Mechanistically, basal and Ang II infusion-induced increase in vascular SOD3-specific activity is significantly inhibited in MNKmut mice. Coimmunoprecipitation analysis reveals that Ang II stimulation promotes association of MNK with SOD3 in cultured vascular smooth muscle cell and in mouse aortas, which may contribute to SOD3-specific activity by increasing copper delivery to SOD3 through MNK. In summary, MNK plays an important role in modulating Ang II–induced hypertension and endothelial function by regulating SOD3 activity and vascular superoxide anion production and becomes a potential therapeutic target for oxidant stress-dependent cardiovascular diseases.
Keywords: angiotensin II, hypertension, MNK protein, norepinephrine, oxidative stress, SOD1, SOD3
Hypertension increases levels of superoxide anion (O2.−) in the vasculature, which contributes to the endothelial dysfunction by limiting the bioavailability of NO and by the formation of peroxynitrite. Superoxide also plays a role in vascular remodeling and vascular tone in hypertension through increased vascular smooth cell proliferation, inflammation, and increased deposition of extracellular matrix protein by activating redox-sensitive transcription factors, ion channels, and by matrix metalloproteinases.1 A major antioxidant defense system against O2.− is the superoxide dismutases (SODs): the cytoplasmic Cu/ZnSOD (SOD1), the mitochondrial MnSOD (SOD2), and the extracellular Cu/ZnSOD (SOD3, ecSOD). SOD3 is a major extracellular antioxidant enzyme highly expressed in the vasculature. Particularly, in angiotensin II (Ang II)–induced hypertension, the excessive vascular O2.−and hypertension are ameliorated by treatment with recombinant SOD3.2 We and others have demonstrated that Ang II–induced increase in blood pressure and vascular O2.− levels in the aorta are increased in SOD3−/− mice,2,3 and gene transfer of SOD3 reduces arterial pressure in a genetic model of hypertension.4 Thus, SOD3 is a potentially important modulator of oxidative stress-dependent hypertension.
We have previously shown that Ang II increases SOD3 activity in mouse aorta and cultured vascular smooth muscle cells.5 However, little is known about how specific activity of SOD3 is regulated in Ang II–induced hypertension in vivo. SOD3 is a secretory enzyme, which requires a catalytic copper at its active site for enzymatic activity in a fashion similar to SOD1.6,7 Under physiological conditions, the intracellular level of free copper is extraordinarily restricted.8 SOD1 obtains its catalytic copper ion through interaction with the cytosolic copper carrier protein CCS (copper chaperone for SOD1).9 In contrast, we have demonstrated that full activation of SOD3 requires the copper transporter Menkes ATPase (ATP7A, MNK), which localizes at the trans-Golgi network where MNK delivers copper to some secretory copper enzymes, including SOD3.10–13 A loss-of-function X-linked mutation of MNK leads to Menkes disease, a disorder with a marked decrease in copper levels in most tissues except for the kidney and small intestine.14 Patients with Menkes disease show multiple abnormalities secondary to deficiencies in the activity of some secretory copper enzymes such as dopamine β-mono-oxygenase (neurological abnormalities), tyrosinase (hypopigmentation), and lysyl oxidase (vascular abnormalities and connective tissue abnormalities), leading to death in infancy.14
We performed the present study to determine the role of MNK in Ang II–induced hypertension using MNK mutant (MNKmut) mice with reduced copper transport function.15–18 We show that Ang II increases systolic blood pressure and vascular O2.− production to a greater extent in MNKmut mice compared with wild-type (WT) mice, which are associated with impaired endothelial function in MNKmut mice. These responses in MNKmut mice are rescued by coinfusion of the SOD mimetic Tempol. By contrast, norepinephrine-induced hypertension, which is not associated with an increase in vascular O2.− production,19 is not affected in MNKmut mice. Mechanistically, the baseline and Ang II–induced increase in specific activity of SOD3, but not SOD1, in aortas from MNKmut mice was abolished. Ang II treatment promoted an association of MNK with SOD3, which is likely required for copper delivery to SOD3 by MNK. Thus, MNK plays an important role as a modulator of vascular O2.− production and blood pressure induced by Ang II and might become a novel target of antioxidant therapy.
Materials and Methods
Animals Studied
Heterozygous blotchy MNK mutant female mice on a C57BL/6JEiJ background and WT males on a C57BL/6JEiJ background were purchased from Jackson Laboratory (Bar Harbor, Maine) and were used for establishing a colony. Mice were maintained on regular chow. In the current study, female heterozygous mice carrying the X-linked blotchy MNK mutation and control littermates were weaned at 4 weeks of age and maintained on regular chow for 3 to 4 months for further studies. Diet and water were provided ad libitum. The Emory University Animal Care and Use Committee approved the protocol for animal use.
Mice were studied between 8 and 12 weeks of age. Ang II was delivered at a rate of 280 ng/min per kilogram for 14 days using osmotic minipumps (Alzet model 2002; Alza Corp) as previously described.3 Norepinephrine was delivered at a rate of 5.6 mg/kg per day for 14 days using osmotic minipumps as previously described.5 At some experiments, 4-hydroxy-2,2,6,6-tetramethylpiperidinyloxy (Tempol) dissolved in saline was administered in separate osmotic minipumps at a rate of 50 mg/kg per day.20 Sham-operated animals underwent an identical surgical procedure, except that no pump was implanted. Blood pressure was measured before and during infusion of Ang II (on Days 14) using the tail-cuff method (BP2000 Visitech System Inc) as previously described.3
Superoxide Dismutase Activity Assays
Tissues were homogenized in 10 vol of 50 mmol/L potassium phosphate (pH 7.4) containing 0.3 mol/L KBr and a cocktail of protease inhibitors (Roche Applied Science, Indianapolis, Ind). SOD activity was measured in 50 mmol/L phosphate buffer by inhibition of the reduction of cytochrome C (50 μmol/L) by superoxide generated by xanthine (0.1 mmol/L) and xanthine oxidase (0.01 U/mL) at pH 7.4 as described previously.21 Cyanide (3 mmol/L) was used to distinguish between the cyanide-sensitive isozymes Cu/Zn SOD and ecSOD and the cyanide-resistant Mn SOD. To isolate SOD3 from vessels of MNKmut mice and their control littermates, Con A-Sepharose chromatography (Pharmacia Biotech, Piscataway, NJ) was used. Unlike Cu/Zn SOD and Mn SOD, the glycoprotein in ecSOD binds to the lectin concanavalin A. The isolation procedure was performed as previously described.22 Briefly, samples were applied to a Con A-Sepharose column equilibrated with 50 mmol/L potassium phosphate buffer (pH 7.4) in 120 mmol/L NaCl. After collecting the eluting fluid, which contains Cu/ZnSOD and MnSOD, ecSOD fraction was eluted with 150 mmol/L α methyl mannoside in 50 mmol/L potassium phosphate buffer (pH 7.4).
Western Blot Analysis
Western blot analysis was performed as previously described.23 The primary antibodies used included a rabbit polyclonal antibody against murine SOD3,23 a sheep antibody against human SOD1, and rabbit polyclonal antisera to MNK. Equal loading of proteins was confirmed by Ponceau or Coomassie blue staining.
Cell Culture and Coimmunoprecipitation Assays
Human aortic smooth muscle cells were purchased from Cascade Biologics, Inc (Portland, Ore). Human aortic smooth muscle cells were cultured in smooth muscle basal medium (Clonetics) and 5% fetal bovine serum as previously described.5 Experiments were performed with 0.5% serum at passages 4 to 8. Coimmunoprecipitation assays were performed as previously described.13
Measurements of Vascular Superoxide Production in Aortas From MNK Mutant Mice and Control Littermates
Animals were euthanized by CO2 inhalation. Vascular O2.− production was determined using lucigenin-enhanced chemiluminescence as described before.24 This method has been validated for O2.− measurements in vascular tissue when low concentrations of lucigenin (5 μmol/L) are used.25
As a second approach to measure reactive oxygen species production in vessels in situ, frozen cross-sections of aortas were stained with dihydroethidium (Molecular Probes Inc, Eugene, Ore) using a previously validated method.26
Studies of Vascular Reactivity
Isometric tension studies of aortic rings were performed as previously described.3 Briefly, thoracic aortas were rapidly removed and cut into 3-mm ring segments. Endothelium-dependent vasorelaxation in response to acetylcholine and endothelium-independent relaxation to nitroglycerin were studied as described in detail previously.3 Both responses were assessed in the absence and presence of the SOD mimetic Tempol (Sigma-Aldrich) to determine the contribution of O2.− to vasorelaxation.
Statistical Analysis
Data are presented as mean±SEM. Data were compared between groups of cells and animals by t test when one comparison was performed or by analysis of variance for multiple comparisons. Comparison of dose–response curves was performed using 1-way analysis of variance for repeated measures. When significance was indicated by analysis of variance, the Tukey-Kramer post hoc test was used to specify between group differences. Values of P<0.05 were considered statistically significant.
Results
Effect of Ang II Infusion on SOD1 and SOD3 Activity in Aortas From MNK Mutant Mice
We have previously shown that Ang II increases SOD3 activity and protein levels in both cultured cells and vessels.5 To examine the role of MNK in regulating Ang II–induced increase in SOD3 activity, we used mice carrying the X-linked blotchy MNK mutation (MNKmut). These mice have a splice site mutation introducing a new stop codon at amino acid residue 794 with impaired copper transport function, but survive to more than 6 months of age.15,16,18 The body weights between heterozygous females and corresponding littermates used in the current study were similar. At baseline, the activity of SOD3 was decreased in MNKmut compared with WT aortas, whereas protein levels of SOD3 were increased by approximately 2-fold (Figure 1A and 1B). In contrast, SOD1 activity and protein levels were not altered in aortas from MNKmut mice (Figure 1A and 1B). Thus, the specific activity of SOD3, as determined by the ratio of activity to protein, was markedly decreased in aortas from MNKmut mice, whereas it was not altered for SOD1 (Figure 1C). Two weeks of Ang II infusion (280 ng/min per kilogram) significantly increased activity, protein levels, and the specific activity of SOD3, but not SOD1, in aortas from WT mice while having little effect on activity of either SOD3 or SOD1 in MNKmut mice (Figure 1A–C). Of note, the Ang II–induced increase in SOD3 protein level was not decreased in MNKmut mice (Figure 1A and 1B), suggesting that the reduction of SOD3 activity in MNKmut mice is due to a decrease in SOD3-specific activity. Moreover, mRNA and protein levels of MNK in aortic homogenates were not significantly changed after Ang II infusion into WT mice (Figure S1; see the online data supplement at http://hyper.ahajournals.org.).
Figure 1.
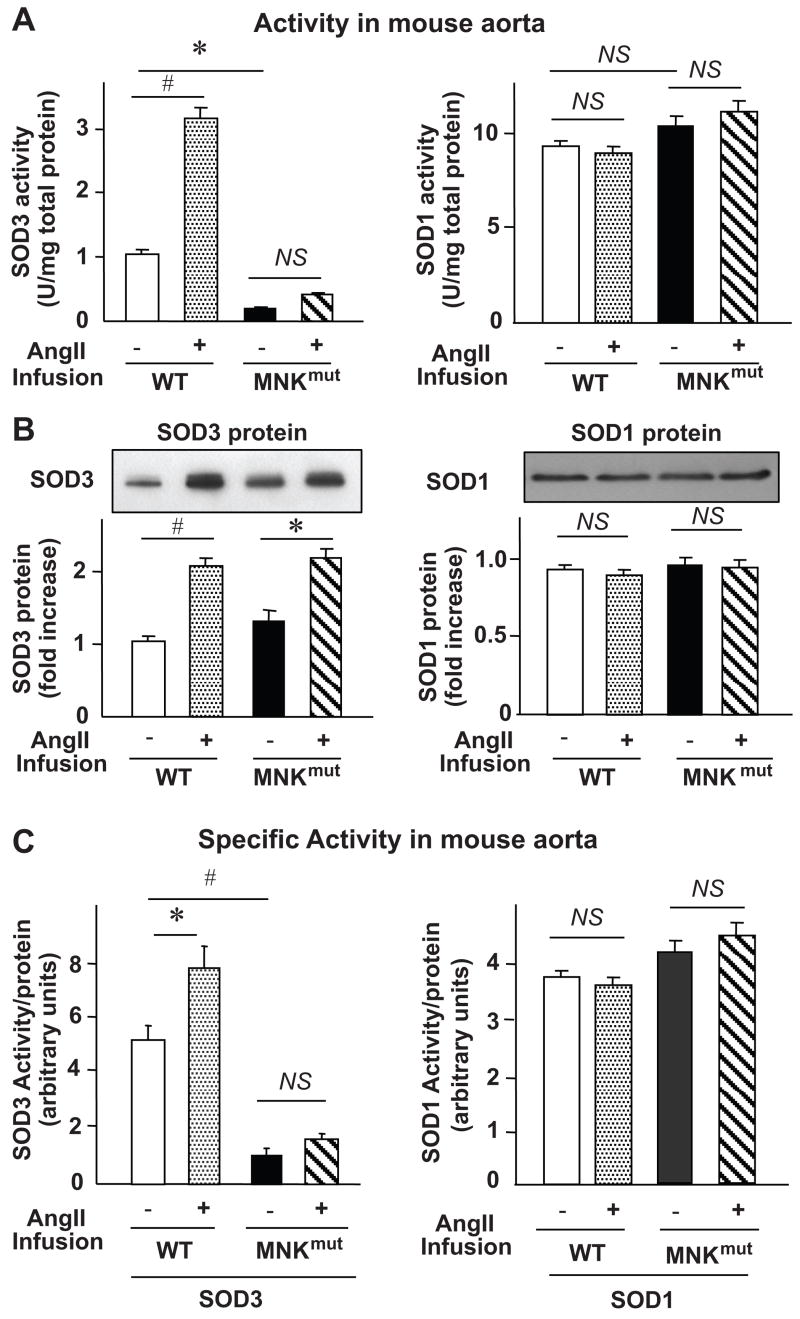
Effect of chronic Ang II infusion on activity (A), protein level (B), and specific activity (C) of SOD3 and SOD1 in aortas from MNKmut mice. Ang II (0.4 mg/kg per day) were chronically infused using osmotic minipumps. Mouse aortas were harvested after a 14-day infusion of Ang II and homogenized, as previously described.23 A and B, Activity of SOD3 and SOD1 in homogenized tissues was analyzed by the inhibition of cytochrome C reduction by xanthine/xanthine oxidase at pH 7.4.23 Con A-Sepharose chromatography was used to isolate SOD3 from tissue homogenates. The results are presented as mean±SEM from 4 separate experiments. Protein levels of SOD1 and SOD3 were determined by Western analysis with antibodies specific to either the SOD1 or the SOD3. Representative blots are from 3 individual experiments. *P<0.05; #P<0.01 vs either not-treated WT mice or not-treated MNKmut mice; NS, not significant. C, Specific activity of SOD1 and SOD3 was determined by the ratio of activity to relative amount of protein as previously described.7,13 The results are presented as mean±SEM from 4 separate experiments. *P<0.05; #P<0.01 vs WT mice with no treatment; NS, not significant.
Effect of Ang II Treatment on Interaction of SOD3 With MNK In Vivo and In Vitro
Because MNK interacts with SOD3 in a copper-dependent manner,13 we sought to determine if Ang II–induced increase in SOD3-specific activity is due to an increase in the association of MNK with SOD3, which would increase copper delivery to SOD3. Coimmunoprecipitation assays revealed that Ang II treatment promoted association of MNK with SOD3 in mice aortas (Figure 2A) and in cultured aortic smooth muscle cells (Figure 2B).
Figure 2.

Effect of Ang II treatment on the interaction of SOD3 with MNK in vivo (A) and in vitro (B). A, Mice were treated as in Figure 1. Equal amounts of lysates from mouse aortas were immunoprecipitated (IP) with either anti-SOD3 antibody or anti-MNK antibody followed by immunoblotting (IB) with either anti-MNK antibody or anti-SOD3 antibody, respectively. IgG was included as a negative control for the immunoprecipitation. The results are presented as mean±SE from 4 separate experiments. *P<0.01 vs nontreatment. B, Cultured human aortic smooth muscle cells were treated with Ang II (100 nmol/L) for 12 hours. Coimmunoprecipitation assays were performed as described for Figure 2A. The results are presented as mean±SE from 4 separate experiments. *P<0.05 vs nontreatment.
Effect of Ang II Infusion on Blood Pressure and O2.− Production in MNK Mutant Mice
Because SOD3 plays an important role in Ang II–induced hypertension,2–4 and MNK is required for the full activation of SOD3,13 we next examined the role of MNK in Ang II-induced hypertension and O2.− levels using MNKmut mice. Chronic low-dose Ang II infusion significantly enhanced systolic blood pressure in MNKmut mice as compared with WT mice (133±4 mm Hg versus 120±4 mm Hg, P<0.01; Figure 3A). Basal and Ang II–induced increase in aortic O2.− production, as assessed by lucigenin chemiluminescence analysis (Figure 3B) and in situ assessed by dihydroethidium staining (Figure 3C), was enhanced in MNKmut mice as compared with WT mice. SOD treatment abolished the dihydroethidium fluorescence,26 confirming specificity of the fluorescent signal for O2.− (data not shown). By contrast, chronic norepinephrine-induced hypertension, which is independent of vascular O2.− production and SOD3 expression,5,19 was not affected in MNKmut mice (Figure 3A). Note that there was no increase in O2.− production in norepinephrine-infused aorta in WT and MNKmut mice (Figure 3B). We also found that the difference in Ang II–induced alterations of blood pressure and O2.− production in MNKmut mice versus WT mice were rescued by coinfusion of the SOD mimetic Tempol (Figure 3A–3B). This result suggests that enhancement of Ang II-induced increase in blood pressure in MNKmut mice is due to increased O2.− levels.
Figure 3.

Effect of Ang II and norepinephrine infusion on blood pressure and vascular O2.− production in MNKmut mice. A, Ang II (0.4 mg/kg per day), Ang II and the SOD mimetic Tempol (50 mg/kg per day), or norepinephrine (5.6 mg/kg per day) were infused subcutaneously with osmotic minipumps. Sham-operated animals underwent an identical surgical procedure, except that either no pump or an empty osmotic pump was implanted. Blood pressure and vascular O2.− production were measured before and 14 days after minipump implantation. (n=5 per group). *P<0.05 vs MNKmut mice with Tempol treatment or WT mice; NS, not significant. B, Aortic O2.− production in control littermates (WT) and MNKmut mice as determined with lucigenin-enhanced chemiluminescence (5 μmol/L). *P<0.05 vs MNKmut mice with Tempol treatment or WT mice; **P<0.01 vs WT mice with no treatment; #P<0.01 vs MNKmut mice with treatment of both Ang II and Tempol or WT mice with Ang II treatment alone; ##P<0.001 vs MNKmut mice with no treatment; NS, not significant. C, In situ detection of superoxide production with dihydroethidium in aortas from WT and MNKmut mice. Fresh-frozen control and MNK mutant mouse aortas were incubated with dihydroethidium for 30 minutes. Data are representative of 3 separate experiments.
Effect of Ang II Infusion on Vascular Function in MNKmut Mice
Because O2.− can alter vascular function, we next examined endothelium-dependent relaxation in WT and MNKmut mice (Figure 4). Under basal conditions, acetylcholine-evoked relaxations were impaired in aortic segments of MNKmut mouse as compared with those of WT mice (Figure 4A). Ang II infusion impaired acetylcholine-evoked relaxation to a greater extent in aortas of MNKmut than WT mice (maximum relaxation 60.6±0.1 versus 77.5±0.1%, P<0.001). Of note, endothelium-independent vasorelaxation induced by nitro-glycerin was not changed in MNKmut mice at both baseline and after Ang II infusion (Figure 4A). The defective acetylcholine-induced vasorelaxation in Ang II–infused MNKmut mice and WT mice were rescued by coinfusion of the SOD mimetic Tempol (Figure 4B).
Figure 4.

Effect of infusions of either Ang II or Ang II and the SOD mimetic Tempol on endothelium-dependent vasodilatation in aortic rings from control littermates (WT) and MNK mutant (MNKmut) mice. A and B, Isometric tension of aortic rings (3 mm) was measured in isolated organ chambers. Vasodilation was evoked by acetylcholine (ACh) and nitroglycerin (NTG). Vasodilator responses were measured after preconstriction with PGF2α (1 μmol/L). Data are expressed as mean±SE (n=4 to 5 per group). A, *P<0.001 vs WT mice with Ang II treatment; **P<0.001; #P<0.05 vs WT mice with no treatment; NS, not significant. B, *P<0.001 vs MNKmut mice with Ang II treatment; **P<0.001 vs WT mice with Ang II treatment.
Discussion
The present studies demonstrate that MNK plays an important role in regulating blood pressure, vascular O2.−production, and endothelial function in Ang II–induced hypertension (Figure 5). We found that (1) the increases in systolic blood pressure and vascular O2.− production caused by Ang II are significantly enhanced in MNKmut mice as compared with WT mice. By contrast, norepinephrine-induced hypertension, which is not associated with an increase in vascular O2.− production, is not affected in MNKmut mice; (2) the specific activity of SOD3 at baseline and after Ang II infusion is significantly decreased in aortas of MNKmut mice as compared with WT mice; (3) the endothelial dysfunction caused by Ang II infusion is further enhanced in MNKmut mice, which is prevented by infusion of the SOD mimetic Tempol. Tempol also reduces systolic blood pressure and vascular O2.− production in MNKmut mice; and (4) coimmunoprecipitation analysis revealed that Ang II stimulation promotes an association of MNK with SOD3 in cultured vascular smooth muscle cells and in mouse aortas.
Figure 5.

Proposed model for the role of MNK in modulating SOD3 activity in Ang II–induced hypertension. Ang II promotes the interaction of MNK with SOD3, thereby enhancing copper delivery to SOD3. Furthermore, Ang II–induced increase in SOD3 activity will decrease O2.− levels, thereby increasing available NO., endothelial function, and reducing blood pressure.
MNK, which is expressed in the majority of tissues except for the liver, is a key regulator of copper homeostasis partially by providing copper to secreted copper-containing enzymes.14,15,27 MNK dysfunction causes a wide range of phenotypes from neurological disorders to connective tissue abnormalities, some of which could be secondary to deficiencies in the activity of secretory copper enzymes involved in these processes.14,15 We and others previously reported that chronic Ang II infusion significantly increases vascular SOD3 activity5 and that mice lacking SOD3 develop more severe hypertension and O2.− production in response to Ang II than control mice.2,3 Thus, SOD3 plays an important role in regulating blood pressure by decreasing extracellular O2.− levels. Our present findings show that the hypertension and vascular O2.− production stimulated by Ang II are significantly enhanced in MNKmut mice as compared with WT mice. Of note, the hypertension in MNKmut mice is ameliorated by infusion of SOD mimetic Tempol, suggesting that this response is due to overproduction of O2.− production in these mice. Norepinephrine-induced hypertension, which is not associated with an increase in vascular O2.− production,19 is not affected in MNKmut mice. Thus, MNK plays an important role in regulating hypertension mediated by increased O2.− production. Mechanistically, we found that basal- and Ang II infusion-induced increases in SOD3 activity, but not SOD3 protein expression, are markedly inhibited in aorta of MNKmut mice. Of note, the increase in SOD3 specific activity caused by Ang II is not due to increase in MNK abundance, because the mRNA and protein levels of vascular MNK were not significantly changed after infusion of Ang II into WT mice. Thus, MNK plays an important role in modulating the increase in vascular O2.− production and blood pressure by regulating SOD3-specific activity.
SOD3 plays a critical role in modulating endothelial function by preventing oxidative inactivation of NO. as it transverses from the endothelium to the smooth muscle.28 In this study, the Ang II–induced impairment of acetylcholine-evoked vasodilatation is further worsened in MNKmut as compared with WT mice, which is rescued by infusion of SOD mimetic. By contrast, endothelium-independent vasorelaxation induced by nitroglycerin was not affected in MNKmut aorta. These results suggest that inhibition of acetylcholine-induced vasodilation in MNKmut mice is not due to a decrease in vascular smooth muscle sensitivity to NO., but due to the decrease in endothelial NO. bioavailability. Despite enhanced endothelial dysfunction and O2.− production in aortas from MNKmut mice, basal blood pressure is not elevated, suggesting that endothelial function and O2.− levels play a minor role at baseline.2–4,29 Given that activity of extracellular SOD3, but not intracellular SOD1, is abolished in MNKmut mouse aortas, MNK may function to protect available NO. from extracellular O2. by promoting SOD3 activity, thereby modulating vascular function in Ang II–induced hypertension.
Levels of O2. in the kidney also plays an important role in regulating renal blood flow and glomerular filtration rate by controlling NO. bioavailability, resulting in altering renal sodium retention, which contributes to hypertension.30 Of note, MNK is highly expressed in the kidney.27 Chu et al reported that gene transfer of SOD3 reduces renal sodium retention in hypertensive rats.4 Thus, it is conceivable that MNK may control hypertension by regulating renal function. MNK might also affect central control of blood pressure.31 The role of the kidney and central nervous system in hypertension in MNKmut mice requires further investigation.
MNK is normally localized in the trans-Golgi network where it supplies copper to copper-dependent enzymes, including SOD3, in the secretory pathway.10–13 The present study using coimmunoprecipitation analysis shows that Ang II increases the interaction of MNK with SOD3 in cultured vascular smooth muscle cells and in mouse aortas. This response could contribute to the increase in SOD3-specific activity caused by Ang II. It has been shown that MNK also constitutively recycles between the plasma membrane and trans-Golgi network.32 The effect of Ang II on MNK trafficking and mechanism for copper delivery to SOD3 requires further investigation.
Previous studies using ecSOD−/− mice have shown that ecSOD functions to prevent oxidative stress-dependent pathological states such as hypertension, ischemia–reperfusion injury, myocardial infarction-induced cardiac hypertrophy, and lung injury induced by hyperoxia.33–36 We recently reported that SOD3 plays an important role in reparative neovascularization in response to hindlimb ischemia37 and that MNK is highly expressed in the intimal lesion of atherosclerosis where SOD3 activity is markedly increased.13 Given that MNK is required for full activation of SOD3, it is conceivable that MNK is involved in these various pathophysiologies in which O2.− production is increased.
Perspectives
In summary, the present study demonstrates that MNK plays a role in regulating blood pressure and endothelial function by regulating SOD3 activity, which scavenges extracellular O2.−, thereby regulating bioavailability of NO. These findings provide novel insight into MNK as a potential therapeutic target for oxidant stress-dependent cardiovascular diseases.
Supplementary Material
Acknowledgments
We thank Dr Lula Hilenski for her technical assistance.
Sources of Funding
This research was supported by National Institutes of Health R01 HL70187 (to T.F.), HL077524 (to M.U.-F.), Program Project Grant HL58000 (to D.G.H. and T.F.), and American Heart Association Grants in Aid 0455242B (to T.F.) and 0755805Z (to M.U.-F.).
Footnotes
Disclosures
None.
References
- 1.Guzik TJ, Harrison DG. Vascular NADPH oxidases as drug targets for novel antioxidant strategies. Drug Discov Today. 2006;11:524–533. doi: 10.1016/j.drudis.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 2.Jung O, Marklund SL, Geiger H, Pedrazzini T, Busse R, Brandes RP. Extracellular superoxide dismutase is a major determinant of nitric oxide bioavailability: in vivo and ex vivo evidence from ecSOD-deficient mice. Circ Res. 2003;93:622–629. doi: 10.1161/01.RES.0000092140.81594.A8. [DOI] [PubMed] [Google Scholar]
- 3.Gongora MC, Qin Z, Laude K, Kim HW, McCann L, Folz JR, Dikalov S, Fukai T, Harrison DG. Role of extracellular superoxide dismutase in hypertension. Hypertension. 2006;48:473–481. doi: 10.1161/01.HYP.0000235682.47673.ab. [DOI] [PubMed] [Google Scholar]
- 4.Chu Y, Iida S, Lund DD, Weiss RM, DiBona GF, Watanabe Y, Faraci FM, Heistad DD. Gene transfer of extracellular superoxide dismutase reduces arterial pressure in spontaneously hypertensive rats: role of heparin-binding domain. Circ Res. 2003;92:461–468. doi: 10.1161/01.RES.0000057755.02845.F9. [DOI] [PubMed] [Google Scholar]
- 5.Fukai T, Siegfried MR, Ushio-Fukai M, Griendling KK, Harrison DG. Modulation of extracellular superoxide dismutase expression by angiotensin II and hypertension. Circ Res. 1999;85:23–28. doi: 10.1161/01.res.85.1.23. [DOI] [PubMed] [Google Scholar]
- 6.McCord JM, Fridovich I. Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein) J Biol Chem. 1969;244:6049–6055. [PubMed] [Google Scholar]
- 7.Jeney V, Itoh S, Wendt M, Gradek Q, Ushio-Fukai M, Harrison DG, Fukai T. Role of antioxidant-1 in extracellular superoxide dismutase function and expression. Circ Res. 2005;96:723–729. doi: 10.1161/01.RES.0000162001.57896.66. [DOI] [PubMed] [Google Scholar]
- 8.Rae TD, Schmidt PJ, Pufahl RA, Culotta VC, O’Halloran TV. Undetectable intracellular free copper: the requirement of a copper chaperone for superoxide dismutase. Science. 1999;284:805–808. doi: 10.1126/science.284.5415.805. [DOI] [PubMed] [Google Scholar]
- 9.Culotta VC, Yang M, O’Halloran TV. Activation of superoxide dismutases: putting the metal to the pedal. Biochim Biophys Acta. 2006;1763:747–758. doi: 10.1016/j.bbamcr.2006.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yuan DS, Dancis A, Klausner RD. Restriction of copper export in Saccharomyces cerevisiae to a late Golgi or post-Golgi compartment in the secretory pathway. J Biol Chem. 1997;272:25787–25793. doi: 10.1074/jbc.272.41.25787. [DOI] [PubMed] [Google Scholar]
- 11.Petris MJ, Strausak D, Mercer JF. The Menkes copper transporter is required for the activation of tyrosinase. Hum Mol Genet. 2000;9:2845–2851. doi: 10.1093/hmg/9.19.2845. [DOI] [PubMed] [Google Scholar]
- 12.El Meskini R, Culotta VC, Mains RE, Eipper BA. Supplying copper to the cuproenzyme peptidylglycine alpha-amidating monooxygenase. J Biol Chem. 2003;278:12278–12284. doi: 10.1074/jbc.M211413200. [DOI] [PubMed] [Google Scholar]
- 13.Qin Z, Itoh S, Jeney V, Ushio-Fukai M, Fukai T. Essential role for the Menkes ATPase in activation of extracellular superoxide dismutase: implication for vascular oxidative stress. FASEB J. 2006;20:334–336. doi: 10.1096/fj.05-4564fje. [DOI] [PubMed] [Google Scholar]
- 14.Mercer JF. The molecular basis of copper-transport diseases. Trends Mol Med. 2001;7:64–69. doi: 10.1016/s1471-4914(01)01920-7. [DOI] [PubMed] [Google Scholar]
- 15.Mercer JF. Menkes syndrome and animal models. Am J Clin Nutr. 1998;67:1022S–1028S. doi: 10.1093/ajcn/67.5.1022S. [DOI] [PubMed] [Google Scholar]
- 16.La Fontaine S, Firth SD, Lockhart PJ, Brooks H, Camakaris J, Mercer JF. Intracellular localization and loss of copper responsiveness of Mnk, the murine homologue of the Menkes protein, in cells from blotchy (Mo blo) and brindled (Mo br) mouse mutants. Hum Mol Genet. 1999;8:1069–1075. doi: 10.1093/hmg/8.6.1069. [DOI] [PubMed] [Google Scholar]
- 17.Kelly EJ, Palmiter RD. A murine model of Menkes disease reveals a physiological function of metallothionein. Nat Genet. 1996;13:219–222. doi: 10.1038/ng0696-219. [DOI] [PubMed] [Google Scholar]
- 18.Starcher B, Madaras JA, Fisk D, Perry EF, Hill CH. Abnormal cellular copper metabolism in the blotchy mouse. J Nutr. 1978;108:1229–1233. doi: 10.1093/jn/108.8.1229. [DOI] [PubMed] [Google Scholar]
- 19.Bech Laursen J, Rajagopalan S, Tarpey M, Freeman BA, Harrison DG. A role of superoxide in angiotensin II- but not catecholamine-induced hypertension. Circulation. 1997;95:588–593. doi: 10.1161/01.cir.95.3.588. [DOI] [PubMed] [Google Scholar]
- 20.Dikalova A, Clempus R, Lassegue B, Cheng G, McCoy J, Dikalov S, San Martin A, Lyle A, Weber DS, Weiss D, Taylor WR, Schmidt HH, Owens GK, Lambeth JD, Griendling KK. Nox1 overexpression potentiates angiotensin II-induced hypertension and vascular smooth muscle hypertrophy in transgenic mice. Circulation. 2005;112:2668–2676. doi: 10.1161/CIRCULATIONAHA.105.538934. [DOI] [PubMed] [Google Scholar]
- 21.Crapo JD, McCord JM, Fridovich I. Preparation and assay of superoxide dismutases. Methods Enzymol. 1978;53:382–393. doi: 10.1016/s0076-6879(78)53044-9. [DOI] [PubMed] [Google Scholar]
- 22.Marklund SL. Extracellular superoxide dismutase in human tissues and human cell lines. J Clin Invest. 1984;74:1398–1403. doi: 10.1172/JCI111550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fukai T, Galis ZS, Meng XP, Parthasarathy S, Harrison DG. Vascular expression of extracellular superoxide dismutase in atherosclerosis. J Clin Invest. 1998;101:2101–2111. doi: 10.1172/JCI2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Landmesser U, Dikalov S, Price SR, McCann L, Fukai T, Holland SM, Mitch WE, Harrison DG. Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. J Clin Invest. 2003;111:1201–1209. doi: 10.1172/JCI14172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Skatchkov MP, Sperling D, Hink U, Mulsch A, Harrison DG, Sindermann I, Meinertz T, Munzel T. Validation of lucigenin as a chemiluminescent probe to monitor vascular superoxide as well as basal vascular nitric oxide production. Biochem Biophys Res Commun. 1999;254:319–324. doi: 10.1006/bbrc.1998.9942. [DOI] [PubMed] [Google Scholar]
- 26.Nguyen AD, Itoh S, Jeney V, Yanagisawa H, Fujimoto M, Ushio-Fukai M, Fukai T. Fibulin-5 is a novel binding protein for extracellular superoxide dismutase. Circ Res. 2004;95:1067–1074. doi: 10.1161/01.RES.0000149568.85071.FB. [DOI] [PubMed] [Google Scholar]
- 27.Lutsenko S, Barnes NL, Bartee MY, Dmitriev OY. Function and regulation of human copper-transporting ATPases. Physiol Rev. 2007;87:1011–1046. doi: 10.1152/physrev.00004.2006. [DOI] [PubMed] [Google Scholar]
- 28.Oury TD, Day BJ, Crapo JD. Extracellular superoxide dismutase: a regulator of nitric oxide bioavailability. Lab Invest. 1996;75:617–636. [PubMed] [Google Scholar]
- 29.Nakazono K, Watanabe N, Matsuno K, Sasaki J, Sato T, Inoue M. Does superoxide underlie the pathogenesis of hypertension? Proc Natl Acad Sci U S A. 1991;88:10045–10048. doi: 10.1073/pnas.88.22.10045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wilcox CS, Welch WJ. Interaction between nitric oxide and oxygen radicals in regulation of tubuloglomerular feedback. Acta Physiol Scand. 2000;168:119–124. doi: 10.1046/j.1365-201x.2000.00668.x. [DOI] [PubMed] [Google Scholar]
- 31.Zimmerman MC, Lazartigues E, Lang JA, Sinnayah P, Ahmad IM, Spitz DR, Davisson RL. Superoxide mediates the actions of angiotensin II in the central nervous system. Circ Res. 2002;91:1038–1045. doi: 10.1161/01.res.0000043501.47934.fa. [DOI] [PubMed] [Google Scholar]
- 32.Petris MJ, Mercer JF, Culvenor JG, Lockhart P, Gleeson PA, Camakaris J. Ligand-regulated transport of the Menkes copper P-type ATPase efflux pump from the Golgi apparatus to the plasma membrane: a novel mechanism of regulated trafficking. Embo J. 1996;15:6084–6095. [PMC free article] [PubMed] [Google Scholar]
- 33.Fattman CL, Schaefer LM, Oury TD. Extracellular superoxide dismutase in biology and medicine. Free Radic Biol Med. 2003;35:236–256. doi: 10.1016/s0891-5849(03)00275-2. [DOI] [PubMed] [Google Scholar]
- 34.Fukai T, Folz RJ, Landmesser U, Harrison DG. Extracellular superoxide dismutase and cardiovascular disease. Cardiovasc Res. 2002;55:239–249. doi: 10.1016/s0008-6363(02)00328-0. [DOI] [PubMed] [Google Scholar]
- 35.Qin Z, Reszka KJ, Fukai T, Weintraub NL. Extracellular superoxide dismutase (ecSOD) in vascular biology: an update on exogenous gene transfer and endogenous regulators of ecSOD. Transl Res. 2008;151:68–78. doi: 10.1016/j.trsl.2007.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.van Deel ED, Lu Z, Xu X, Zhu G, Hu X, Oury TD, Bache RJ, Duncker DJ, Chen Y. Extracellular superoxide dismutase protects the heart against oxidative stress and hypertrophy after myocardial infarction. Free Radic Biol Med. 2008;44:1305–1313. doi: 10.1016/j.freeradbiomed.2007.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim HW, Lin A, Guldberg RE, Ushio-Fukai M, Fukai T. Essential role of extracellular SOD in reparative neovascularization induced by hindlimb ischemia. Circ Res. 2007;101:409–419. doi: 10.1161/CIRCRESAHA.107.153791. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.