

Abstract
Cocaine sensitization is produced by repeated exposure to the drug and is thought to reflect neuroadaptations that contribute to addiction. Here, we identify the Ca2+/calmodulin-stimulated adenylyl cyclases, type 1 (AC1) and type 8 (AC8), as novel regulators of this behavioral plasticity. We show that, whereas AC1 and AC8 single knock-out mice (AC1−/− and AC8−/−) exhibit Ca2+-stimulated adenylyl cyclase activity in striatal membrane fractions, AC1/8 double-knock-out (DKO) mice do not. Furthermore, DKO mice are acutely supersensitive to low doses of cocaine and fail to display locomotor sensitization after chronic cocaine treatment. Because of the known role for the extracellular signal-regulated kinase (ERK)/mitogen-activated protein kinase signaling pathway in cocaine-induced behavioral plasticity and its coupling to calcium-stimulated cAMP signaling in the hippocampus, we measured phosphorylated ERK (pERK) levels in the striatum. Under basal conditions, pERK is upregulated in choline acetyltransferase-positive interneurons in DKO mice relative to wild-type (WT) controls. After acute cocaine treatment, pERK signaling is significantly suppressed in medium spiny neurons (MSNs) of DKO mice relative to WT mice. In addition to the lack of striatal ERK activation by acute cocaine, signaling machinery downstream of ERK is uncoupled in DKO mice. We demonstrate that AC1 and AC8 are necessary for the phosphorylation of mitogen and stress-activated kinase-1 (pMSK1) at Ser376 and Thr581 and cAMP response element-binding protein (pCREB) at Ser133 after acute cocaine treatment. Our results demonstrate that the Ca2+-stimulated adenylyl cyclases regulate long-lasting cocaine-induced behavioral plasticity via activation of the ERK/MSK1/CREB signaling pathway in striatonigral MSNs.
Keywords: adenylate cyclase, cocaine, cAMP, sensitization, ERK, MSK1
Introduction
Chronic exposure to drugs of abuse causes molecular and cellular neuroadaptations that ultimately lead to addiction. An attractive model for this process is behavioral sensitization, the progressive and persistent enhancement of behavioral responses to a drug after repeated, intermittent exposure (Robinson and Berridge, 1993, 2001). Importantly, behavioral sensitization to psychostimulants appears to be accompanied by enhancement of the motivational and rewarding effects of these drugs, because cocaine-sensitized animals acquire cocaine self-administration more rapidly and at lower doses (Horger et al., 1990; Di Ciano, 2008; Zhang and Kosten, 2007). Sensitization during the early stages of drug-taking may be a critical determinant of long-term self-administration patterns (Morgan et al., 2006; Roberts et al., 2007).
Numerous neural mechanisms for psychostimulant sensitization have been proposed. Early research focused on the observation that sensitization enhances dopaminergic transmission from the midbrain ventral tegmental area to the striatum (Woolverton and Johnson, 1992). However, locomotor sensitization has also been reported in the absence of increased striatal dopamine release (Segal and Kuczenski, 1992; Kalivas and Duffy, 1993; Heidbreder et al., 1996), suggesting a postsynaptic component. Potential postsynaptic mechanisms include enhanced AMPA receptor-mediated neurotransmission (Boudreau and Wolf, 2005; Boudreau et al., 2007; Kourrich et al., 2007), upregulation of the extracellular signal-regulated kinase (ERK)/MSK1/cAMP response element-binding protein (CREB) signaling pathway (Brami-Cherrier et al., 2005; Ferguson et al., 2006; Lu et al., 2006), and upregulation of the cAMP/protein kinase A (PKA)/CREB pathway (Miserendino and Nestler, 1995; Schroeder et al., 2004; Dong et al., 2006). Most of these mechanisms were first shown to be important to long-term memory formation in other brain regions, a parallel that strongly influences current theories of addiction (Kelley, 2004; Hyman et al., 2006; Kauer and Malenka, 2007).
Although cAMP signaling pathways are known to be involved in cocaine-induced neural and behavioral plasticity, the identities of specific adenylyl cyclases (ACs) responsible for initiating these pathways remain unknown. There are 10 adenylyl cyclases, each with distinct regulatory properties, and nine of these enzymes are expressed in the brain. In this study, we used a genetic strategy to discern the contributions of the Ca2+-stimulated adenylyl cyclases, type 1 (AC1) and type 8 (AC8), in cocaine-dependent behavioral and neuronal plasticity. AC1−/−], AC8−/−, and double knock-out (DKO) mice were tested for striatal Ca2+-stimulated cyclase activity, cocaine-induced locomotion, and cocaine-dependent activation of the ERK/MSK1/CREB signaling pathway. These experiments revealed that expression of AC1 or AC8 is critical for the development of psychomotor sensitization resulting from chronic cocaine exposure. We also discovered that acute cocaine treatment results in a robust coactivation of the ERK/MSK1/CREB pathway in medium spiny neurons (MSNs) in wild-type (WT) mice but not DKO animals. This transcriptional signaling pathway has been shown to be critical in regulating long-lasting cocaine-induced behavioral plasticity (Carlezon et al., 1998; Nestler, 2004; Brami-Cherrier et al., 2005; Valjent et al., 2006b). These findings mirror those we have reported in the hippocampus after contextual fear conditioning and provide another common mechanism of drug- and learning-induced neuroplasticity (Sindreu et al., 2007).
Materials and Methods
Animals.
Male mice 2–4 months of age were used for all experiments. AC1−/−, AC8−/−, and DKO mice were generated as described previously (Wu et al., 1995; Wong et al., 1999). All knock-out strains have been backcrossed into a C57BL/6 background for at least 10 generations. AC1−/− littermate control mice were used as WT controls for behavioral and DAB immunohistochemistry experiments. Commercial C57BL/6 mice were used in other experiments. All experiments were approved by the University of Washington animal care committee and performed in accordance with their guidelines.
Adenylyl cyclase assay.
Mice were killed by cervical dislocation, and striata [includes caudate and nucleus accumbens (NAcc)] and hippocampi were dissected and homogenized in buffer containing 50 mm Tris-HCl, pH 7.4, 2 mm MgCl2, 1 mm EDTA, 0.5 mm DTT, and protease inhibitor mixture (Roche). Homogenate was centrifuged at 1000 × g (4°C) for 5 min, and the supernatant was centrifuged at high speed (100,000 × g, 4°C) for 1 h to pellet membrane fractions. Pellets were resuspended and homogenized in buffer. Adenylyl cyclase assays were performed with 95 μl of membrane homogenate in a total volume of 250 μl containing 39.2 mm Tris-HCl, pH 7.4, 1 mm ATP, 1.38 mm EDTA, 0.2 mm EGTA, 5.76 mm MgCl2, 0.1 mm DTT, protease inhibitor mixture, 1 mm β-mercaptoethanol, 0.2 mm IBMX, 0.1% BSA, and trace amounts of [α-32P]ATP (∼5.0 × 106 counts per reaction; PerkinElmer Life and Analytical Sciences). To monitor sample recovery, [3H]cAMP (10,000 counts per reaction; PerkinElmer Life and Analytical Sciences) and 2 mm cAMP were also included. When measuring calcium-stimulated activity, calmodulin (50 units per reaction; Sigma) was added with 300 μm calcium (4 μm free calcium, calculated using the Maxchelator algorithm; http://www.stanford.edu/∼cpatton/maxc.html). Reactions were performed for 15 min at 30°C and stopped with 1.5% SDS and heating to 100°C for 2 min. Samples were cooled and loaded onto ionic (H+), analytical grade, 200–400 mesh columns packed with Dowex AG 50W-X4 resin (Bio-Rad). Samples were eluted onto alumina columns (WN3 type; Sigma) with ddH20. A final elution step from the alumina columns was performed with 0.1 m imidazole. cAMP produced was determined by scintillation counting of [32P]cAMP and normalized to [3H]cAMP counts. A Pierce BCA assay was performed on membrane homogenates to obtain results in picomoles of cAMP per minute per milligram protein. Experiments were repeated three separate times, pooling three to five samples for each repeat, and analyzed with three technical replicate for each experimental replicate.
Locomotor activity and sensitization.
Locomotor activity was measured in a 25 × 25 cm arena using TruScan 99/2.0 software (Coulbourn Instruments). Ambulatory distance is defined as the sum total of vector coordinate changes less stereotypic movement distance (comprising moves of <2.25 cm, followed by a return to the original coordinate). In acute cocaine experiments, mice were habituated to the arena for 1 h before injection (0, 5, 10, and 20 mg/kg, i.p.) and returned to the arena for 1 h after injection. Locomotor activity was measured in 5 min intervals. In cocaine sensitization experiments, mice received a 10 mg/kg acute cocaine injection as described above on day 1. On days 2–7, mice received a 20 mg/kg cocaine injection and were returned to their home cage. On day 8, after 48 h of withdrawal, mice received a 10 mg/kg challenge dose, and locomotor activity was measured as described above.
Immunoblotting.
Mice were killed by cervical dislocation 15 min after treatment, and striatum was dissected out and homogenized with 50 strokes of a Kontes “B” pestle in 200 μl of homogenization buffer [1% sodium deoxycholate, 10 mm HEPES, pH 7.5, 5 mm EDTA, pH 8.0, 10% glycerol, 1% NP-40, 1.5 mm MgCl2, 150 mm NaCl, 50 mm NaF, 20 mm Na4P2O7, 1 mm Na3VO4, 1:100 dilutions of phosphatase 1 and phosphatase 2 inhibitor mixtures (Sigma), and 1 tablet/10 ml Complete Mini EDTA-free protease inhibitor (Roche)]. Homogenates were spun at 14,000 × g for 10 min, and supernatant was retained as the whole-cell soluble fraction. An equal amount of 4× SDS-PAGE sample buffer was added to each sample, and samples were heated for 5 min at 90°C. Samples were loaded onto a 4–20% Tris-HCl gel (Bio-Rad) for SDS-PAGE analysis. Protein was transferred on to polyvinylidene difluoride membranes (Immobilon-P; Millipore) and blocked with 5% BSA in 0.05% Tween 20 in PBS (PBSTw). Primary antibody used were Phospho-(Ser/Thr) PKA Substrate Antibody at 1:2000 dilution (catalog #9621; Cell Signaling Technology) and MAB1501R mouse anti-actin monoclonal antibody at 1:10,000 (Millipore Bioscience Research Reagents) in 5% BSA in PBSTw overnight at 4°C. Blots were then incubated with alkaline phosphatase-conjugated secondary antibodies in 5% BSA in PBSTw for 2 h at room temperature (Sigma). Immunoblots were developed using CDP-Star (Tropix). When necessary, immunoblots were stripped in 25 mm glycine, pH 2, and 1% SDS for 30 min at room temperature. Bands were visualized with Eastman Kodak X-Omat Blue film and digitized with a scanner calibrated to measure optical density. NIH ImageJ was used to measure the integrated optical density of the bands. Integrated optical density of protein bands resulting from anti-phosphorylated PKA substrates (pPKA-s) antibody were normalized to the integrated optical density of corresponding actin bands.
DAB immunohistochemistry.
Mice were killed by cervical dislocation, and cerebral hemispheres were immersion fixed in 50 ml of 4% formaldehyde in 0.1 m phosphate buffer, pH 7.4, at 4°C for 24 h, with rocking and one change of fixative solution after 2 h. Striatal sections, spanning between +1.7 and +1.0 mm from bregma, were cut, stained, and analyzed for pERK cell counts as described previously (Sindreu et al., 2007).
Immunofluorescent confocal microscopy.
Mice were transcardially perfused with isotonic saline containing 50 mm NaF and 1 mm Na3VO4, followed by 4% formaldehyde solution. Dissected brains were cryoprotected in 30% sucrose, frozen in dry ice, and cut in a cryostat. Sections were sequentially pretreated (rinses between) with 0.5% NaBH4, 1% H2O2 in 10% methanol, and 10% serum in 0.1 m Tris-HCl, pH 7.4, 0.9% NaCl, 0.1% Tween 20. Sections were first reacted with rabbit anti-pERK (1:10,000; Cell Signaling Technology) or anti-phospho-Ser376 MSK1 (1:6000; Millipore) antibodies and visualized by deposition of cyanine-3–tyramide complexes according to the instructions of the manufacturer for tyramide signal amplification (PerkinElmer Life and Analytical Sciences). They were then rinsed, blocked again, and incubated in one (or two) of the following antibodies: mouse anti-striatal enriched phosphatase (STEP) (1:1000; Novus Biologicals), rabbit anti-DARPP32 (1:200), rabbit anti-phospho-Thr581 MSK1 (1:300), and rabbit anti-phospho-CREB (1:100; clone 87G3), from Cell Signaling Technology; mouse anti-neuronal-specific nuclear protein (1:1000), rabbit anti-phospho-Ser376 MSK1 (1:250), and rabbit anti-choline acetyltransferase (ChAT) (1;1000) from Millipore; and rabbit anti-dynorphin (1:500; Peninsula Labs). Second primary antibodies were detected with corresponding secondary antibodies conjugated to Alexa Fluor 488 or 647 (1:500). Sections were Hoescht counterstained and mounted in anti-fading medium (Gelmount). Absence of cross-reactivity between the first primary antibody and the second secondary antibody was confirmed in experiments in which the second primary antibody was omitted (data not shown).
Images were captured on a Leica SP1 confocal microscope and analyzed with MetaMorph software (Universal Imaging Corporation). Density filter, pinhole aperture, detector gain, and offset were initially set to obtain pixel densities within a linear range and were then kept constant for experimental comparisons. Signals were acquired using sequential line scanning. Z-series stacks (z-step size, 2 μm) were collected as 512 × 512 pixel images (average of four frames per image) with a 25× objective. To estimate the percentage of activated neurons, Z-series stacks 8–10 μm high of the dorsomedial caudate–putamen were acquired from at least three sections per mouse. Analysis of average somatic intensity was performed at the midplane of every neuron based on the nuclear counterstaining. Because of basal levels of pCREB and pMSK1T581 labeling in control groups, only those cells with signal intensities within the top 20% were considered immunopositive for these antibodies. Colocalization of immunosignals was confirmed in x, y, and z dimensions. Labeling intensity was represented on a gray scale of 0 to 255.
RNA extraction and reverse transcription-quantitative PCR.
Striata were dissected from adult WT and AC1/AC8 double knock-out mice, and RNA was extracted using the Absolutely RNA kit (catalog #400800; Stratagene). RNA was treated with DNase I (catalog #EN0521; Fermentas), and total RNA was reverse transcribed using the TaqMan Gold RT-PCR kit per the instructions of the manufacturer (catalog #N808-0233; Applied Biosystems). TaqMan gene expression assays were obtained from Applied Biosystems. The AC1 primers amplified the region spanning exons 1–2 (catalog #Mm01187830_m1). AC8 was amplified in the region spanning exons 14–15 (catalog #Mm01194290_m1). Glyceraldehyde-3-phosphate dehydrogenase was amplified and used as a within-sample normalizing control (catalog #Mm99999915_g1). Comparisons made between genotypes were analyzed using the standard curve method, in which all standard curves were constructed with six twofold dilutions. All standard curves had r2 values of >0.990.
Statistical analysis.
Measures are expressed as mean ± SEM. Data were analyzed by two-tailed t test and one-way or two-way ANOVA, as indicated, with post hoc pairwise comparisons performed using Tukey's honestly significant difference test or the Bonferroni's test as indicated. Significance was set at p < 0.05.
Results
Striatal Ca2+-stimulated adenylyl cyclase activity in wild-type, AC1−/−, AC8−/−, and DKO mice
Striatal Ca2+-stimulated adenylyl cyclase activity has been demonstrated previously in rodent and human striatal membrane homogenates, but the individual contribution of AC1 and AC8 to this activity has not been reported (Gnegy et al., 1980; Treisman et al., 1985; Ahlijanian and Cooper, 1988; Tong et al., 2001). We first confirmed the presence of AC1 and AC8 mRNA in the striatum by reverse transcription (RT)-quantitative PCR (qPCR) (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). We then measured the functional contribution of these enzymes to Ca2+-stimulated adenylyl cyclase activity in the striatum by performing adenylyl cyclase activity assays on WT, AC1−/−, AC8−/−, and DKO mice. Ca2+-stimulated adenylyl cyclase activity increased 2.5-fold in striatal membrane fractions from WT mice obtained from Charles River Laboratories and a comparable 2.2-fold in wild-type AC1−/− littermates (AC1+/+), which were used for subsequent behavioral studies (Fig. 1A). For comparison, Ca2+-stimulated adenylyl cyclase activity in hippocampal membranes increased 4.2-fold over basal activity (Fig. 1A,B). Ca2+ also significantly stimulated adenylyl cyclase activity 1.7-fold in AC1−/− mice and 1.5-fold in AC8−/− mice (Fig. 1C). However, Ca2+-stimulated adenylyl cyclase activity in DKO mice was abolished in the hippocampus and striatum (Fig. 1B,C).
Figure 1.
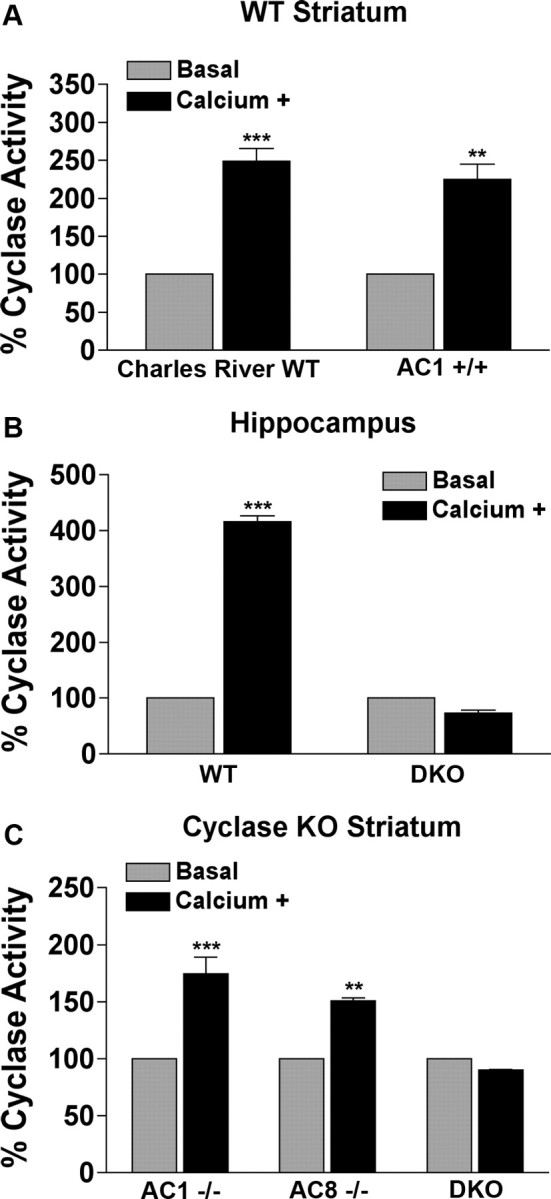
Ca2+-stimulated adenylyl cyclase activity as a percentage of basal adenylyl cyclase activity in the striatum depends on AC1 and AC8. A, Striatal adenylyl cyclase activity in naive WT mice from Charles River Laboratories and AC1+/+ littermate controls under baseline conditions and in the presence of 4 μm free calcium. B, Hippocampal cyclase activity under same conditions as in A. C, Striatal adenylyl cyclase activity in AC1−/−, AC8−/−, and DKO mice under baseline and calcium+ conditions. All values are means ± SEM of three separate experiments. Individual experiments were performed on tissue pooled from three to five animals with samples analyzed in triplicate. Two-way ANOVA, **p < 0.001, ***p < 0.001 (Bonferroni's test).
Acute supersensitivity to cocaine in DKO mice
To determine whether Ca2+-stimulated adenylyl cyclases play a functional role in behavioral responses to cocaine, we tested three knock-out mouse lines: AC1−/−, AC8−/−, and DKO. We first monitored acute locomotor responses by injecting animals with cocaine after 1 h of habituation to the testing chamber. AC1 and AC8 single knock-out animals demonstrated hyperactivity comparable with wild-type (AC1+/+) controls at a 10 mg/kg dose (n = 10–15 mice per group) (Fig. 2A,B). However, DKO mice ambulated significantly more than WT mice during the half hour after injection (n = 9–15 mice per group) at both the 5 and 10 mg/kg doses (Fig. 2C,D). DKO mice displayed no difference in locomotor activity in response to saline or a higher dose of cocaine (Fig. 2D).
Figure 2.

Enhanced locomotor responses to acute cocaine in DKO mice. A–C, Mice were habituated to the testing chamber for 60 min before injection with 10 mg/kg cocaine injected intraperitoneally at time 0. Ambulatory distance traveled is shown for AC1−/−, AC8−/−, DKO, and WT (AC1+/+) mice. D, Ambulatory distance traveled over the first 30 min after injection for saline and three doses of cocaine in WT and DKO mice. n = 9–15 mice per group. Two-way ANOVA, **p < 0.01 (Tukey's post hoc test).
AC1 and AC8 are required for locomotor sensitization to cocaine
Because DKO mice have impaired hippocampus-dependent neural and behavioral plasticity (Wong et al., 1999), we were interested in examining their cocaine-induced behavioral plasticity. Locomotor sensitization to cocaine occurs in response to repeated, spaced injections and is potentiated by context association but not dependent on it (Partridge and Schenk, 1999). Because of the DKO animal's context-dependent memory deficits, we thought it important to choose a context-independent cocaine sensitization paradigm in which animals are sensitized in a context distinct from the testing chamber (Yao et al., 2004). This protocol produced significant sensitization in WT (AC1+/+), AC1−/−, and AC8−/− mice but not DKO mice (n = 10–13 mice per group) (Fig. 3). It is important to note that this lack of sensitization in DKOs was not attributable to a ceiling effect on cocaine-induced locomotion, because DKO mice given a 20 mg/kg acute dose (Fig. 2D) ambulated significantly more than mice subjected to the sensitization protocol (n = 13; two-tailed t test; p = 0.011).
Figure 3.

Absence of locomotor sensitization in DKO mice after chronic cocaine treatment. A–D, Mice were tested on day 1 with a 10 mg/kg acute dose of cocaine. On days 2–6, they received 20 mg/kg intraperitoneal cocaine injections in their home cage. On day 8, they received a 10 mg/kg challenge dose of cocaine. Ambulatory distance traveled on days 1 and 8 is shown for WT(AC1+/+), AC1−/−, AC8−/−, and DKO mice. E, Summary data. WT, AC1−/−, and AC8−/− mice sensitize significantly to chronic cocaine, whereas DKO mice do not. n = 10–13 mice per group analyzed by two-way ANOVA, **p < 0.01 (Tukey's post hoc test).
Altered phosphorylation of PKA substrates after cocaine treatment in DKO striatum
It has been shown that the upregulation of the PKA/cAMP signaling pathway modulates cocaine-dependent behavior, including enhancement of locomotion during the development of behavioral sensitization (Miserendino and Nestler, 1995; Schroeder et al., 2004; Dong et al., 2006). Because DKO mice lack a potential source of cocaine-induced cAMP production, we hypothesized that levels of phosphorylated downstream targets of PKA would be lower in these animals relative to WT controls after acute cocaine treatment.
To test this hypothesis, WT (Charles River Laboratories) and DKO mice were injected with either saline or cocaine (15 mg/kg) and killed 15 min later, and striatal tissue was processed for analysis of pPKA-s by Western blot. The anti-pPKA-s antibody used in this experiment recognizes proteins phosphorylated at a conserved PKA phosphorylation motif. Its ability to specifically monitor PKA activation in vivo has been confirmed previously (Sindreu et al., 2007). The antibody detects a number of PKA substrates resulting in an array of protein bands by Western analysis (Fig. 4A). We chose bands representing three PKA substrates for analysis and reported as the average percentage increase in band intensity of the cocaine-treated samples compared with the saline-treated samples (Fig. 4B,C). In both WT and DKO mice, acute cocaine treatment increased the phosphorylation of PKA substrates compared with saline-treated animals. In two of the three bands analyzed, WT mice exhibited significantly higher levels of PKA activation than DKO mice. The increase in phospho-PKA substrate labeling in DKO animals after acute cocaine treatment reflects that calcium-stimulated adenylyl cyclases are not the only source of PKA activation in the striatum.
Figure 4.

Altered PKA activation after acute cocaine treatment in DKO. A, B, Full image of immunoblot probed with pPKA-s antibody. Individual bands from pPKA-s blot and actin shown in B. Bands labeled 1–3 were analyzed. C, Data obtained by recording the integrated density of cocaine-treated WT (Charles River Laboratories) and DKO pPKA-s bands normalized to the integrated density of corresponding actin band, reported as average percentage change over saline-treated controls. Saline control groups for WT and DKO were set to 100%. n = 4 per condition. One-way ANOVA, *p < 0.05 (Tukey's post hoc test).
Basal and cocaine-stimulated pERK levels are altered in DKO striatum
cAMP impinges on several downstream signal transduction pathways that play roles in short- and long-term neural plasticity. We chose to examine the ERK/mitogen-activated protein kinase signaling pathway because it has been implicated in cocaine sensitization, and we have shown previously that DKO mice have ERK signaling deficits in the hippocampus associated with contextual fear conditioning (Sindreu et al., 2007). As reported previously (Brami-Cherrier et al., 2005; Valjent et al., 2005), acute cocaine injection increased pERK cell counts in WT (AC1+/+) animals in all three striatal subregions: the dorsal striatum, NAcc core, and NAcc shell (Fig. 5B–D). DKO mice had elevated basal pERK cell counts in both the dorsal striatum and NAcc core compared with WT (Fig. 5B,C). In addition, DKO mice only showed a significant increase in pERK cell counts after cocaine injection in one striatal subregion, the NAcc core (Fig. 5C). These data indicate that the loss of Ca2+-stimulated adenylyl cyclase activity results in elevated baseline numbers of pERK-positive (pERK+) cells in the striatum and limited additional increase after acute cocaine injection. It has been shown previously that, after chronic cocaine treatment in WT mice, there is no, or little, increase in pERK+ cells compared with acute treatment (Valjent et al., 2005; Bertran-Gonzalez et al., 2008). We show that DKO mice chronically treated with cocaine do not have a significant increase in pERK+ cells compared with the saline-treated condition. Furthermore, after chronic treatment, WT mice maintain a significant increase in pERK+ cells compared with DKO mice (supplemental Fig. 2, available at www.jneurosci.org as supplemental material).
Figure 5.

Elevated basal striatal pERK levels and impaired activation in response to acute cocaine in DKO mice. A, Immunohistochemistry for pERK in the dorsal striatum from sections representative of WT (AC1+/+) and DKO mice 30 min after injection with saline or 10 mg/kg cocaine. Scale bar, 50 μm. B–D, pERK-positive cell count summary data for the dorsal striatum, nucleus accumbens core, and nucleus accumbens shell. Values are means ± SEM for five to seven animals, whose means were determined from three to six sections per subregion. Data analyzed by two-way ANOVA, *p < 0.05, **p < 0.01 (Tukey's post hoc test).
DKO mice exhibit a selective upregulation of pERK in non-MSNs under basal conditions
Previous research has shown that, after acute cocaine treatment, ERK is activated specifically in striatonigral, dopamine D1 receptor-containing MSNs (Bertran-Gonzalez et al., 2008). Also, activation of this kinase is necessary for the development of cocaine-induced locomotor sensitization (Ferguson et al., 2006; Lu et al., 2006; Valjent et al., 2006b). Because DKO mice displayed hyperlocomotion in response to an acute cocaine injection and levels of pERK appear to be globally upregulated in these animals under control conditions, we hypothesized that this upregulation of pERK was occurring in MSNs and contributing to the enhanced locomotor activity after acute cocaine treatment.
To characterize the cells in which pERK is increased in control DKO mice, the striatum was double labeled for pERK and STEP (an ERK1/2 phosphatase and MSN marker) (Lombroso et al., 1993; Valjent et al., 2005). Although basal pERK colocalized with STEP in ∼80% cells in WT (Charles River Laboratories) mice, it was mostly found in cells devoid of STEP in DKO mice (∼90% cells) (Fig. 6A). Double labeling for pERK and DARPP32 (a dual-function protein kinase/phosphatase inhibitor and specific MSN marker) (Ouimet et al., 1984; Fienberg et al., 1998) confirmed the selective upregulation of basal pERK in non-MSNs (DARPP32-negative) of DKO mice (p = 0.006 between control WT and DKO; n = 4–5 mice per group) (Fig. 6C). The shape and size of the neurons suggested that the increased basal ERK activation in DKO mice may occur, at least in part, in cholinergic interneurons located throughout the striatum. Indeed, significantly more ChAT+ neurons stained for pERK in DKO mice than in WT mice, under basal conditions (Fig. 6B; p = 0.009). This contrasts with the pERK increase observed after cocaine in WT mice, which is not only more widespread at its peak (10–15 min after injection) but also specific to MSNs. Thus, basal ERK activation in DKO mice is different in nature and magnitude from the one normally brought about by cocaine. Furthermore, cocaine-induced ERK activation in DARPP32+ cells was significantly reduced in DKO mice compared with WT mice (p = 0.017 between cocaine-treated groups; n = 4 mice per group) (Fig. 6C). Thus, contrary to our hypothesis, the locomotor “presensitization” seen in DKO mice (after acute cocaine treatment) is not the result of elevated pERK signaling in MSNs.
Figure 6.

Basal pERK levels are elevated in non-MSNs in DKO striatum. Images are three-dimensional reconstructions of optical z-stacks taken from the rostral, dorsomedial caudate–putamen. A, Immunodetection of pERK and STEP in naive DKO mice. Arrowheads indicate pERK+ neurons that lacked STEP expression. Bar graph summarizes the percentage distribution of pERK+ cells relative to the expression of STEP in control WT and DKO mice (n = 5 per group). A total of 1547 cells were analyzed from DKO mice, and 898 cells were analyzed from WT mice. Scale bar, 40 μm. B, Immunodetection of pERK and ChAT in naive WT (Charles River Laboratories) and DKO mice. Arrowheads indicate neurons in which pERK+ neurons colocalized with ChAT+ neurons. Bar graph displays the percentage of pERK+, cholinergic cells in WT and DKO mice; p = 0.009 between WT and DKO. Scale bar, 80 μm. C, Immunodetection of pERK and DARPP32. Arrowheads indicate some pERK+ cells. Note that, although pERK was segregated from DARPP-32 in control DKO mice, they strictly colocalized after cocaine injection in WT (Charles River Laboratories) mice. Scale bar, 80 μm. n = 4–5 mice per group. Data analyzed by two-tailed t test (p = 0.006 between DARPP32− cells in saline-treated groups; p = 0.017 between DARPP32+ cells in cocaine-treated groups).
MSK1 and CREB activation is impaired in DKO mice and robustly coactivated in WT mice
We next asked whether activation of nuclear regulators of CREB-dependent transcription was also impaired in DKO mice after cocaine injection. The ERK-dependent CREB kinase MSK1 becomes activated after dual phosphorylation at Ser376 and Thr581 (McCoy et al., 2005). MSK1 phosphorylation at both sites increased after acute cocaine treatment (15 mg/kg, 15 min) in WT (Charles River Laboratories) striatum (p < 0.01 for both sites, between saline- and cocaine-treated WT), but it was suppressed in DKO mice (p < 0.02 for both sites, between cocaine-treated WT and DKO mice) (Fig. 7). In keeping with this, cocaine-induced CREB phosphorylation was also impaired in DKO mice (p = 0.014, between cocaine-treated WT and DKO mice). Notably, the percentages of neurons showing ERK1/2, MSK1, and CREB phosphorylation were remarkably similar within each genotype, indicating a causal relationship.
Figure 7.
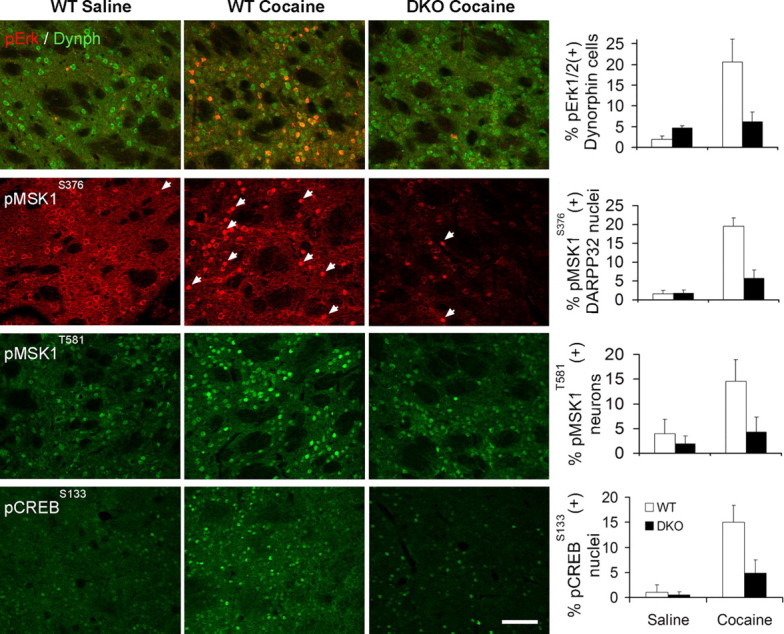
Increases in ERK, MSK1, and CREB phosphorylation after cocaine injection are suppressed in DKO mice. Immunodetection of pERKThr183/Tyr185, pMSK1Ser376, pMSK1Thr581, and pCREBSer133 in WT (Charles River Laboratories) and DKO mice. Images are three-dimensional reconstructions of optical z-stacks taken from the rostral, dorsomedial caudate–putamen. Arrows indicate neurons with nuclear pMSK1Ser376 immunolabeling. Scale bar, 80 μm. A significant interaction between treatment and genotype was found for all four phosphorylation events (one-way ANOVA, p < 0.0001; pairwise comparisons with Tukey's post hoc test: p < 0.05 between saline pERK groups, as well as between all cocaine groups; n = 4–5 mice per group). Dynph, Dynorphin.
To establish a firmer link between ERK activation and downstream MSK1 and CREB activation, we quantified the extent of their coactivation in single MSNs after cocaine. As shown in Figure 8, nuclear pERK, pMSK1, and pCREB signals all colocalized with each other in >90% of neurons analyzed, which is clearly above the incidence of colocalization predicted by randomness. Interestingly, MSK1 phosphorylation at Ser376 was accompanied by a sharp intracellular redistribution, from a rim-like perinuclear localization to an intranuclear accumulation (Fig. 8). In contrast, pThr581 signal appeared invariably nuclear regardless of changes in intensity, suggesting that it is a later phosphorylation event.
Figure 8.
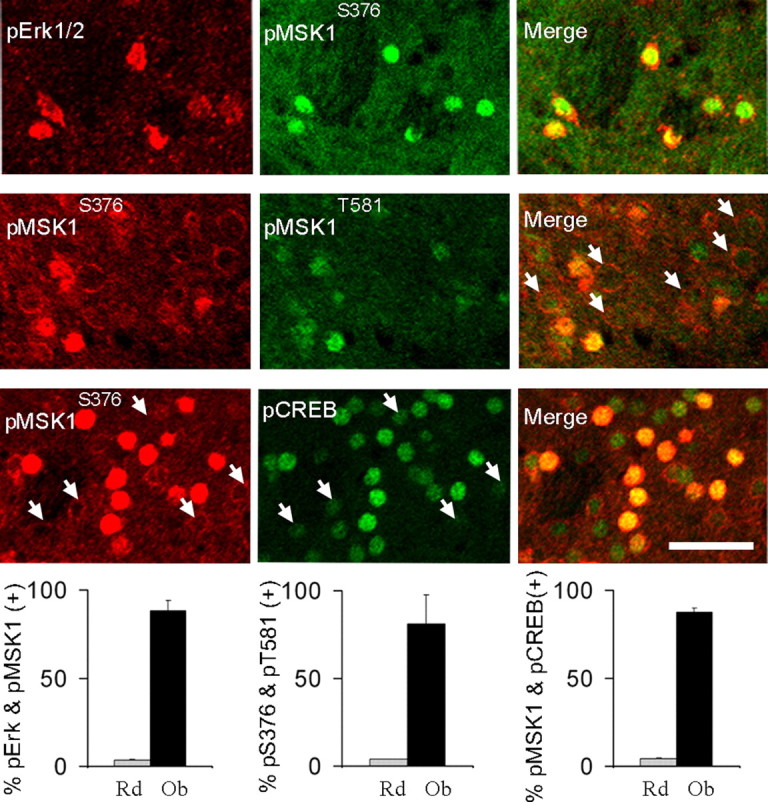
Cocaine treatment coactivates ERK, MSK1, and CREB in the same neurons. Images are three-dimensional reconstructions of optical z-stacks taken from the rostral, dorsomedial caudate–putamen of WT (Charles River Laboratories) mice. Representative examples of neurons showing nuclear colocalization of pERK and pMSK1Ser376 immunosignals (first row), pMSK1Ser376 and pMSK1Thr581 immunosignals (second row; arrows illustrate low pMSK1Thr581 levels in neurons lacking nuclear pMSK1Ser376), and pMSK1Ser376 and pCREB immunosignals (third row; arrows illustrate low pCREB levels in neurons lacking nuclear pMSK1). Bar graphs summarize the observed (Ob) percentage of colocalization versus colocalization predicted by randomness (Rd) for each pair of antibodies (the latter estimated by multiplying the individual percentages of immunopositive MSNs after cocaine). Between 150 and 200 neurons from four mice were sampled per staining group. Two-tailed t test: p < 0.000001 between random and observed values for all groups. Scale bar, 40 μm.
In summary, the above data indicate that AC1/8-dependent cAMP increases provide a major signal regulating the activation of the PKA/ERK/MSK1/CREB pathway by cocaine in the striatum. Additionally, the deletion of both AC1/AC8 blocks the development of cocaine-dependent locomotor sensitization.
Discussion
The cAMP signaling pathway has long been implicated in drug addiction, but the source of the cAMP signal has received very little attention. In this study, we show that the striatum expresses Ca2+-stimulated adenylyl cyclase activity, AC1/AC8 mRNA, and that genetic deletion of both AC1 and AC8 prevents the development of cocaine-induced psychomotor sensitization. To our knowledge, this is the first evidence of a role for specific adenylyl cyclases in cocaine sensitization. In addition to the behavioral phenotype of the DKO mice, these mice also exhibited deficits in the activation of signaling pathways typically induced by cocaine. In particular, activation of PKA and the ERK/MSK1/CREB transcriptional pathway, which are known to augment behavioral responses to cocaine, were significantly suppressed in DKO mice (Miserendino and Nestler, 1995; Carlezon et al., 1998; Nestler, 2004; Schroeder et al., 2004; Brami-Cherrier et al., 2005; Lu et al., 2006; Valjent et al., 2006b).
The major goal of this research was to determine the role of calcium-stimulated adenylyl cyclases in cocaine-dependent locomotor sensitization. We initially show that Ca2+-stimulated adenylyl cyclase activity is expressed in the striatum. The presence of Ca2+-stimulated adenylyl cyclase activity in the striatum has been reported in rodent and human striatal tissue by various other laboratories (Gnegy et al., 1980; Treisman et al., 1985; Ahlijanian and Cooper, 1988; Tong et al., 2001). In line with recognition of functional AC1 and AC8 protein seen with the cyclase activity data, AC1 mRNA has been shown to be expressed in the striatum and to be enriched in the dorsal striatum compared with the ventral striatum (Yao et al., 2004; Olsen et al., 2008). We show that mRNA for AC1 and AC8 is expressed in the striatum. On the basis of the adenylyl cyclase activity data in the wild-type mice and lack of activity in the DKO mice, as well as our RT-qPCR data, we feel that the presence of Ca2+-stimulated adenylyl cyclases in the striatum is indisputable.
Downstream of cAMP, several targets have been implicated in cocaine-induced neural plasticity, including AMPA receptors (Boudreau and Wolf, 2005; Boudreau et al., 2007; Kourrich et al., 2007) and the ERK/MSK1/CREB transcriptional pathway (Brami-Cherrier et al., 2005; Ferguson et al., 2006; Lu et al., 2006). We focused on the ERK/MSK1/CREB pathway because of our recent observations that its activation in the hippocampus after contextual fear conditioning is dependent on AC1 and AC8 (Sindreu et al., 2007). We found that basal striatal pERK levels were elevated in DKO mice, although additional analysis revealed that this increase was not localized to MSNs. Investigation of the particular neuronal subtype expressing increased basal levels of pERK in DKO mice indicated that large aspiny cholinergic interneurons are accounting, at least in part, for this increase. We also observed that cocaine administration to DKO mice did not stimulate increases in pERK levels in MSNs compared with those observed in WT controls.
This suppression of ERK activation could be responsible for the DKO animal's lack of locomotor sensitization to cocaine, because ERK activity is required for the development of cocaine sensitization (Ferguson et al., 2006; Valjent et al., 2006b). We hypothesize that ERK activation likely leads to the initiation of a transcriptional program necessary for the development of this long-term behavioral response but is not required for the acute response. This hypothesis is further supported by our data showing that, in DKO mice, an acute cocaine injection results in increased locomotion with suppressed phosphorylation of ERK compared with WT mice. DKO mice also lack locomotor sensitization, a typical consequence of chronic cocaine treatment. However, it is unlikely that the level of ERK activity has any relationship with the acute locomotor response to cocaine. Previous work has shown that suppression of ERK phosphorylation, via intraperitoneal injections of the MEK inhibitor SL327 before an acute injection of cocaine, does not reduce locomotor activity in mice. Additionally, SL327 does not block the expression of a previously acquired psychomotor sensitization (Valjent et al., 2006b). It has been reported recently that a critical factor in determining the magnitude of locomotion induced by an acute cocaine injection is the level of NMDA-type glutamate receptors present in the striatum (Ramsey et al., 2008).
As postulated by Girault et al. (2007), long-lasting cocaine-dependent behavior may be predicted by the status of ERK phosphorylation after acute cocaine exposure. Phosphorylated ERK, resulting from cocaine-induced glutamatergic and dopaminergic neurotransmission, may result in switching neurons into a plasticity permissive state (Girault et al., 2007). This would allow for the cellular and behavioral alterations evident in WT mice that receive chronic cocaine treatment. We hypothesize that activation of calcium-stimulated cyclases are essential for ERK activation and the resulting long-term behavioral adaptations. Our data, showing the lack of cocaine-dependent ERK activation and lack of psychomotor sensitization in the DKO mice, strongly corroborates our hypothesis.
In addition to the suppressed activation of ERK in DKO mice after cocaine, activation of MSK1 and CREB was also diminished. MSK1 is a direct substrate of ERK and has been shown to be particularly essential for the development of cocaine sensitization (Brami-Cherrier et al., 2005). Typically, ERK phosphorylates MSK1 at Thr581. Phosphorylation of this residue after acute cocaine treatment is reported here and has been shown by others (Brami-Cherrier et al., 2005; Bertran-Gonzalez et al., 2008). We extended the analysis of in vivo MSK1 phosphorylation by showing that cocaine treatment leads to the intranuclear accumulation of MSK1 phosphorylated at Ser376, a residue required for MSK1 activation (McCoy et al., 2005). Interestingly, under control conditions, pSer376 is retained in a distinct perinuclear distribution pattern in MSNs. However, after cocaine treatment, pSer376 dramatically shifts to a primarily intranuclear localization. In this condition, MSK1 phosphorylated at Ser376 colocalizes in ∼90% of activated neurons with MSK1 phosphorylated at Thr581. This interesting phenomenon indicates that phosphorylation of MSK1 at both of these essential sites is required for nuclear accumulation, likely allowing MSK1 to phosphorylate CREB. MSK1 has been shown to be the major kinase for the transcription factor CREB after context-dependent learning and cocaine exposure (Brami-Cherrier et al., 2005; Sindreu et al., 2007). In addition to our finding that ∼90% of neurons displaying activation of pERK colocalize with nuclear pMSK1 at Ser376, ∼90% of neurons exhibiting nuclear pMSK1 at Ser376 colocalize with nuclear pCREB. Upregulation of this signaling cascade in the same neurons supports the idea that there is a causal relationship between the activation of these signaling molecules after cocaine treatment. Because DKO lack MSK1 activation, it is not surprising that CREB phosphorylation is attenuated in DKO mice after cocaine treatment. The data presented here support the prevailing view that the ERK/MSK1/CREB transcriptional pathway is critical to neuronal adaptations leading to cocaine-induced behavioral plasticity. AC1 and AC8 appear to be indispensable for activation of this pathway in the striatum after cocaine, mirroring our observations in the hippocampus after the acquisition of contextual fear memory.
Drug addiction has been referred to as a disease of learning and memory (Hyman, 2005). Addicts form such strong associations between drug-related cues and reward that they are unable to break them despite negative consequences. This comparison has been strengthened by copious research identifying common properties and molecular mechanisms of drug-induced and associative learning-induced neural plasticity (Kelley, 2004; Hyman et al., 2006; Kauer and Malenka, 2007). Cocaine sensitization is no exception and shares several features with long-term hippocampus-dependent fear memory: persistence (Kalivas and Duffy, 1993; Belin et al., 2007), dependence on protein synthesis (Valjent et al., 2006a), dependence on the ERK signaling pathway (Brami-Cherrier et al., 2005; Valjent et al., 2006b), and involvement of the cAMP/PKA/CREB signaling pathway (Gelowitz and Berger, 2001; Nestler, 2001). Based on our current study, we can add dependence on Ca2+-stimulated adenylyl cyclases to this list.
The functional relevance of cocaine sensitization to addiction remains a subject of debate. Two recent studies dissociate locomotor sensitization from addictive behavior in rats as measured by drug-primed reinstatement of self-administration after withdrawal (Ahmed and Cador, 2006; Knackstedt and Kalivas, 2007), whereas two others report that cocaine-sensitized rats self-administered more drug and acquired the behavior at lower doses than saline-injected controls (Di Ciano, 2008; Zhang and Kosten, 2007). We examined cocaine conditioned place preference (CPP) in DKO mice and found it to be normal (data not shown), which suggests that AC1 and AC8 may not be required for the rewarding properties of cocaine. The DKO animal is not unique in demonstrating dissociation between locomotor sensitization to cocaine and CPP; several other mouse models have deficits or enhancements in one but not the other (Dong et al., 2004; Hall et al., 2004; Brami-Cherrier et al., 2005). Human data is scarce, but some small clinical studies have demonstrated sensitization to amphetamine in humans (Sax and Strakowski, 2001). Interestingly, a recent study reports higher levels of forskolin-stimulated adenylyl cyclase activity in nucleus accumbens tissue from deceased human smokers compared with former smokers and nonsmokers (Hope et al., 2007), which raises the possibility that AC1 and AC8 play a role in human addiction.
In summary, our data demonstrate that the Ca2+-stimulated adenylyl cyclases AC1 and AC8 contribute to cocaine-induced behavioral plasticity. We show that their deletion results in supersensitivity to acute cocaine injection, without locomotor sensitization to chronic cocaine. Molecularly, loss of AC1 and AC8 leads to suppressed activation of the ERK/MSK1/CREB transcriptional pathway in response to cocaine, providing a possible mechanism for the observed behavioral phenotypes. This mirrors our previous findings in the hippocampus using contextual fear conditioning, providing another example of addictive drugs usurping the natural experience-dependent mechanisms of the brain for neural plasticity.
Footnotes
This work was supported by National Institutes of Health (NIH) Grants NS20498 and MH 073601. D.P.D and Z.S.S. were supported in part by NIH Predoctoral Training Grants T32 DA07278 and T32 GM07108, respectively. C.B.S. was supported by NIH Postdoctoral Training Grant T32 AG000057. We thank members of the Storm laboratory for suggestions and critical reading of this manuscript and Mike Garelick and Aaron Nudelman for technical discussion.
References
- Ahlijanian MK, Cooper DM. Distinct interactions between Ca2+/calmodulin and neurotransmitter stimulation of adenylate cyclase in striatum and hippocampus. Cell Mol Neurobiol. 1988;8:459–469. doi: 10.1007/BF00711229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed SH, Cador M. Dissociation of psychomotor sensitization from compulsive cocaine consumption. Neuropsychopharmacology. 2006;31:563–571. doi: 10.1038/sj.npp.1300834. [DOI] [PubMed] [Google Scholar]
- Belin D, Deroche-Gamonet V, Jaber M. Cocaine-induced sensitization is associated with altered dynamics of transcriptional responses of the dopamine transporter, tyrosine hydroxylase, and dopamine D2 receptors in C57Bl/6J mice. Psychopharmacology (Berl) 2007;193:567–578. doi: 10.1007/s00213-007-0790-3. [DOI] [PubMed] [Google Scholar]
- Bertran-Gonzalez J, Bosch C, Maroteaux M, Matamales M, Hervé D, Valjent E, Girault JA. Opposing patterns of signaling activation in dopamine D1 and D2 receptor-expressing striatal neurons in response to cocaine and haloperidol. J Neurosci. 2008;28:5671–5685. doi: 10.1523/JNEUROSCI.1039-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudreau AC, Wolf ME. Behavioral sensitization to cocaine is associated with increased AMPA receptor surface expression in the nucleus accumbens. J Neurosci. 2005;25:9144–9151. doi: 10.1523/JNEUROSCI.2252-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudreau AC, Reimers JM, Milovanovic M, Wolf ME. Cell surface AMPA receptors in the rat nucleus accumbens increase during cocaine withdrawal but internalize after cocaine challenge in association with altered activation of mitogen-activated protein kinases. J Neurosci. 2007;27:10621–10635. doi: 10.1523/JNEUROSCI.2163-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brami-Cherrier K, Valjent E, Hervé D, Darragh J, Corvol JC, Pages C, Arthur SJ, Simon AJ, Girault JA, Caboche J. Parsing molecular and behavioral effects of cocaine in mitogen- and stress-activated protein kinase-1-deficient mice. J Neurosci. 2005;25:11444–11454. doi: 10.1523/JNEUROSCI.1711-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlezon WA, Jr, Thome J, Olson VG, Lane-Ladd SB, Brodkin ES, Hiroi N, Duman RS, Neve RL, Nestler EJ. Regulation of cocaine reward by CREB. Science. 1998;282:2272–2275. doi: 10.1126/science.282.5397.2272. [DOI] [PubMed] [Google Scholar]
- Di Ciano P. Facilitated acquisition but not persistence of responding for a cocaine-paired conditioned reinforcer following sensitization with cocaine. Neuropsychopharmacology. 2008;33:1426–1431. doi: 10.1038/sj.npp.1301542. [DOI] [PubMed] [Google Scholar]
- Dong Y, Saal D, Thomas M, Faust R, Bonci A, Robinson T, Malenka RC. Cocaine-induced potentiation of synaptic strength in dopamine neurons: behavioral correlates in GluRA(-/-) mice. Proc Natl Acad Sci U S A. 2004;101:14282–14287. doi: 10.1073/pnas.0401553101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Y, Green T, Saal D, Marie H, Neve R, Nestler EJ, Malenka RC. CREB modulates excitability of nucleus accumbens neurons. Nat Neurosci. 2006;9:475–477. doi: 10.1038/nn1661. [DOI] [PubMed] [Google Scholar]
- Ferguson SM, Fasano S, Yang P, Brambilla R, Robinson TE. Knockout of ERK1 enhances cocaine-evoked immediate early gene expression and behavioral plasticity. Neuropsychopharmacology. 2006;31:2660–2668. doi: 10.1038/sj.npp.1301014. [DOI] [PubMed] [Google Scholar]
- Fienberg AA, Hiroi N, Mermelstein PG, Song W, Snyder GL, Nishi A, Cheramy A, O'Callaghan JP, Miller DB, Cole DG, Corbett R, Haile CN, Cooper DC, Onn SP, Grace AA, Ouimet CC, White FJ, Hyman SE, Surmeier DJ, Girault J, Nestler EJ, Greengard P. DARPP-32: regulator of the efficacy of dopaminergic neurotransmission. Science. 1998;281:838–842. doi: 10.1126/science.281.5378.838. [DOI] [PubMed] [Google Scholar]
- Gelowitz DL, Berger SP. Signal transduction mechanisms and behavioral sensitization to stimulant drugs: an overview of cAMP and PLA2. J Addict Dis. 2001;20:33–42. doi: 10.1300/J069v20n03_04. [DOI] [PubMed] [Google Scholar]
- Girault JA, Valjent E, Caboche J, Hervé D. ERK2: a logical AND gate critical for drug-induced plasticity? Curr Opin Pharmacol. 2007;7:77–85. doi: 10.1016/j.coph.2006.08.012. [DOI] [PubMed] [Google Scholar]
- Gnegy ME, Hultin T, Treisman G. Effect of calmodulin on catecholamine-linked adenylate cyclase activity in rat striatum and cerebral cortex. Adv Biochem Psychopharmacol. 1980;21:125–131. [PubMed] [Google Scholar]
- Hall FS, Goeb M, Li XF, Sora I, Uhl GR. micro-Opioid receptor knockout mice display reduced cocaine conditioned place preference but enhanced sensitization of cocaine-induced locomotion. Brain Res Mol Brain Res. 2004;121:123–130. doi: 10.1016/j.molbrainres.2003.10.024. [DOI] [PubMed] [Google Scholar]
- Heidbreder CA, Thompson AC, Shippenberg TS. Role of extracellular dopamine in the initiation and long-term expression of behavioral sensitization to cocaine. J Pharmacol Exp Ther. 1996;278:490–502. [PubMed] [Google Scholar]
- Hope BT, Nagarkar D, Leonard S, Wise RA. Long-term upregulation of protein kinase A and adenylate cyclase levels in human smokers. J Neurosci. 2007;27:1964–1972. doi: 10.1523/JNEUROSCI.3661-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horger BA, Shelton K, Schenk S. Preexposure sensitizes rats to the rewarding effects of cocaine. Pharmacol Biochem Behav. 1990;37:707–711. doi: 10.1016/0091-3057(90)90552-s. [DOI] [PubMed] [Google Scholar]
- Hyman SE. Addiction: a disease of learning and memory. Am J Psychiatry. 2005;162:1414–1422. doi: 10.1176/appi.ajp.162.8.1414. [DOI] [PubMed] [Google Scholar]
- Hyman SE, Malenka RC, Nestler EJ. Neural mechanisms of addiction: the role of reward-related learning and memory. Annu Rev Neurosci. 2006;29:565–598. doi: 10.1146/annurev.neuro.29.051605.113009. [DOI] [PubMed] [Google Scholar]
- Kalivas PW, Duffy P. Time course of extracellular dopamine and behavioral sensitization to cocaine. I. Dopamine axon terminals. J Neurosci. 1993;13:266–275. doi: 10.1523/JNEUROSCI.13-01-00266.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauer JA, Malenka RC. Synaptic plasticity and addiction. Nat Rev Neurosci. 2007;8:844–858. doi: 10.1038/nrn2234. [DOI] [PubMed] [Google Scholar]
- Kelley AE. Memory and addiction: shared neural circuitry and molecular mechanisms. Neuron. 2004;44:161–179. doi: 10.1016/j.neuron.2004.09.016. [DOI] [PubMed] [Google Scholar]
- Knackstedt LA, Kalivas PW. Extended access to cocaine self-administration enhances drug-primed reinstatement but not behavioral sensitization. J Pharmacol Exp Ther. 2007;322:1103–1109. doi: 10.1124/jpet.107.122861. [DOI] [PubMed] [Google Scholar]
- Kourrich S, Rothwell PE, Klug JR, Thomas MJ. Cocaine experience controls bidirectional synaptic plasticity in the nucleus accumbens. J Neurosci. 2007;27:7921–7928. doi: 10.1523/JNEUROSCI.1859-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombroso PJ, Naegele JR, Sharma E, Lerner M. A protein tyrosine phosphatase expressed within dopaminoceptive neurons of the basal ganglia and related structures. J Neurosci. 1993;13:3064–3074. doi: 10.1523/JNEUROSCI.13-07-03064.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu L, Koya E, Zhai H, Hope BT, Shaham Y. Role of ERK in cocaine addiction. Trends Neurosci. 2006;29:695–703. doi: 10.1016/j.tins.2006.10.005. [DOI] [PubMed] [Google Scholar]
- McCoy CE, Campbell DG, Deak M, Bloomberg GB, Arthur JS. MSK1 activity is controlled by multiple phosphorylation sites. Biochem J. 2005;387:507–517. doi: 10.1042/BJ20041501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miserendino MJ, Nestler EJ. Behavioral sensitization to cocaine: modulation by the cyclic AMP system in the nucleus accumbens. Brain Res. 1995;674:299–306. doi: 10.1016/0006-8993(95)00030-t. [DOI] [PubMed] [Google Scholar]
- Morgan D, Liu Y, Roberts DC. Rapid and persistent sensitization to the reinforcing effects of cocaine. Neuropsychopharmacology. 2006;31:121–128. doi: 10.1038/sj.npp.1300773. [DOI] [PubMed] [Google Scholar]
- Nestler EJ. Molecular basis of long-term plasticity underlying addiction. Nat Rev Neurosci. 2001;2:119–128. doi: 10.1038/35053570. [DOI] [PubMed] [Google Scholar]
- Nestler EJ. Molecular mechanisms of drug addiction. Neuropharmacology. 2004;47(Suppl 1):24–32. doi: 10.1016/j.neuropharm.2004.06.031. [DOI] [PubMed] [Google Scholar]
- Olsen CM, Huang Y, Goodwin S, Ciobanu DC, Lu L, Sutter TR, Winder DG. Microarray analysis reveals distinctive signaling between the bed nucleus of the stria terminalis, nucleus accumbens, and dorsal striatum. Physiol Genomics. 2008;32:283–298. doi: 10.1152/physiolgenomics.00224.2006. [DOI] [PubMed] [Google Scholar]
- Ouimet CC, Miller PE, Hemmings HC, Jr, Walaas SI, Greengard P. DARPP-32, a dopamine- and adenosine 3′:5′-monophosphate-regulated phosphoprotein enriched in dopamine-innervated brain regions. III. Immunocytochemical localization. J Neurosci. 1984;4:111–124. doi: 10.1523/JNEUROSCI.04-01-00111.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Partridge B, Schenk S. Context-independent sensitization to the locomotor-activating effects of cocaine. Pharmacol Biochem Behav. 1999;63:543–548. doi: 10.1016/s0091-3057(99)00013-1. [DOI] [PubMed] [Google Scholar]
- Ramsey AJ, Laakso A, Cyr M, Sotnikova TD, Salahpour A, Medvedev IO, Dykstra LA, Gainetdinov RR, Caron MG. Genetic NMDA receptor deficiency disrupts acute and chronic effects of cocaine but not amphetamine. Neuropsychopharmacology. 2008;33:2701–2714. doi: 10.1038/sj.npp.1301663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts DC, Morgan D, Liu Y. How to make a rat addicted to cocaine. Prog Neuropsychopharmacol Biol Psychiatry. 2007;31:1614–1624. doi: 10.1016/j.pnpbp.2007.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson TE, Berridge KC. The neural basis of drug craving: an incentive-sensitization theory of addiction. Brain Res Brain Res Rev. 1993;18:247–291. doi: 10.1016/0165-0173(93)90013-p. [DOI] [PubMed] [Google Scholar]
- Robinson TE, Berridge KC. Incentive-sensitization and addiction. Addiction. 2001;96:103–114. doi: 10.1046/j.1360-0443.2001.9611038.x. [DOI] [PubMed] [Google Scholar]
- Sax KW, Strakowski SM. Behavioral sensitization in humans. J Addict Dis. 2001;20:55–65. doi: 10.1300/J069v20n03_06. [DOI] [PubMed] [Google Scholar]
- Schroeder JA, Hummel M, Unterwald EM. Repeated intracerebroventricular forskolin administration enhances behavioral sensitization to cocaine. Behav Brain Res. 2004;153:255–260. doi: 10.1016/j.bbr.2003.12.005. [DOI] [PubMed] [Google Scholar]
- Segal DS, Kuczenski R. Repeated cocaine administration induces behavioral sensitization and corresponding decreased extracellular dopamine responses in caudate and accumbens. Brain Res. 1992;577:351–355. doi: 10.1016/0006-8993(92)90297-m. [DOI] [PubMed] [Google Scholar]
- Sindreu CB, Scheiner ZS, Storm DR. Ca2+-stimulated adenylyl cyclases regulate ERK-dependent activation of MSK1 during fear conditioning. Neuron. 2007;53:79–89. doi: 10.1016/j.neuron.2006.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong J, Ross BM, Sherwin AL, Kish SJ. Dopamine D1-stimulated adenylyl cyclase activity in cerebral cortex of autopsied human brain. Neurochem Int. 2001;39:117–125. doi: 10.1016/s0197-0186(01)00016-x. [DOI] [PubMed] [Google Scholar]
- Treisman GJ, Muirhead N, Iwaniec L, Gnegy ME. Inhibition of a low Km GTPase activity in rat striatum by calmodulin. J Neurochem. 1985;44:518–525. doi: 10.1111/j.1471-4159.1985.tb05444.x. [DOI] [PubMed] [Google Scholar]
- Valjent E, Pascoli V, Svenningsson P, Paul S, Enslen H, Corvol JC, Stipanovich A, Caboche J, Lombroso PJ, Nairn AC, Greengard P, Hervé D, Girault JA. Regulation of a protein phosphatase cascade allows convergent dopamine and glutamate signals to activate ERK in the striatum. Proc Natl Acad Sci U S A. 2005;102:491–496. doi: 10.1073/pnas.0408305102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valjent E, Corbillé AG, Bertran-Gonzalez J, Hervé D, Girault JA. Inhibition of ERK pathway or protein synthesis during reexposure to drugs of abuse erases previously learned place preference. Proc Natl Acad Sci U S A. 2006a;103:2932–2937. doi: 10.1073/pnas.0511030103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valjent E, Corvol JC, Trzaskos JM, Girault JA, Hervé D. Role of the ERK pathway in psychostimulant-induced locomotor sensitization. BMC Neurosci. 2006b;7:20. doi: 10.1186/1471-2202-7-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong ST, Athos J, Figueroa XA, Pineda VV, Schaefer ML, Chavkin CC, Muglia LJ, Storm DR. Calcium-stimulated adenylyl cyclase activity is critical for hippocampus-dependent long-term memory and late phase LTP. Neuron. 1999;23:787–798. doi: 10.1016/s0896-6273(01)80036-2. [DOI] [PubMed] [Google Scholar]
- Woolverton WL, Johnson KM. Neurobiology of cocaine abuse. Trends Pharmacol Sci. 1992;13:193–200. doi: 10.1016/0165-6147(92)90063-c. [DOI] [PubMed] [Google Scholar]
- Wu ZL, Thomas SA, Villacres EC, Xia Z, Simmons ML, Chavkin C, Palmiter RD, Storm DR. Altered behavior and long-term potentiation in type I adenylyl cyclase mutant mice. Proc Natl Acad Sci U S A. 1995;92:220–224. doi: 10.1073/pnas.92.1.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao WD, Gainetdinov RR, Arbuckle MI, Sotnikova TD, Cyr M, Beaulieu JM, Torres GE, Grant SG, Caron MG. Identification of PSD-95 as a regulator of dopamine-mediated synaptic and behavioral plasticity. Neuron. 2004;41:625–638. doi: 10.1016/s0896-6273(04)00048-0. [DOI] [PubMed] [Google Scholar]
- Zhang XY, Kosten TA. Previous exposure to cocaine enhances cocaine self-administration in an alpha 1-adrenergic receptor dependent manner. Neuropsychopharmacology. 2007;32:638–645. doi: 10.1038/sj.npp.1301120. [DOI] [PubMed] [Google Scholar]