

Abstract
Ca2+ release from cardiac sarcoplasmic reticulum (SR) via ryanodine receptors (RyRs) is regulated by dyadic cleft [Ca2+] and intra-SR free [Ca2+] ([Ca2+]SR). Robust SR Ca2+ release termination is important for stable excitation–contraction coupling, and partial [Ca2+]SR depletion may contribute to release termination. Here, we investigated the regulation of SR Ca2+ release termination of spontaneous local SR Ca2+ release events (Ca2+ sparks) by [Ca2+]SR, release flux, and intra-SR Ca2+ diffusion. We simultaneously measured Ca2+ sparks and Ca2+ blinks (localized elementary [Ca2+]SR depletions) in permeabilized ventricular cardiomyocytes over a wide range of SR Ca2+ loads and release fluxes. Sparks terminated via a [Ca2+]SR-dependent mechanism at a fixed [Ca2+]SR depletion threshold independent of the initial [Ca2+]SR and release flux. Ca2+ blink recovery depended mainly on intra-SR Ca2+ diffusion rather than SR Ca2+ uptake. Therefore, the large variation in Ca2+ blink recovery rates at different release sites occurred because of differences in the degree of release site interconnection within the SR network. When SR release flux was greatly reduced, long-lasting release events occurred from well-connected junctions. These junctions could sustain release because local SR Ca2+ release and [Ca2+]SR refilling reached a balance, preventing [Ca2+]SR from depleting to the termination threshold. Prolonged release events eventually terminated at a steady [Ca2+]SR, indicative of a slower, [Ca2+]SR-independent termination mechanism. These results demonstrate that there is high variability in local SR connectivity but that SR Ca2+ release terminates at a fixed [Ca2+]SR termination threshold. Thus, reliable SR Ca2+ release termination depends on tight RyR regulation by [Ca2+]SR.
Keywords: heart, sarcoplasmic reticulum, Ca2+ sparks, Ca2+-induced Ca2+ release, ryanodine receptor
In cardiac muscle, excitation–contraction coupling is initiated by membrane depolarization and Ca2+ entry via voltage-gated Ca2+ channels that activates sarcoplasmic reticulum (SR) Ca2+ release channels (ryanodine receptors [RyRs]). This process is known as Ca2+-induced Ca2+ release (CICR).1,2 Elementary SR Ca2+ release events, which arise from the simultaneous opening of clustered RyRs in a single SR Ca2+ release site, can be measured as Ca2+ sparks,3–5 a locally restricted increase in cytosolic Ca2+; or Ca2+ blinks,6 the corresponding local intra-SR Ca2+ ([Ca2+]SR) depletion that accompanies a spark. Following the synchronized initiation of elementary Ca2+ release during excitation–contraction coupling, sparks summate in space and time and give rise to the global Ca2+ transient required for contraction. Ca2+ sparks also occur spontaneously because RyRs have a stochastic open probability in a resting myocyte. Despite the inherent positive feedback of CICR that may lead to the complete depletion of the SR Ca2+ store, compelling data have shown that during global Ca2+ transients and Ca2+ sparks, intra-SR [Ca2+] only partially depletes.6–8 This raises the critical question of what terminates SR Ca2+ release.
RyRs underlie a complex regulation by both [Ca2+] in the dyadic cleft ([Ca2+]cleft), as well as intra-SR free [Ca2+].9,10 SR Ca2+ release can occur in response to an increase in [Ca2+]cleft (eg, during excitation–contraction coupling) but can also occur spontaneously when [Ca2+]SR is high (eg, during SR Ca2+ overload), which can lead to triggered arrhythmias. Partial depletion of [Ca2+]SR is considered a key factor involved in SR release termination.7,11–13 In fact, Terentyev et al14 have recently shown that release termination of Ca2+ blinks occurs at a relatively constant [Ca2+]SR level even after calsequestrin overexpression. However, dynamic changes in [Ca2+]cleft, which occur during Ca2+ release, may also modulate the mechanism of release termination.15–18 The aim of this study was: (1) to investigate the functional importance of [Ca2+]SR depletion for the termination of elementary Ca2+ release; (2) to investigate whether the SR Ca2+ release flux (and therefore [Ca2+] in the dyadic cleft) regulates the termination mechanism; and (3) to test whether Ca2+-independent SR Ca2+ release termination can occur when local [Ca2+]SR remains constant during release over a prolonged period of time.
We simultaneously measured Ca2+ sparks and corresponding [Ca2+]SR depletions (Ca2+ blinks6) in permeabilized rabbit ventricular myocytes over a wide range of SR Ca2+ loads and release fluxes. We found that Ca2+ sparks terminated at a particular absolute [Ca2+]SR depletion threshold (at ≈60% of resting [Ca2+]SR under control conditions), independent of either SR Ca2+ load or release flux. This suggests that Ca2+ spark termination is mainly determined by a [Ca2+]SR-dependent mechanism and not by release flux or [Ca2+]cleft. Local [Ca2+]SR refilling after a blink was mainly attributable to intra-SR Ca2+ diffusion from nearby regions rather than SR Ca2+ reuptake. Despite a consistent [Ca2+]SR nadir and highly reproducible blink kinetics at any given site, there was remarkably wide variation in blink recovery kinetics among sites. This is consistent with high variation in how well a given junction is connected within the SR network. When SR Ca2+ release flux was substantially reduced by partial RyR inhibition, well-connected SR release sites exhibited prolonged SR Ca2+ release events. At these sites, local [Ca2+]SR was maintained above the termination threshold as local SR Ca2+ release and replenishment reached a steady state. These long events eventually terminated by a slower, [Ca2+]SR-independent termination mechanism. These results provide direct experimental evidence for dynamic regulation of release termination by [Ca2+]SR, release flux, and intra-SR Ca2+ diffusion in a cellular environment.
Materials and Methods
Ventricular myocytes were isolated from New Zealand White rabbits as previously described.7 All procedures were approved by the Institutional Animal Care and Use Committee. For simultaneous recording of [Ca2+]SR and cytosolic [Ca2+], we used the low-affinity Ca2+ indicator Fluo-5N and the high-affinity Ca2+ indicator Rhod-2, respectively. Following the incubation of cardiomyocytes with Fluo-5N/acetoxymethylester (AM) under conditions that promote dye accumulation within the SR,7,19 the sarcolemma of Fluo-5N/AM-loaded myocytes was permeabilized with saponin as described before.20 The saponin-free internal solution was composed of (in mmol/L unless otherwise indicated): K aspartate 100; KCl 15; KH2PO4 5; MgATP 5; EGTA 0.35; CaCl2 0.12; MgCl2 0.75; phosphocreatine 10; HEPES 10; Rhod-2 pentapotassium salt 0.04; creatine phosphokinase 5 U/mL; dextran (Mr 40 000) 8% and pH 7.2 (KOH). Free [Ca2+] and [Mg2+] of this solution were 150 nmol/L and 1 mmol/L, respectively. For the experiments shown in Figure 5A, the free [Ca2+] was adjusted to 200 nmol/L by adding appropriate amounts of CaCl2. Membrane permeabilization has been used extensively in the study of cardiac SR Ca2+ release20–22 and provides several advantages in the context of our experiments: the cytosolic milieu is tightly controlled (eg, ion concentrations and energy supply) and can easily be manipulated during the experiments. Furthermore, any cytosolic Fluo-5N is washed out after permeabilization providing a purely SR-derived Fluo-5N fluorescence signal. All experiments were performed at room temperature (20 to 23°C).
Figure 5.
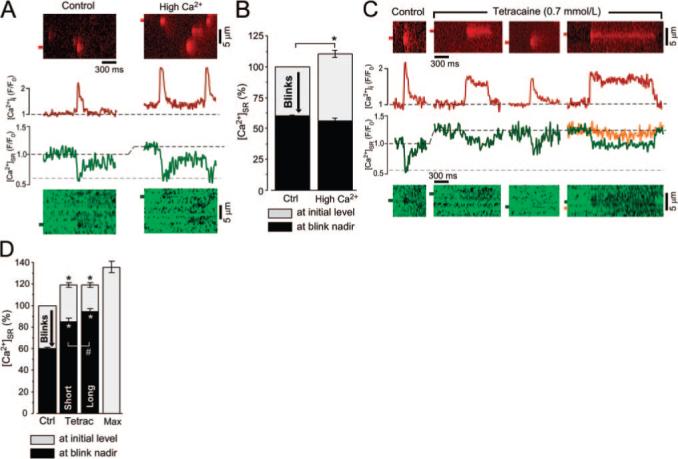
Ca2+ sparks and blinks at increased [Ca2+]SR and at inhibited SR release flux. A, Line scan images and corresponding profiles of Ca2+ sparks and blinks under control conditions and during increased [Ca2+]SR. In these experiments, [Ca2+]SR was increased by elevation of cytosolic [Ca2+] from 150 (Control) to 200 nmol/L (High Ca2+). B, Effect of increased initial [Ca2+]SR on blink nadir. C, Ca2+ sparks and blinks under control conditions and during application of 0.7 mmol/L tetracaine. The region marked in orange shows [Ca2+]SR depletion in a neighboring, quiescent region of the SR. Recordings of different release sites were made from the same cell. D, Effect of release flux inhibition with tetracaine (Tetrac) on blink nadir. Maximum SR Ca2+ load (Max) was obtained during stimulation of SR Ca2+ uptake by the catalytic subunit of PKA (5 U/mL) and complete inhibition of RyR with ruthenium red (20 μmol/L). SR Ca2+ load shown in gray with blink nadir shown in black (B and C). *P<0.05.
Changes in cytosolic [Ca2+] and [Ca2+]SR were measured simultaneously with a laser-scanning confocal microscope (Radiance 2000 MP, Bio-Rad) equipped with a ×40 oil immersion objective lens (NA=1.3). Fluo-5N was excited with the 488-nm laser line of an argon ion laser, and fluorescence was measured at 515±15 nm. Rhod-2 was excited with the 543-nm laser line of a HeNe laser and fluorescence was measured at wavelengths >600 nm. Images were acquired in line scan mode (3 ms per scan; pixel size, 0.12 μm). Ca2+ sparks were detected and analyzed using SparkMaster.23 For each detected Ca2+ spark, corresponding changes of the Fluo-5N signal were analyzed. The profiles of local [Ca2+]SR depletions (Ca2+ blinks) were fitted with the product of 2 exponential functions to the decay and recovery phase, respectively, as described previously for spark24 and blink analysis.6 Blink amplitudes and kinetic parameters were obtained from the fit of the experimental data. Amplitudes of sparks and blinks are expressed as F/F0, where F0 is the initial fluorescence before release. All Fluo-5N signals (blinks) were corrected for the Ca2+-insensitive component of the Fluo-5N fluorescence after complete SR Ca2+ depletion by 10 mmol/L caffeine. This concentration of caffeine fully activates RyRs25 and leads to the synchronized release of the total Ca2+ stored in the SR. Thapsigargin was used to block the sarcoplasmic Ca2+ ATPase (SERCA). The complete inhibition of SERCA was confirmed by repeated SR Ca2+ load measurements using rapid application of 10 mmol/L caffeine. Thapsigargin (10 μmol/L) completely inhibited SERCA in permeabilized cells within 2 minutes (data not shown).
The maximum SR Ca2+ release flux was calculated from the peak of the first derivative of the cytosolic fluorescence intensity and expressed as d(ΔF/F0)/ms13,20,26,27 where ΔF=F–F0. Although this approach lacks the accuracy of methods previously used to calculate release flux,17,28 the waveform of directly measured SR Ca2+ release using the “Ca2+ spike” method is very similar to the d(ΔF/F0)/dt waveform.27 This method therefore provides a valid measure of the SR Ca2+ release flux.
To assess the degree of interconnection between individual SR Ca2+ release sites, the rate of Fluo-5N photobleaching was measured. [Ca2+]SR and cytosolic [Ca2+] were simultaneously measured as described above but the excitation intensity of the 488-nm laser line was 10 times higher than for normal scans to induce Fluo-5N photobleaching. No obvious cellular damage was induced using this protocol. We only compared photobleaching in SR release sites that showed approximately the same Fluo-5N fluorescence intensity before bleaching. The resulting rate of Fluo-5N signal decay is determined by Fluo-5N photobleaching and Fluo-5N diffusion from neighboring SR regions that replenish bleached Fluo-5N. Because the rate of Fluo-5N photobleaching is considered identical for all bleached release sites in an image, differences in the decay rate of the fluorescence intensity result from differences in Fluo-5N diffusion that occurs during the photobleaching. The decay rate can therefore be considered an index of SR connectivity for individual SR release sites (fast decay rates denoting little interconnection and slow decay rates denoting extensive interconnection, respectively). The time course of the Fluo-5N fluorescence intensity decline during photo-bleaching could be well-fitted using a monoexponential function.
Global cytosolic Ca2+ transients and [Ca2+]SR depletions were measured in intact, field-stimulated rabbit ventricular myocytes after loading with Fluo-5N/AM or Rhod-2/AM, respectively.19 To avoid motion artifacts, the scan line was positioned along the short axis in the central region of the cell where cell motion is minimal during contraction. Stimulation frequency was 0.5 Hz.
All chemicals were purchased from Sigma-Aldrich (St Louis, Mo). Fluo-5N/AM and Rhod-2 were purchased from Molecular Probes/Invitrogen (Carlsbad, Calif).
Data are presented as means±SEM of n measurements. Statistical comparisons between groups were performed with the Student t test. Differences were considered statistically significant at P<0.05. Coefficient of variation was calculated as SD/mean.
Results
Properties of Ca2+ Sparks and Blinks Under Control Conditions
To investigate the role of [Ca2+]SR depletion in terminating local SR Ca2+ release (Ca2+ sparks), we simultaneously measured cytosolic [Ca2+] and concurrent intra-SR [Ca2+] depletions (Ca2+ blinks6) in isolated ventricular myocytes that were permeabilized using standard protocols (Figure 1A).20–22
Figure 1.
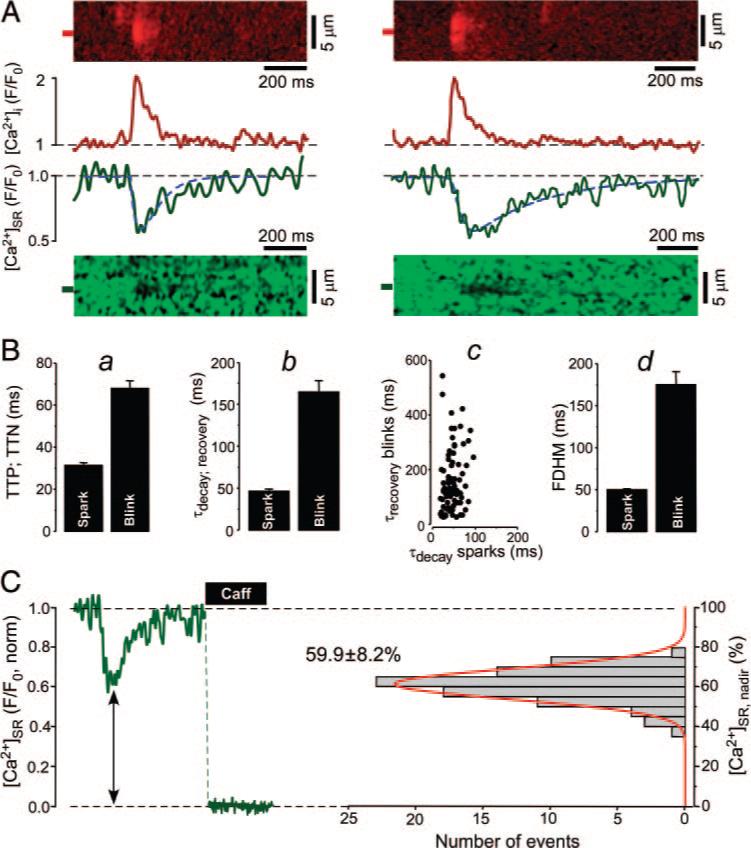
Properties of Ca2+ sparks and corresponding Ca2+ blinks. A, Two examples (left and right) of simultaneously measured Ca2+ sparks and Ca2+ blinks recorded from different release sites. Top, Normalized (F/F0) line scan images of Rhod-2 fluorescence and corresponding profiles of Ca2+ sparks obtained by averaging fluorescence from the 1-μm-wide regions marked by red boxes. Bottom, Normalized line scan images of Fluo-5N fluorescence and corresponding profiles of Ca2+ blinks obtained by averaging fluorescence from the regions marked by green boxes. Blink profiles were fitted by exponential functions to the rising and decay phase, respectively (dashed blue line). B, a, Average time-to-peak (TTP) of sparks and time-to-nadir (TTN) of blinks. b, Average time constants of spark decay and blink recovery. c, Relationship between the time constants of spark decay and blink recovery. d, Average FDHM of sparks and blinks. C, Left, Fluo-5N signal during Ca2+ blink and complete depletion of the SR after the application of 10 mmol/L caffeine. Right, Distribution and mean±SD of [Ca2+]SR at the nadir of blinks ([Ca2+]SR, nadir). Histogram was fitted with a single Gaussian function (red line).
On average, the time-to-nadir of Ca2+ blinks was approximately twice as long as the time-to-peak of the associated Ca2+ sparks (68±4 versus 31±1 ms; 86 events; P<0.05; Figure 1B, a). This demonstrates that SR Ca2+ release terminates well after the peak of a Ca2+ spark (also see elsewhere6,12). The average time constant of local SR Ca2+ refilling after release was ≈3.5 times slower than the cytosolic [Ca2+] decline of the corresponding Ca2+ spark (161±13 versus 46±2 ms; 86 events; P<0.05; Figure 1B, b) and did not correlate with spark decay time (R2=0.14; Figure 1B, c). This is also evident in Figure 1A where the 2 Ca2+ sparks had essentially the same decay time constants (48 and 50 ms), whereas the recovery time constants of the blinks were 101 and 351 ms, respectively. The longer time-to-nadir and slower [Ca2+]SR refilling resulted in a ≈3.5 times longer Ca2+ blink duration (full duration at half-maximum [FDHM]) compared to Ca2+ sparks (175±15 versus 50±1 ms; 86 events; P<0.05; Figure 1B, d). The blink duration had a much higher variability than the more stereotypical Ca2+ spark (ie, the coefficient of variation [SD/mean] was 4 times higher for Ca2+ blink duration than for Ca2+ spark duration).
Despite the high variability in Ca2+ blink kinetics, [Ca2+]SR at the blink nadir showed remarkably little variation (59.9±0.9% of caffeine releasable Ca2+; 86 events; Figure 1C). Indeed, there was no correlation between blink nadir [Ca2+]SR and either SR Ca2+ release flux [d(ΔF/F0)/dt]or spark width (μm), but larger release flux correlated significantly with faster blink recovery rates (data not shown). Here, and elsewhere blink nadir is reported as percentage [Ca2+]SR with respect to diastolic [Ca2+]SR under control conditions (taken as 100%).
Global [Ca2+]SR depletions in intact cardiomyocytes during excitation–contraction coupling had a duration that was only slightly longer than Ca2+ blinks (FDHM, 323±14 ms) and also exhibit a relatively similar constant nadir at 63.4±2.8% (Figure 2, n=6 cells; also see elsewhere7,19). In contrast, Ca2+ sparks are much briefer and smaller in amplitude compared to global cytosolic Ca2+ transients. The finding that a substantial amount of Ca2+ remains in the SR at the end of the release rules out total SR Ca2+ depletion as the mechanism of Ca2+ spark termination. Rather, the consistent [Ca2+]SR at the blink nadir and the known luminal Ca2+-sensitivity of RyR gating29,30 suggests that release terminates when [Ca2+]SR decreases to a critical threshold.
Figure 2.
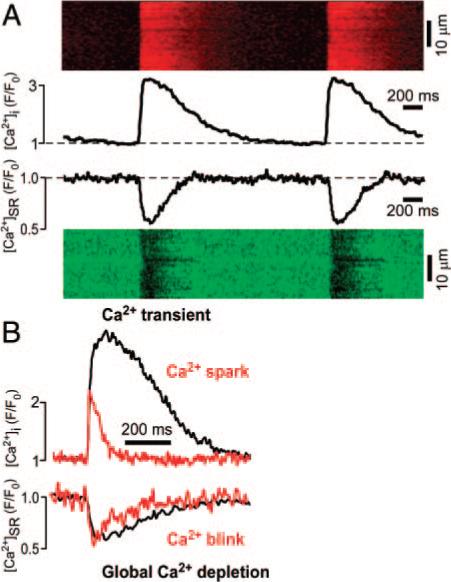
Global and local SR Ca2+ release events. A, Global cytosolic Ca2+ transients and [Ca2+]SR depletions measured in field-stimulated, intact rabbit ventricular cardiomyocytes at a stimulation frequency of 0.5 Hz. Measurements were performed in different cells and time-aligned to illustrate the relationship between Ca2+ transients and global SR Ca2+ depletions. Line scan images and profile plots (obtained by averaging the entire width of the line scan) are shown. B, Comparison between cytosolic [Ca2+] and [Ca2+]SR depletion on a global (cytosolic Ca2+ transients and global intra-SR Ca2+ depletion, black traces) and local level (Ca2+ spark and Ca2+ blink, red traces). Original traces representative for average experimental values are shown.
To test whether the residual variability of [Ca2+]SR depletion during Ca2+ sparks was characteristic of an individual release site or whether variability occurred randomly, we recorded repeated release events from the same site. At a given release site termination occurred at a very consistent [Ca2+]SR (Figure 3A and 3B) even when release started from a lower initial [Ca2+]SR early after a preceding event, eg, second event in Figure 3B. From release site to release site, however, the [Ca2+]SR at the nadir of a blink varied over a modest range (between 43% and 73% in 48 different sites, average depletion was to 59.2±1.0%; Figure 3C). Notably, despite some variation in [Ca2+]SR at the nadir, the blink nadir of repeated release events at a given site was highly correlated (R2=0.85; n=48). This is consistent with a critical [Ca2+]SR threshold being the main factor responsible for SR Ca2+ release termination.
Figure 3.
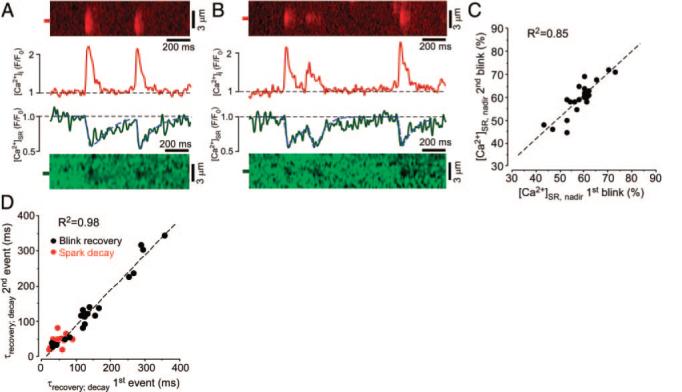
Ca2+ blinks originating from the same release site. A, Line scan images and corresponding profiles of 2 consecutive Ca2+ sparks and blinks originating from the same release site. Profiles of sparks and blinks were obtained by averaging fluorescence from the regions marked by the 1-μm-wide red and green boxes, respectively. B, Similar recordings of multiple release events and corresponding blinks from a different SR Ca2+ release site. C, Relationship between [Ca2+]SR at the nadir of consecutive blinks ([Ca2+]SR, nadir), originating from the same release site. D, Relationship between blink recovery time constants (black data points) and spark decay time constants (red data points) of consecutive release events from the same release site. Time constants were measured from concurrent sparks and blinks.
In contrast, the Ca2+ refilling time constants of Ca2+ blinks varied at different release sites over more than a 10-fold range (values varied from 30 to 358 ms in 48 successively measured blinks; average time constant, 139±14 ms, SD/mean=0.668) but were remarkably consistent at each individual release site (R2=0.98; n=48; Figure 3D, black data points). In comparison, the decay time constants of the simultaneously measured Ca2+ sparks varied over a much narrower range (average time constant, 44±2 ms; SD/mean=0.318; Figure 3D, red data points).
These results show that the rate of local [Ca2+]SR replenishment varies throughout the SR but is constant for any given release site. The variability of blink recovery among release sites (despite a similar extent of depletion) and the consistency of blink recovery at a given site, could either result from differences of Ca2+ uptake into the SR via the SERCA or could result from differences in intra-SR diffusion between release sites. These 2 possibilities were tested in the following experiments.
Ca2+ Sparks and Blinks at Decreased SR Ca2+ Load
To test whether the [Ca2+]SR threshold for SR Ca2+ release termination depends on the initial [Ca2+]SR before release occurs, we performed experiments in which we varied SR Ca2+ load over a wide range. To decrease SR Ca2+ load, we applied the selective SERCA inhibitor thapsigargin (10 μmol/L), which completely inhibits SR Ca2+ uptake within 2 minutes (see Materials and Methods). On thapsigargin application, [Ca2+]SR and Ca2+ spark frequency progressively decreased until Ca2+ sparks ceased when [Ca2+]SR had declined to ≈50% (data not shown). Interestingly, this [Ca2+]SR was close to the termination threshold of Ca2+ blinks and may be an additional manifestation of the same RyR-gating regulation by [Ca2+]SR.
As resting [Ca2+]SR declined after thapsigargin application, the amplitudes of sparks and blinks, as well as the peak SR Ca2+ release flux, progressively decreased. Average values of release flux under different experimental conditions are presented in the Table. However, complete SERCA inhibition by thapsigargin did not prevent local [Ca2+]SR recovery during blinks. [Ca2+]SR at the blink nadir remained constant at 62.3±0.9% of the initial SR Ca2+ load under control conditions, consistent with a critical absolute [Ca2+]SR threshold for release termination that is independent of the initial [Ca2+]SR and release flux (Figure 4A). At release sites where repeated sparks and blinks could be measured at different initial [Ca2+]SR (Figure 4B, abscissa), [Ca2+]SR at the nadir did not change (Figure 4B, ordinate) as the initial SR Ca2+ load decreased. When data were grouped according to the initial SR Ca2+ load (Figure 4C), the [Ca2+]SR at the nadir during blinks was constant (black bars in Figure 4C). These results are in line with the results from the previous experiments showing that termination occurred at the same [Ca2+]SR threshold when [Ca2+]SR had not fully recovered after a preceding release (Figure 3B; second event). Thus, even when the initial SR Ca2+ load is low and release flux is low, release terminates at the same absolute [Ca2+]SR as under control conditions.
Table.
SR Ca2+ Release Flux and Corresponding [Ca2+]SR at the Blink Nadir Measured Under Different Experimental Conditions
| SR Ca2+ Release Flux [d (ΔF/F0)/ms] | [Ca2+]SR at the Blink Nadir (% of Initial [Ca2+]SR at Control) | No. of Events | |
|---|---|---|---|
| Control | 0.049±0.002 | 59.9±0.9 | 89 |
| Thapsigargin | 0.036±0.03* | 62.3±0.9 | 17 |
| High Ca2+ | 0.070±0.05* | 55.8±2.4 | 14 |
| Tetracaine | 0.029±0.02*† | 90.5±2.0*† | 24 |
Thapsigargin: 10 μmol/L; high Ca2+: 200 nmol/L; tetracaine: 0.7 mmol/L.
P<0.05 vs control
P<0.05 vs Thapsigargin
Figure 4.
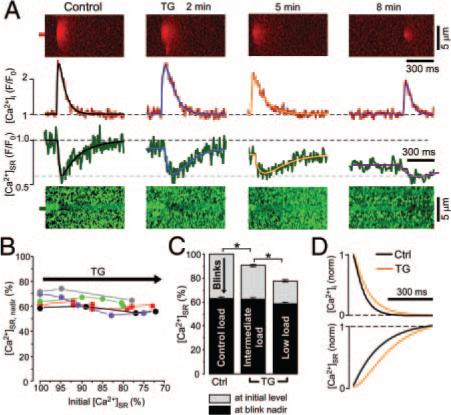
Ca2+ sparks and blinks at decreased [Ca2+]SR. A, Line scan images and corresponding profiles of Ca2+ sparks and blinks originating from the same release site under control conditions and at different times following thapsigargin (TG) (10 μmol/L) application. Spark and blink profiles were fitted with a double exponential function (black, blue, orange, and purple lines). B, Relationship between the initial [Ca2+]SR and the [Ca2+]SR at the blink nadir ([Ca2+]SR, nadir). Data were obtained from different Ca2+ release sites (indicated by different colors) during thapsigargin application and were normalized to the initial SR Ca2+ load before thapsigargin application (100%). C, Summary of results shown in B with data grouped into control (100%), intermediate (85% to 95%), and low (70% to 80%) initial [Ca2+]SR level. Initial [Ca2+]SR (SR Ca2+ load before release) represented in gray with [Ca2+]SR at the blink nadir shown in black. D, Normalized decay of Ca2+ sparks and corresponding recovery of blinks under control conditions (black) and in the presence of 10 μmol/L thapsigargin (orange). The profiles were obtained from exponential fits of experimental traces in A (control and thapsigargin, 5 minutes). *P<0.05.
Contribution of Ca2+ Uptake and Intra-SR Diffusion to Ca2+ Blink Recovery
With SR Ca2+ uptake blocked by thapsigargin, local [Ca2+]SR recovery must be attributable to diffusion of Ca2+ from neighboring SR regions. Figure 4D shows the normalized Ca2+ spark decay and the corresponding blink recovery under control conditions and in the presence of 10 μmol/L thapsigargin (exponential fits taken from Figure 4A; control and thapsigargin, 5 minutes). SERCA blockade increased the time constant of spark decline by 36.2±7.2% (n=5), in agreement with previous data indicating that the cytosolic [Ca2+] decline of sparks is dominated by cytosolic Ca2+ diffusion rather than Ca2+ reuptake into the SR.31 Furthermore, thapsigargin increased the time constant of local [Ca2+]SR refilling by a similar 33.4±7.6% (n=5; Figure 4D). These data indicate that diffusion of Ca2+ within the SR network rather than SR Ca2+ uptake dominates recovery of local [Ca2+]SR. Therefore, the large variation in [Ca2+]SR recovery kinetics between individual release sites results mainly from differences in local intra-SR Ca2+ diffusion. Better interconnected SR release sites within the SR (ie, where diffusion is faster between that junction and the rest of the SR) would therefore exhibit faster blink recovery as intra-SR Ca2+ diffusion is facilitated. Conversely, more isolated SR release sites would show slower local [Ca2+]SR refilling.
Ca2+ Sparks and Blinks at Increased SR Ca2+ Load
To complement the experiments with decreased [Ca2+]SR, we next measured Ca2+ blinks at increased SR Ca2+ load. For this purpose, cytosolic [Ca2+] was elevated from 150 to 200 nmol/L (thereby enhancing SERCA activity32). This increased [Ca2+]SR to 110.4±2.8% (n=12; P<0.05; Figure 5A and 5B), increased Ca2+ spark frequency by 46% [from 9.7±0.9 to 14.2±1.4 sparks×(100 μm)−1×sec−1; n=12; P<0.05], and increased maximal SR Ca2+ release flux by 43% [from 0.049±0.002 (n=89) to 0.070±0.05 d(ΔF/F0)/ms; n=14; P<0.05; Table]. Despite the increased [Ca2+]SR and maximal SR release flux, release still terminated at a similar [Ca2+]SR (56±2%; n=14) as control (60±1%; n=89; Figure 5B and Table). These results argue for a stable absolute [Ca2+]SR termination threshold that is not affected by the initial SR Ca2+ load and not regulated by the SR release flux which was altered in these experiments over a wide range (Table).
Ca2+ Sparks and Blinks at Partially Inhibited RyRs
We have shown previously that partial inhibition of SR Ca2+ release flux in combination with increased SR Ca2+ load induces long-lasting release events in some SR Ca2+ release sites.20 These prolonged release events can sustain a constant SR Ca2+ release flux for several hundred milliseconds before termination occurs. We simultaneously measured cytosolic [Ca2+] and intra-SR [Ca2+] at partially inhibited RyRs to: (1) test the hypothesis that additional releasable Ca2+ can become available from neighboring SR regions via intra-SR Ca2+ diffusion, when the release site is well connected within the SR network; and (2) test whether the termination of prolonged release events is [Ca2+]SR-dependent or -independent.
We partially inhibited RyRs using tetracaine, Mg2+, or ruthenium red at concentrations that decrease the open probability (Po) of RyRs reconstituted in lipid bilayers by 80% to 90%.20 We and others have shown previously that these RyR inhibitors decrease RyR Po without changing mean open time, single-channel amplitude or inducing subconductance states.20,33,34 SR Ca2+ release flux during local release events in the presence of tetracaine was significantly reduced compared to control conditions or to experiments when SR Ca2+ load was depleted with thapsigargin (Table).
Partial RyR inhibition initially abolished SR Ca2+ release, but as [Ca2+]SR increased, spark frequency gradually recovered to a new steady state within 4 to 6 minutes because of RyR activation via higher [Ca2+]SR [spark frequency decreased from 9.3±1.2 sparks×sec−1×(100 μm)−1 under control conditions to 4.0±1.2 sparks×sec−1×(100 μm)−1 6 minutes after tetracaine application; n=10; P<0.05]. Under these conditions, elementary release events with durations similar to sparks under control conditions occurred, but additional long-lasting Ca2+ release events originating from some of the SR Ca2+ release sites were observed.20 Figure 5C shows Ca2+ sparks from different release sites and corresponding local [Ca2+]SR depletions before and after tetracaine application, including long-lasting release events. Tetracaine (0.7 mmol/L, 6 minutes) increased SR Ca2+ load to 119±2% (n=8; Figure 5D). Intra-SR Fluo-5N was not saturated under these conditions because fluorescence could be further increased to 136±5% (n=10) by SERCA stimulation using the PKA catalytic subunit (5 U/mL) and complete RyR inhibition with 20 μmol/L ruthenium red (Figure 5D).
When the release flux was decreased by partial RyR inhibition, the decrease in [Ca2+]SR during release failed to reach the nadir seen for control blinks (Figure 5C and 5D). Short and prolonged release events in the presence of tetracaine terminated at 85±4% (n=10) and 94±3% (n=14) of the initial [Ca2+]SR under control conditions, respectively, much higher than for blinks observed in the absence of RyR inhibition (59.9±0.9%; n=86; P<0.05). We infer that the threshold [Ca2+]SR for release termination may be higher in the presence of tetracaine (which is known to alter RyR Ca2+ sensitivity35). During long-lasting release events (second and fourth image in Figure 5C), [Ca2+]SR decreased to a relatively stable level from which termination eventually occurred, suggesting a [Ca2+]SR- and [Ca2+]i-independent mechanism of release termination (because neither [Ca2+] is changing). The sites that showed long-lasting SR Ca2+ release events in the presence of tetracaine could have maintained local [Ca2+]SR during release because at a reduced SR Ca2+ release flux, there may be sufficient luminal Ca2+ diffusion from neighboring SR regions to prevent [Ca2+]SR from decreasing to the critical [Ca2+]SR threshold that terminates release. This could occur if these release sites are especially well connected within the SR. Direct evidence for intra-SR diffusion and Ca2+ supply from neighboring release sites is presented in the right image of Figure 5C, which shows [Ca2+]SR depletion in a quiescent SR region ≈1.8 μm lateral to a release site where prolonged Ca2+ release occurred (marked in orange). The distance at which the depletion in the neighboring SR region was observed was close to the sarcomere length and indicates that the depletion was indeed spreading to a neighboring release site. Similar [Ca2+]SR depletion in quiescent neighboring junctions was observed during 5 long-lasting release events. Additionally, we found that the time constant of [Ca]SR recovery was much faster after long release events (τ=73.4±11.8 ms; n=9) than after short events (τ=113.4±29.2 ms; n=10; P<0.05) in the presence of tetracaine. This provides evidence that faster diffusional SR refilling occurs at sites where long-lasting events occur. Next, we tested directly whether SR release sites that show long-lasting release events with partial RyR inhibition are especially well connected within the SR network.
Heterogeneity of SR Ca2+ Release Site Interconnection Within the SR
If the long-lasting events evoked by partial RyR inhibition correlate with more extensive Ca2+ release site interconnection within the SR (ie, limiting local depletion), then Fluo-5N should also diffuse more rapidly into these sites during local Fluo-5N photobleaching. To test this hypothesis, we locally photobleached intra-SR Fluo-5N and compared the fluorescence decline kinetics of release sites which showed prolonged release events with sites that exhibited short release events during partial RyR inhibition.
The 2 traces at the top of Figure 6A show the cytosolic fluorescence at 2 different release sites along the same scan line in a permeabilized myocyte. Release flux was partially inhibited with 0.7 mmol/L tetracaine (Table). The long-lasting release event originating from site 1 (s1, black trace) had a duration of ≈500 ms, whereas the 2 short release events occurring at site 2 (s2, gray trace) were comparable to typical sparks (FDHM≈80 ms). The bottom of Figure 6A shows the simultaneously measured Fluo-5N fluorescence intensity, which was similar at both sites at the beginning of the measurement. When the laser power in these experiments was increased to induce photobleaching, the Fluo-5N fluorescence decreased with an exponential time course at both sites. Both the time course and the extent of bleaching were different at the 2 sites. Despite the photobleaching, the [Ca2+]SR depletions, which were associated with the release events shown in the top trace are easily identifiable. The SR Ca2+ release site exhibiting the long-lasting release event (s1, black) had slower and less extensive decay of fluorescence than the site from which the short release events originated (s2, gray). The relationship between the time constant of Fluo-5N fluorescence decline and the duration of cytosolic release events measured in 35 different release sites is presented in Figure 6B. Both parameters correlated remarkably well (R2=0.78), and a similar correlation was seen for the extent of photo-bleaching during a 5 second period (data not shown). A potential explanation for the Fluo-5N photobleaching variability is presented schematically in Figure 6A (right). The SR is organized as a network of release junctions connected by free SR. It has been shown that the typical SR junction connects to the free SR at 4 or 5 locations along its periphery and that some junctions may have fewer connections,6 thus indicating variability in connectivity of individual SR junctions. A release site that is well connected within the SR network (s1, top) shows a slower and less extensive decrease in fluorescence because diffusion from neighboring sites replenish the dye into the release site that is being bleached. In contrast, if a release site is relatively isolated within the SR (s2, bottom), diffusion of Fluo-5N into this site is restricted, resulting in a faster and more extensive decline in fluorescence during photobleaching. These results suggest that the wide variation in Ca2+ spark/blink duration at partial RyR inhibition (Figure 6B) and that the variation in the Ca2+ blink recovery time constant (Figures 1 and 3) reflects variance in the extent of individual SR Ca2+ release site interconnection within the SR network.
Figure 6.
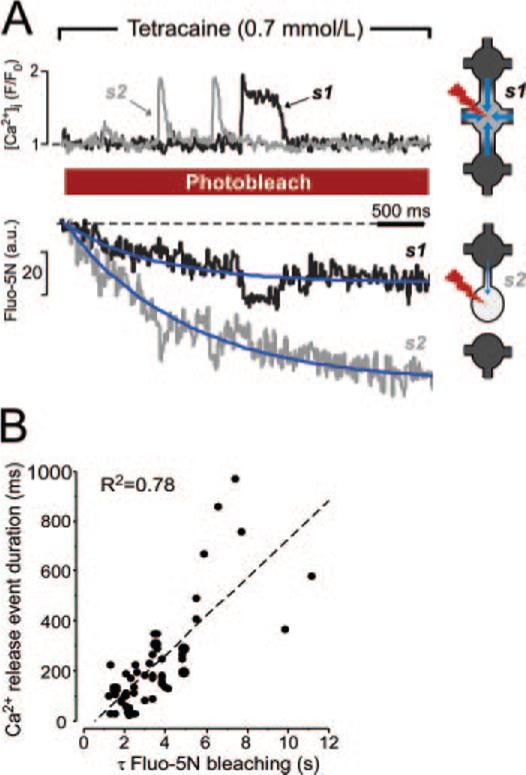
Local Fluo-5N photobleaching in permeabilized myocytes. A, Left, Ca2+ sparks and corresponding blinks during the Fluo-5N bleaching protocol. Recordings were made from 2 different SR Ca2+ release sites (s1, black trace; s2, gray trace) along the same scan line during 0.7 mmol/L tetracaine application. Experimental data were fitted by a monoexponential function (blue lines). Right, Potential mechanism of Fluo-5N bleaching variability. Fluorescence in a well-connected SR release site (s1) decays slowly because of fast diffusion of dye (blue arrows) from adjacent regions of the SR. An isolated SR release site (s2) bleaches fast because of restricted diffusion of the dye. The degree of intra-SR connectivity was symbolized by the number of free SR connections and diameter of free SR. B, The relationship between Ca2+ spark duration and the rate of Fluo-5N bleaching after tetracaine application.
Discussion
Robust termination of SR Ca2+ release is required to ensure stable physiological excitation–contraction coupling in the heart. Failure of SR Ca2+ release termination has been linked to arrhythmogenesis in both acquired (eg, hypertrophy and heart failure)36,37 and inherited pathological conditions (eg, catecholaminergic polymorphic ventricular tachycardia).38 However, despite intensive study, the mechanism of release termination in cardiac muscle is still unclear.
In the present study, we investigated the importance of local [Ca2+]SR depletion and release flux in the termination of elementary SR Ca2+ release in permeabilized rabbit ventricular cardiomyocytes. The main findings were: (1) local [Ca2+]SR depletions during sparks were similar in amplitude and duration to global SR Ca2+ depletions; (2) under control conditions, SR Ca2+ release terminated when the local [Ca2+]SR declined to a fixed absolute termination threshold; (3) this termination threshold was independent of the initial [Ca2+]SR and release flux; and (4) decreasing the release flux by inhibiting RyR led to the occurrence of long-lasting SR Ca2+ release events that originated from especially well-connected SR release sites and terminated independently of [Ca2+]SR depletion.
Intra-SR Ca2+ Diffusion Shapes the Kinetics of Ca2+ Blinks
A remarkable feature of Ca2+ blinks is that on average they are similar in amplitude and duration to global SR Ca2+ depletions, which occur when release is triggered simultaneously at all release sites during excitation–contraction coupling (Figure 2 and elsewhere6,7,19). This contrasts sharply with Ca2+ sparks, which are much smaller in amplitude and duration than Ca2+ transients. The explanation may be that Ca2+ sparks summate extensively in space and time during the synchronized activation of SR Ca2+ release during excitation–contraction coupling, forming the high-amplitude cell-wide Ca2+ transient. In contrast, most of the Ca2+ released during excitation–contraction coupling or during a spark is available locally from individual SR release sites. Because Ca2+ diffusion in the SR is slower than in the cytosol,39–41 and [Ca2+]SR is buffered by calsequestrin only minor spatiotemporal summation or overlap of intra-SR release events occurs, resulting in the relative similarity of local and global SR Ca2+ depletions. Also, Ca2+ spark decline and Ca2+ blink recovery kinetics are substantially influenced by cytosolic and intra-SR Ca2+ buffering, respectively. Increased intra-SR Ca2+ buffering (eg, by overexpressing calsequestrin) leads to a slower [Ca2+]SR recovery after the nadir of a blink.14 Ca2+ blink recovery was only modestly prolonged when SERCA activity was completely inhibited (Figure 4), clearly showing that intra-SR Ca2+ diffusion rather than Ca2+ uptake into the SR is the main factor responsible for local [Ca2+]SR recovery. Although the entire SR network and nuclear envelope are part of one large Ca2+ compartment throughout which diffusion occurs,39,41 the large variability in blink recovery kinetics (Figures 1A, 1B, and 3D) indicates that there is substantial variation in how isolated a particular SR release site is and how well diffusion can occur between neighboring release sites. A junction that is well connected within the SR and allows for faster intra-SR Ca2+ diffusion would then replenish faster than a more isolated junction. The fact that we found consistent recovery kinetics for any given release site (Figure 3D) suggests that the variation in release site recovery is based on relatively fixed structural inhomogeneities rather than on alternating functional or stochastic differences. The importance of release site interconnection for release termination is discussed below.
The properties of the Ca2+ blinks described here differ substantially from the initial description of Ca2+ blinks by Brochet et al,6 who described blinks with much faster kinetics similar to the kinetics of cytosolic Ca2+ sparks. Several factors may be responsible for these differences: the present study was performed in permeabilized myocytes, whereas the study by Brochet et al was performed in intact myocytes. Although certain cytosolic proteins may be washed out during permeabilization, it is unlikely that the fundamental differences in blink properties can be attributed to the cell permeabilization because Ca2+ spark kinetics are virtually identical in intact and permeabilized ventricular cardiomyocytes.21 It is also possible that Brochet et al preferentially detected smaller and shorter depletions, especially in experiments in which Ca2+ blinks were not measured simultaneously with Ca2+ sparks. All Ca2+ blinks in our study were measured simultaneously with cytosolic [Ca2+], which enabled us to unequivocally prevent false-positive Ca2+ blink detections (which may be of short duration). Finally, the Ca2+ blink characteristics here agree closely with other published data.14,36
[Ca2+]SR Depletion to a Fixed Threshold Terminates SR Ca2+ Release
A major finding of the present study is that SR Ca2+ release robustly terminated at a seemingly fixed [Ca2+]SR threshold of ≈60% of the diastolic [Ca2+]SR value under control conditions. The termination threshold was stable for individual SR Ca2+ release sites but varied to some extent from site to site. These results confirm previous experimental findings6,8,14 and predictions from mathematical modeling12,42 that the termination involves changes in RyR gating rather than exhaustion of releasable Ca2+. Our findings also make random release termination (such as stochastic attrition43) unlikely, because this would result in varying termination levels.
Both cytosolic [Ca2+] and [Ca2+]SR are known to regulate RyR gating9,10 through modulation of activation, as well as termination of SR Ca2+ release.18,29,44,45 Therefore, it is likely that termination is the consequence of a dynamic balance between luminal and cytosolic [Ca2+] regulation of RyRs during Ca2+ release. The [Ca2+] in the dyadic cleft is mainly determined by the SR Ca2+ release flux,18 which, in turn, is determined by SR Ca2+ loading and the number of active channels in a release site. Our results, however, argue against an important dynamic regulation of spark termination by [Ca2+]cleft because the threshold [Ca2+]SR for SR release termination remained constant when the SR Ca2+ load and therefore the release flux was varied over a wide range (Figures 4 and 5A and Table). These findings suggest that partial depletion of intra-SR [Ca2+] to a fixed threshold represents the main mechanisms of cardiac Ca2+ spark termination via a [Ca2+]SR-dependent mechanism.
Release Termination at Partial RyR Inhibition
Partial RyR inhibition has 2 important consequences that influence the termination process. Firstly, the number of active RyRs (or total RyR Po) in a SR release site is substantially reduced, leading to a decreased SR Ca2+ release flux and [Ca2+]cleft for any given SR Ca2+ load in comparison to control conditions. Secondly, because SERCA activity remains unaltered by selective RyR inhibition, the SR Ca2+ content increases.
As in the experiments with elevated cytosolic Ca2+ (Figure 5A), release started from a similarly elevated SR Ca2+ load. However, release did not terminate at the [Ca2+]SR threshold that we consistently observed in the absence of RyRs inhibition (Figures 1, 3, 4, and 5A). Rather, termination occurred at a much higher [Ca2+]SR in both short and prolonged release events. Tetracaine acts primarily by inhibiting RyR activation rather than by promoting channel closing in reconstituted RyRs in lipid bilayers (prolonging closings without changing mean open time).33,35 Thus, it is likely that tetracaine directly alters the threshold at which SR Ca2+ release terminates. This may be why brief Ca2+ blinks under this condition terminated at higher [Ca2+]SR in tetracaine versus control conditions. We cannot exclude the possibility that the reduced SR Ca2+ release flux with tetracaine causes less RyR stimulation by [Ca2+]cleft (which could contribute to release termination at a smaller [Ca2+]SR depletion). However, this would require a very steep relationship between release flux and RyR stimulation by [Ca2+]cleft, because the reduction in SR Ca2+ release flux that occurred at low SR Ca2+ loads after SERCA inhibition did not influence the termination threshold.
Intra-SR Ca2+ Diffusion Provides [Ca2+]SR for Prolonged Release Events
A remarkable feature of prolonged release events is that [Ca2+]SR decreases to a stable level from which termination occurs (Figure 5C, fourth image). The first question that arises is how a single release site can supply sufficient Ca2+ to maintain ongoing SR Ca2+ release over several hundred milliseconds without getting depleted. The answer likely involves the extent of interconnection that individual release sites have within the SR. As discussed above, differences in intra-SR interconnection determine the kinetics of [Ca2+]SR refilling after the nadir of a blink. In addition, a well connected release site may also draw Ca2+ from a neighboring SR region and limit local [Ca2+]SR decline, allowing continued release because the [Ca2+]SR termination threshold is not reached when the release flux is low. Supporting data for this hypothesis is shown in the fourth image of Figure 5C: the long-lasting release event does not only deplete its own junction but also partially depletes a neighboring junction. Although this local intra-SR Ca2+ diffusion would also be expected to occur in well-connected release sites under control conditions, it may be difficult to detect experimentally because of the brevity of corresponding blinks and the smaller depletion expected at that time and distance. Under control conditions there was a significant correlation between release flux and spark width, such that some sites release more total Ca2+ (despite unaltered [Ca2+]SR nadir). These sites of higher release flux exhibited faster blink recovery rates, consistent with the idea that larger amounts of Ca2+ release are in fact enabled by better connected SR (ie, greater diffusive flux through the region before the release threshold is reached). Otherwise greater Ca2+ release might be expected to result in slower refilling.
The photobleach experiments further test whether the sites that exhibit long-lasting release events (with tetracaine) are especially well connected within the SR network. Similar to intra-SR Ca2+, Fluo-5N can diffuse freely within the sarcoplasmic reticulum.39,46 The rate of local Fluo-5N photo-bleaching provides direct insight into the functional interconnection of individual release sites within the SR: isolated release sites would exhibit faster and more extensive fluorescence decrease during photobleaching, whereas SR release sites that are well connected with neighboring SR would show slower and less extensive decrease in fluorescence because intra-SR Fluo-5N diffuses into the release sites that are being bleached (see schematic diagram in Figure 6A, right). The junctions where partial RyR inhibition led to long-lasting release events had photobleaching characteristics expected for well-connected release sites. On the other hand, the bleaching kinetics of release sites that showed short release events in the presence of partial RyR inhibition had the hallmarks of isolated domains. These data support our previous conclusion that there are substantial differences in the degree of release site interconnection within the SR which was based on the large variation of Ca2+ blink recovery kinetics and the relatively small contribution of Ca2+ uptake to local [Ca2+]SR refilling (Figures 1, 3, and 4). Well-connected release sites therefore have a functionally higher [Ca2+]SR reserve because Ca2+ can be supplied from neighboring release sites. In this sense, the prolongation of SR Ca2+ release with partially inhibited RyRs and increased [Ca2+]SR resembles the prolongation of release at enhanced intra-SR Ca2+ buffering.13,47 At enhanced intra-SR buffering, additional Ca2+ is available for release from the buffers in the SR release site and this delays approach to the termination [Ca2+]SR termination threshold.
Termination of Prolonged SR Release Events: [Ca2+]SR-Independent Termination of Release
The remaining question is how long-lasting release events terminate. The long duration of a relatively sustained [Ca2+]SR depletion from which termination occurs (Figure 5C, fourth image) would suggest a slower, [Ca2+]SR- and [Ca]i-independent mechanism of release termination. One such mechanism is stochastic attrition, where the simultaneous closure of all RyRs in a release cluster leads to release termination by breaking the inherent positive feedback of Ca2+-induced Ca2+ release. Because stochastic attrition is very sensitive to the number of RyRs available for activation,43 it is probably not relevant for the termination of SR Ca2+ release under control conditions. However, this mechanism may become relevant for Ca2+ spark termination during RyR inhibition when a smaller than normal number of RyRs are active. Additionally, RyR adaptation15 may be involved in the termination of prolonged SR Ca2+ release events because the RyRs in a junction that exhibits a long-lasting release event are exposed to a relatively high cleft [Ca2+] for a long period of time. Adaptation was initially described to occur with a very slow time constant (>1 second) but can also occur on a timescale relevant for the termination of prolonged release events.48
In conclusion, this study demonstrates that SR Ca2+ release terminates at a critical [Ca2+]SR threshold that is not dynamically regulated by SR Ca2+ release flux or cleft [Ca2+]. Intra-SR Ca2+ diffusion can supply Ca2+ from neighboring SR regions and thereby maintain SR Ca2+ release when the release flux is low. Multiple release termination mechanisms exist in the heart contributing to the stability of cardiac excitation–contraction coupling.
Sources of Funding
This work was supported by NIH grants HL80101 (to L.A.B. and D.M.B.), HL62231 (to L.A.B.), and HL30077 (to D.M.B.) and American Heart Association Grant AHA0530309Z (to A.V.Z.).
Footnotes
Disclosures None.
References
- 1.Fabiato A. Rapid ionic modifications during the aequorin-detected calcium transient in a skinned canine cardiac Purkinje cell. J Gen Physiol. 1985;85:189–246. doi: 10.1085/jgp.85.2.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fabiato A. Time and calcium dependence of activation and inactivation of calcium-induced release of calcium from the sarcoplasmic reticulum of a skinned canine cardiac Purkinje cell. J Gen Physiol. 1985;85:247–289. doi: 10.1085/jgp.85.2.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cheng H, Lederer WJ, Cannell MB. Calcium sparks: elementary events underlying excitation-contraction coupling in heart muscle. Science. 1993;262:740–744. doi: 10.1126/science.8235594. [DOI] [PubMed] [Google Scholar]
- 4.Lopez-Lopez JR, Shacklock PS, Balke CW, Wier WG. Local calcium transients triggered by single L-type calcium channel currents in cardiac cells. Science. 1995;268:1042–1045. doi: 10.1126/science.7754383. [DOI] [PubMed] [Google Scholar]
- 5.Bridge JH, Ershler PR, Cannell MB. Properties of Ca2+ sparks evoked by action potentials in mouse ventricular myocytes. J Physiol. 1999;518(pt 2):469–478. doi: 10.1111/j.1469-7793.1999.0469p.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brochet DX, Yang D, Di Maio A, Lederer WJ, Franzini-Armstrong C, Cheng H. Ca2+ blinks: rapid nanoscopic store calcium signaling. Proc Natl Acad Sci U S A. 2005;102:3099–3104. doi: 10.1073/pnas.0500059102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shannon TR, Guo T, Bers DM. Ca2+ scraps: local depletions of free [Ca2+] in cardiac sarcoplasmic reticulum during contractions leave substantial Ca2+ reserve. Circ Res. 2003;93:40–45. doi: 10.1161/01.RES.0000079967.11815.19. [DOI] [PubMed] [Google Scholar]
- 8.Trafford AW, Diaz ME, Sibbring GC, Eisner DA. Modulation of CICR has no maintained effect on systolic Ca2+: simultaneous measurements of sarcoplasmic reticulum and sarcolemmal Ca2+ fluxes in rat ventricular myocytes. J Physiol. 2000;522(pt 2):259–270. doi: 10.1111/j.1469-7793.2000.t01-2-00259.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fill M, Copello JA. Ryanodine receptor calcium release channels. Physiol Rev. 2002;82:893–922. doi: 10.1152/physrev.00013.2002. [DOI] [PubMed] [Google Scholar]
- 10.Meissner G. Molecular regulation of cardiac ryanodine receptor ion channel. Cell Calcium. 2004;35:621–628. doi: 10.1016/j.ceca.2004.01.015. [DOI] [PubMed] [Google Scholar]
- 11.DelPrincipe F, Egger M, Niggli E. Calcium signalling in cardiac muscle: refractoriness revealed by coherent activation. Nat Cell Biol. 1999;1:323–329. doi: 10.1038/14013. [DOI] [PubMed] [Google Scholar]
- 12.Sobie EA, Dilly KW, dos Santos CJ, Lederer WJ, Jafri MS. Termination of cardiac Ca(2+) sparks: an investigative mathematical model of calcium-induced calcium release. Biophys J. 2002;83:59–78. doi: 10.1016/s0006-3495(02)75149-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Terentyev D, Viatchenko-Karpinski S, Valdivia HH, Escobar AL, Gyorke S. Luminal Ca2+ controls termination and refractory behavior of Ca2+-induced Ca2+ release in cardiac myocytes. Circ Res. 2002;91:414–420. doi: 10.1161/01.res.0000032490.04207.bd. [DOI] [PubMed] [Google Scholar]
- 14.Terentyev D, Kubalova Z, Valle G, Nori A, Vedamoorthyrao S, Terentyeva R, Viatchenko-Karpinski S, Bers DM, Williams SC, Volpe P, Gyorke S. Modulation of SR Ca release by luminal ca and calsequestrin in cardiac myocytes: effects of CASQ2 mutations linked to sudden cardiac death. Biophys J. 2008;95:2037–2048. doi: 10.1529/biophysj.107.128249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gyorke S, Fill M. Ryanodine receptor adaptation: control mechanism of Ca(2+)-induced Ca2+ release in heart. Science. 1993;260:807–809. doi: 10.1126/science.8387229. [DOI] [PubMed] [Google Scholar]
- 16.Sham JS, Song LS, Chen Y, Deng LH, Stern MD, Lakatta EG, Cheng H. Termination of Ca2+ release by a local inactivation of ryanodine receptors in cardiac myocytes. Proc Natl Acad Sci U S A. 1998;95:15096–15101. doi: 10.1073/pnas.95.25.15096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lukyanenko V, Wiesner TF, Gyorke S. Termination of Ca2+ release during Ca2+ sparks in rat ventricular myocytes. J Physiol. 1998;507(pt 3):667–677. doi: 10.1111/j.1469-7793.1998.667bs.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xu L, Meissner G. Regulation of cardiac muscle Ca2+ release channel by sarcoplasmic reticulum lumenal Ca2+. Biophys J. 1998;75:2302–2312. doi: 10.1016/S0006-3495(98)77674-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Picht E, DeSantiago J, Blatter LA, Bers DM. Cardiac alternans do not rely on diastolic sarcoplasmic reticulum calcium content fluctuations. Circ Res. 2006;99:740–748. doi: 10.1161/01.RES.0000244002.88813.91. [DOI] [PubMed] [Google Scholar]
- 20.Zima AV, Picht E, Bers DM, Blatter LA. Partial inhibition of sarcoplasmic reticulum ca release evokes long-lasting ca release events in ventricular myocytes: role of luminal ca in termination of ca release. Biophys J. 2008;94:1867–1879. doi: 10.1529/biophysj.107.114694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lukyanenko V, Gyorke S. Ca2+ sparks and Ca2+ waves in saponin-permeabilized rat ventricular myocytes. J Physiol. 1999;521(pt 3):575–585. doi: 10.1111/j.1469-7793.1999.00575.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Smith GL, O'Neill SC. A comparison of the effects of ATP and tetracaine on spontaneous Ca(2+) release from rat permeabilised cardiac myocytes. J Physiol. 2001;534:37–47. doi: 10.1111/j.1469-7793.2001.00037.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Picht E, Zima AV, Blatter LA, Bers DM. SparkMaster: automated calcium spark analysis with ImageJ. Am J Physiol Cell Physiol. 2007;293:C1073–C1081. doi: 10.1152/ajpcell.00586.2006. [DOI] [PubMed] [Google Scholar]
- 24.Lacampagne A, Klein MG, Ward CW, Schneider MF. Two mechanisms for termination of individual Ca2+ sparks in skeletal muscle. Proc Natl Acad Sci U S A. 2000;97:7823–7828. doi: 10.1073/pnas.97.14.7823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rousseau E, Meissner G. Single cardiac sarcoplasmic reticulum Ca2+-release channel: activation by caffeine. Am J Physiol. 1989;256:H328–H333. doi: 10.1152/ajpheart.1989.256.2.H328. [DOI] [PubMed] [Google Scholar]
- 26.Sipido KR, Wier WG. Flux of Ca2+ across the sarcoplasmic reticulum of guinea-pig cardiac cells during excitation-contraction coupling. J Physiol. 1991;435:605–630. doi: 10.1113/jphysiol.1991.sp018528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Song LS, Sham JS, Stern MD, Lakatta EG, Cheng H. Direct measurement of SR release flux by tracking ‘Ca2+ spikes’ in rat cardiac myocytes. J Physiol. 1998;512(pt 3):677–691. doi: 10.1111/j.1469-7793.1998.677bd.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Blatter LA, Huser J, Rios E. Sarcoplasmic reticulum Ca2+ release flux underlying Ca2+ sparks in cardiac muscle. Proc Natl Acad Sci U S A. 1997;94:4176–4181. doi: 10.1073/pnas.94.8.4176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gyorke I, Gyorke S. Regulation of the cardiac ryanodine receptor channel by luminal Ca2+ involves luminal Ca2+ sensing sites. Biophys J. 1998;75:2801–2810. doi: 10.1016/S0006-3495(98)77723-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sitsapesan R, Williams AJ. Regulation of the gating of the sheep cardiac sarcoplasmic reticulum Ca(2+)-release channel by luminal Ca2+. J Membr Biol. 1994;137:215–226. doi: 10.1007/BF00232590. [DOI] [PubMed] [Google Scholar]
- 31.Gomez AM, Cheng H, Lederer WJ, Bers DM. Ca2+ diffusion and sarcoplasmic reticulum transport both contribute to [Ca2+]i decline during Ca2+ sparks in rat ventricular myocytes. J Physiol. 1996;496(pt 2):575–581. doi: 10.1113/jphysiol.1996.sp021708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hove-Madsen L, Bers DM. Sarcoplasmic reticulum Ca2+ uptake and thapsigargin sensitivity in permeabilized rabbit and rat ventricular myocytes. Circ Res. 1993;73:820–828. doi: 10.1161/01.res.73.5.820. [DOI] [PubMed] [Google Scholar]
- 33.Gyorke S, Lukyanenko V, Gyorke I. Dual effects of tetracaine on spontaneous calcium release in rat ventricular myocytes. J Physiol. 1997;500(pt 2):297–309. doi: 10.1113/jphysiol.1997.sp022021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lukyanenko V, Gyorke I, Subramanian S, Smirnov A, Wiesner TF, Gyorke S. Inhibition of Ca(2+) sparks by ruthenium red in permeabilized rat ventricular myocytes. Biophys J. 2000;79:1273–1284. doi: 10.1016/S0006-3495(00)76381-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xu L, Tripathy A, Pasek DA, Meissner G. Potential for pharmacology of ryanodine receptor/calcium release channels. Ann N Y Acad Sci. 1998;853:130–148. doi: 10.1111/j.1749-6632.1998.tb08262.x. [DOI] [PubMed] [Google Scholar]
- 36.Kubalova Z, Terentyev D, Viatchenko-Karpinski S, Nishijima Y, Gyorke I, Terentyeva R, da Cunha DN, Sridhar A, Feldman DS, Hamlin RL, Carnes CA, Gyorke S. Abnormal intrastore calcium signaling in chronic heart failure. Proc Natl Acad Sci U S A. 2005;102:14104–14109. doi: 10.1073/pnas.0504298102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.George CH. Sarcoplasmic reticulum Ca2+ leak in heart failure: mere observation or functional relevance? Cardiovasc Res. 2008;77:302–314. doi: 10.1093/cvr/cvm006. [DOI] [PubMed] [Google Scholar]
- 38.Liu N, Priori SG. Disruption of calcium homeostasis and arrhythmogenesis induced by mutations in the cardiac ryanodine receptor and calsequestrin. Cardiovasc Res. 2008;77:293–301. doi: 10.1093/cvr/cvm004. [DOI] [PubMed] [Google Scholar]
- 39.Wu X, Bers DM. Sarcoplasmic reticulum and nuclear envelope are one highly interconnected Ca2+ store throughout cardiac myocyte. Circ Res. 2006;99:283–291. doi: 10.1161/01.RES.0000233386.02708.72. [DOI] [PubMed] [Google Scholar]
- 40.Michailova A, DelPrincipe F, Egger M, Niggli E. Spatiotemporal features of Ca2+ buffering and diffusion in atrial cardiac myocytes with inhibited sarcoplasmic reticulum. Biophys J. 2002;83:3134–3151. doi: 10.1016/S0006-3495(02)75317-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Swietach P, Spitzer KW, Vaughan-Jones RD. Ca2+-mobility in the sarcoplasmic reticulum of ventricular myocytes is low. Biophys J. 2008;95:1412–1427. doi: 10.1529/biophysj.108.130385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hinch R. A mathematical analysis of the generation and termination of calcium sparks. Biophys J. 2004;86:1293–1307. doi: 10.1016/S0006-3495(04)74203-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stern MD. Theory of excitation-contraction coupling in cardiac muscle. Biophys J. 1992;63:497–517. doi: 10.1016/S0006-3495(92)81615-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Qin J, Valle G, Nani A, Nori A, Rizzi N, Priori SG, Volpe P, Fill M. Luminal Ca2+ regulation of single cardiac ryanodine receptors: insights provided by calsequestrin and its mutants. J Gen Physiol. 2008;131:325–334. doi: 10.1085/jgp.200709907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Laver DR. Ca2+ stores regulate ryanodine receptor Ca2+ release channels via luminal and cytosolic Ca2+ sites. Biophys J. 2007;92:3541–3555. doi: 10.1529/biophysj.106.099028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Duncan AM, Picht E, Bers DM. Dynamic Ca movement within cardiac sarcoplasmic reticulum. Circulation. 2005:112–157. [Google Scholar]
- 47.Terentyev D, Viatchenko-Karpinski S, Gyorke I, Volpe P, Williams SC, Gyorke S. Calsequestrin determines the functional size and stability of cardiac intracellular calcium stores: mechanism for hereditary arrhythmia. Proc Natl Acad Sci U S A. 2003;100:11759–11764. doi: 10.1073/pnas.1932318100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Valdivia HH, Kaplan JH, Ellis-Davies GC, Lederer WJ. Rapid adaptation of cardiac ryanodine receptors: modulation by Mg2+ and phosphorylation. Science. 1995;267:1997–2000. doi: 10.1126/science.7701323. [DOI] [PMC free article] [PubMed] [Google Scholar]