

Abstract
The ClC transport protein family comprises both Cl− ion channel and H+/Cl− and H+/NO3− exchanger members. Structural studies on a bacterial ClC transporter reveal a pore obstructed at its external opening by a glutamate side-chain which acts as a gate for Cl− passage and in addition serves as a staging post for H+ exchange. This same conserved glutamate acts as a gate to regulate Cl− flow in ClC channels. The activity of ClC-2, a genuine Cl− channel, has a biphasic response to extracellular pH with activation by moderate acidification followed by abrupt channel closure at pH values lower than ∼7. We have now investigated the molecular basis of this complex gating behaviour. First, we identify a sensor that couples extracellular acidification to complete closure of the channel. This is extracellularly-facing histidine 532 at the N-terminus of transmembrane helix Q whose neutralisation leads to channel closure in a cooperative manner. We go on to show that acidification-dependent activation of ClC-2 is voltage dependent and probably mediated by protonation of pore gate glutamate 207. Intracellular Cl− acts as a voltage-independent modulator, as though regulating the pKa of the protonatable residue. Our results suggest that voltage dependence of ClC-2 is given by hyperpolarisation-dependent penetration of protons from the extracellular side to neutralise the glutamate gate deep within the channel, which allows Cl− efflux. This is reminiscent of a partial exchanger cycle, suggesting that the ClC-2 channel evolved from its transporter counterparts.
Flow of ions across membranes is central in many processes, including the building of intra- to extracellular gradients, generation of electrical signals, control of cell volume and epithelial transport. Transporters are capable of building up gradients by coupling ion movement to ATP hydrolysis or the downhill movement of other ions. Ion channels, on the other hand, mediate passive downhill ion movement often at very high ion conduction rates. Although ion channels and transporters share the property of ion selectivity, they are believed to function in fundamentally different ways (Gadsby, 2004). Ion channel function requires a continuous aqueous pore through the protein to ensure ion migration by electrodiffusion. To allow for regulated flux, there must be also a mechanism for opening and closing the permeation pathway, a process termed the gating of the channel. Transporters, on the other hand, require at least two gates never simultaneously open that temporarily trap the ion within the pore. As might be expected from their functional differences, ion channels and transporters belong to different protein families. Notable exceptions to this rule are the cystic fibrosis transmembrane conductance regulator (CFTR) Cl− channel, which belongs to the ATP-binding cassette ABC family of transporters (Gadsby et al. 2006), and the ClC transport protein family, which has both Cl− ion channel and H+-coupled Cl− (or NO3−) transporter members (Accardi & Miller, 2004; Picollo & Pusch, 2005; Scheel et al. 2005; De et al. 2006; De Angeli et al. 2006; Graves et al. 2008).
ClC transporters and channels are well represented in prokaryotes and in various organs and tissues of eukaryotes, where they perform important roles in normal physiology and disease (Jentsch et al. 2005). Available bacterial ClC structures reveal a selectivity filter providing a transmembrane pathway for chloride ions which is obstructed at its external opening by a highly conserved glutamate residue side-chain (Dutzler et al. 2002). Extrapolating from the bacterial structure, a displacement of the conserved selectivity filter glutamate to clear the pathway for ion passage is believed to be the process gating the pore in ClC channels (Dutzler et al. 2003; Niemeyer et al. 2003; Estévez et al. 2003). In the transporters, the equivalent residue serves to capture a proton translocated in exchange for Cl− across the plasma membrane in a cycle of conformational changes not yet well defined (Accardi & Miller, 2004; Accardi et al. 2005).
ClC-2 is an inwardly rectifying plasma membrane Cl− channel expressed in various epithelia, the brain and in the heart (Thiemann et al. 1992). The gating of ClC-2 by membrane hyperpolarisation, mild extracellular acidification and intracellular Cl− has been the object of several studies (Gründer et al. 1992; Jordt & Jentsch, 1997; Pusch et al. 1999; Niemeyer et al. 2003; Zúñiga et al. 2004; de Santiago et al. 2005; Yusef et al. 2006). The conserved glutamate of the selectivity filter plays a central role in the activation of the ClC-2 chloride channel by intracellular Cl− and hyperpolarisation (Niemeyer et al. 2003). Extracellular pH, on the other hand, affects ClC-2 gating in a complex manner, with activation by mild acidification superimposed on a complete inhibition by stronger acidification (Arreola et al. 2002; Niemeyer et al. 2003). Here we show, firstly, that the complete inhibition of ClC-2 by extreme acidification is sensed by extracellular-facing histidine 532 located at the N-terminus end of transmembrane helix Q. And secondly, that the voltage-dependent gating step in channel opening is the protonation, probably of the conserved pore gate glutamate, promoted by H+ influx into the selectivity filter. In this process intracellular Cl− acts in a permissive role, consistent with modulation of the gate glutamate pKa in a voltage-independent manner. This model is reminiscent of a partial reaction of an H+/Cl− exchanger, suggesting the idea that ClC channels might have evolved from their transporter counterparts utilising relicts of the exchanger cycle in their gating.
Methods
Complementary DNA constructs and transfections
The ClC-2 cDNA was ClC-2Δ77-86 from Cavia porcellus (Cid et al. 2000). Numbering corresponds to GenBank sequence no. AF113529. Mutagenesis was done using PCR and confirmed by sequencing. HEK-293 cells were grown and transiently transfected with expression plasmids for the various ClC-2 constructs and πH3-Cd8 to identify effectively transfected cells as described previously (Cid et al. 2000). Experiments were performed on cells in 35 mm cell culture plastic Petri dishes mounted directly on the microscope stage.
Electrophysiology and solutions
Standard whole cell patch-clamp recordings were performed as described elsewhere (Yusef et al. 2006). The composition of the solutions used is given in Table 1. The pH of solutions was checked at the end of each experiment.
Table 1.
Composition of solutions used in electrophysiological experiments
| [Cl−] (mm) of intracellular solutions |
Bath solutions |
||||||
|---|---|---|---|---|---|---|---|
| 10 | 35 | 60 | 135 | 200 | Standard | 200 [Cl−]i | |
| NaCl | — | — | — | — | — | 140 | 140 |
| Na gluconate | 125 | 100 | 75 | — | — | — | — |
| CsCl | 8 | 33 | 58 | 133 | 188 | — | — |
| CaCl2 | — | — | — | — | — | 2 | 2 |
| MgCl2 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| sucrose | — | — | — | — | — | 22 | 85 |
| EGTA | 2 | 2 | 2 | 2 | 2 | — | — |
| ATP | 1 | 1 | 1 | 1 | 1 | — | — |
| HEPES-TRIS | 10 | 10 | 10 | 10 | 10 | 10 | 10 |
| pH | 7.4 | 7.4 | 7.4 | 7.4 | 7.4 | 7.4 | 7.4 |
All concentrations are given in mm. To obtain the solutions with different extracellular pH values the following buffers were used: pH 4.5–6.5, 2-morpholinoethanesulfonic acid (Mes); pH 7.0–8.0, N-(2-hydroxyethyl)-piperazine-N′-2-ethanesulfonic acid (Hepes), both adjusted with Tris; pH 8.5–9.0, N-(1,1-dimethyl-2-hydroxyethyl)-3-amino-2-hydroxypropanesulfonic acid (Ampso), adjusted with Hepes; pH 10.0–11.0, 3-(cyclohexylamino)-1-propanesulfonic acid (Caps) adjusted with NaOH. The pH of solutions was checked at the end of each experiment. Bath solution labelled 200 [Cl−]i is a hypertonic medium used in conjunction with the 200 mm intracellular Cl− pipette solution.
Data analysis
To analyse the biphasic titration curves, a double Hill equation (Arreola et al. 2002) was fitted to the data:
| (1) |
Where I is the current at a given extracellular pH, [H+] is the external proton concentration and actKa and inhKa are the dissociation constants of protonation, nact and ninh are Hill coefficients.
To account for the voltage-dependent concentration of protons within the electrical field of the membrane, a model developed by Woodhull (Woodhull, 1973) was used. The observed pK1/2 as a function of membrane potential is given by:
| (2) |
where pK1/2 (0 mV) is the pK1/2 value in the absence of an applied voltage, z is the valence of the ion, V is the voltage across the membrane, δ is the fraction of the membrane voltage at the binding site and F, R and T have their usual meaning. In this analysis we have assumed that there is no H+ permeation (‘punch-through’) or competition for Cl− binding sites in ClC-2 channels.
pKa shift calculations
To get some insight into the effects of the intracellular chloride on the gating glutamate we performed quantum-chemical energy calculations (Jensen et al. 2005) on reduced chemical models of the gating glutamate in order to estimate the pKa shift (ΔpKa) due to the presence of Cl− at Scen (ΔpKa1) (see equation below) and the extra pKa shift due to a second Cl− at Sint (ΔpKa2) (Jensen et al. 2005).
 |
(3) |
For this purpose we need to estimate the energy change of the glutamate protonation reaction in the presence (Cl) and absence (no Cl) of chlorides. Assuming that these energy differences ΔΔE are a reasonable approximation to the differences in free energies ΔΔG, we estimated the pKa shifts of the gating glutamate for the two chloride configurations mentioned above. These calculations were made on EcClC and a model of ClC-2.
The solvated energies 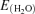 of the unprotonated (EGlu−) or protonated (EGluH) glutamate species were calculated with the widely used Density Functional Theory (DFT) quantum-chemical approach. The self consistent reaction field (SCRF) model was used considering a water dielectric medium. All computed energies were performed with the Gaussian 03 (B.04) software package (Gaussian, Inc., Pittsburgh, PA, USA) on the structures without any geometry relaxation. For the pKa shifts a temperature of 298 K was assumed.
of the unprotonated (EGlu−) or protonated (EGluH) glutamate species were calculated with the widely used Density Functional Theory (DFT) quantum-chemical approach. The self consistent reaction field (SCRF) model was used considering a water dielectric medium. All computed energies were performed with the Gaussian 03 (B.04) software package (Gaussian, Inc., Pittsburgh, PA, USA) on the structures without any geometry relaxation. For the pKa shifts a temperature of 298 K was assumed.
ClC structures completed with hydrogens at standard distances were taken from: 1OTS (chain A) ClC PDB structure from Escherichia coli (EcClC); a ClC-2 monomer model structure from Cavia porcellus (cpClC-2) was built with the homology modelling software MODELLER (Sali & Blundell, 1993). The alignment used is similar to the one reported by Dutzler et al. (2002). In the absence of direct structural information for ClC-2, we have resorted to homology modelling based on the available bacterial protein structure. In spite of the amino acid differences between the bacterial and the ClC-2 proteins, the gating-glutamate zone considered in the calculations is highly conserved within the ClC family. Therefore, in principle one should not expect major structural differences in this zone where the uncertainties are lower. Nevertheless, the conclusions drawn must necessarily remain speculative but are a valuable basis for further experimental work.
These structures were chemically simplified, deleting atoms and conserving the amino acids conformations to the following. (a) Minimal models (∼17 atoms), which consider only the gating glutamate including its backbone amide nitrogen (Model 1). The chlorides were placed at equivalent distances in Scen and Sint. (b) Models considering ∼100 selectivity filter atoms around the glutamate (SF100). In this way we could take into account the contribution of the local protein environment to the glutamate pKa shifts and to the chloride–glutamate interactions.
We created two hypothetical protonated states of the glutamate adding the proton to the carboxylic oxygen more accessible from the extracellular side of the models. The proton geometry was left coplanar to the carboxylate-group plane. We considered as the closed conformations those where the glutamate occupies the Sext position. The open conformations were created translating the glutamine side-chain dihedrals of the E148Q ClC structure (PDB: 1OTU) to the corresponding glutamate. Chlorides within ClC-2 models at Scen and Sin positions were placed after a backbone geometrical fitting over 1OTS crystallographic structure.
Results
Non-additive activation of ClC-2 by hyperpolarisation and extracellular acidification
The widely distributed mammalian ClC-2 Cl− channels are gated open by hyperpolarisation, intracellular Cl− and moderate extracellular acidification (Jordt & Jentsch, 1997; Niemeyer et al. 2003; Zúñiga et al. 2004). The steady-state activity of ClC-2 displays a bell-shaped dependence on extracellular pH with a maximum at pH ∼7, compatible with the presence of two independent protonatable sites with opposing effects on channel gating (Arreola et al. 2002). Our previous work (Niemeyer et al. 2003) suggested that protonation of the side chain of selectivity filter E207 (which was erroneously referred to as E217 in Niemeyer et al. 2003) accounts for the activation site and, accordingly, its neutralisation in mutant E207V gives an active channel at alkaline pH but with unaltered inhibition by acid pH. ClC-2 channels are inhibited by extracellular acidification with a pK1/2 of around 6.2, and lowering extracellular pH to 5.5 leads to complete blockade of ClC-2 mediated currents (Jordt & Jentsch, 1997; Arreola et al. 2002; Niemeyer et al. 2003). Nevertheless it is possible to see a fast transient activation of ClC-2 channels with pH 5.5 before the much slower inhibition takes place (Arreola et al. 2002; Niemeyer et al. 2003). We argued that this fast activation is due to protonation of E207 as it is absent in the ClC-2-E207V mutant (Niemeyer et al. 2003). If protonation of E207 and hyperpolarisation were not resulting in full opening, i.e. not achieving maximal Po, we would expect their effects to be additive. To test this, we examined the degree of transient activation by fast acidification to pH 5.5 at different voltages. As seen in Fig. 1A–D the more negative the voltage, and higher the apparent Po, the less the effect of pH 5.5. This effect is not due to a H+/Cl− exchange process with a significantly high stoichiometry, as the reversal potential was always close to the equilibrium potential for Cl− independently of pHo. This is documented by the current–voltage curves in Fig. 1E and F, where data with ClC-2 and a mutant, which lacks inhibition by protons (see below), are shown. The results might imply that effects of hyperpolarisation and acidification are detected by independent sensors either of which can effect full channel opening. On the other hand, they are also consistent with the idea of channel opening by hyperpolarisation entailing protonation of the selectivity filter E207. An increase in the apparent pK1/2 for E207 neutralisation by hyperpolarisation could account for this effect. To test this more directly we constructed titration curves for ClC-2 activity at different potentials.
Figure 1. Hyperpolarisation potentiates transient activation of ClC-2 Cl− channel by extracellular acidification.

A–D, current recordings in a HEK-293 cell expressing ClC-2 channels. The cell was held at a potential of 0 mV and channel opening was elicited by pulses to −65, −95, −120 or −140 mV, as indicated. These main activating pulses were followed by a depolarisation to 30 mV to evoke channel closure. The records shown were taken under continuous superfusion with extracellular solutions of the pH values indicated, except for control records (shown in grey) that were maintained at pH 7.4. Intracellular Cl− and pH were 35 mm and 7.4 throughout. Inward, negative current induced by the hyperpolarising pulses corresponds to efflux of Cl− from the cell into the bath. The measured fold increase in current is shown next to each peak stimulation evoked by pH 5.5 superfusion. E–F, current–voltage relationships for ClC-2 and ClC-2-H532F mutant at various extracellular pH values. The points were obtained during rapid (50 ms) voltage ramps given after activating the channels to −170 (E) or −130 (F) mV. The arrows point to the value of the equilibrium potential for Cl− in the abscissa. Results shown are typical of 5 similar sets of experiments. Notice that in E triangles are obscured by circles, with which they coincide.
The apparent pK1/2 of acidification-dependent activation of ClC-2, but not its inhibition, is dependent upon voltage
Figure 2A and B are current traces showing ClC-2 channel opening in response to hyperpolarisation pulses at −70 and −170 mV followed by pulses to 30 mV during which channels close. Acidification up to pH 7.0 evoked a marked increase in current. Sizable currents were seen at pH 9.0 and 8.5 at −170 mV but not at −70 mV. At pH 5.5 there was full inhibition of channel activity at both potentials. Figure 2C shows titration curves at −70 and −170 mV. The biphasic nature of the curves was maintained at both voltages and inhibition at acid pH occurred with the same pK1/2 value at −70 and −170 mV respectively. Activation by protons, on the other hand, took place with widely different pK1/2 values. The more negative the potential, the more alkaline the apparent activation pK1/2: 8.6 at −170 mV and 7.1 at −70 mV. Two explanations could account for this result: if activation were due to neutralisation of E207, hyperpolarisation might lead to a conformational change that alters the pKa of the E207 side chain. Alternatively, a change in the concentration of protons in the microenvironment of the residue driven by hyperpolarisation might be taking place. To be able to study this phenomenon in more detail, and particularly to estimate the pK1/2 activation values more accurately, we searched for the sensor responsible for proton inhibition of ClC-2 in order to inactivate it.
Figure 2. Hyperpolarisation shifts the dependence of ClC-2 Cl− channel activation upon moderate extracellular acidification but does not affect inhibition by strong acidification.

A–B, current recordings in a HEK-293 cell expressing ClC-2 channels. The cell was held a potential of 0 mV and channel opening was elicited by pulses to −70 (A) or −170 mV (B) at the indicated extracellular pH values. The main pulses were followed by a depolarisation to 30 mV to evoke channel closure. C, summary of results for experiments as in A and B showing the pH dependence of the currents at −170 (triangles) and −70 mV (circles). The lines are fits of a double Hill model (eqn (1)) to the data (n= 4 each) and yielded the constants (pKa) shown. The Hill coefficients were nact 0.74 and 0.97 and ninh 1.94 and 1.81 for data at −170 and −70 mV respectively. Intracellular Cl− and pH were 35 mm and 7.4 throughout. Error bars indicate s.e.m.
Identification of the molecular sensor responsible for ClC-2 inhibition by extracellular acidification
The inhibition of ClC-2 currents by extracellular acidification occurs with a pK1/2 of 6.1–6.2, which would be consistent with the protonation/deprotonation of a histidine residue. An examination of a topology model for ClC-2 based on the structure of a bacterial ClC protein revealed three potentially outside-facing histidine residues (Fig. 3A), H212, H487 and H532. Mutation of H212 and H487 to asparagine (N) had no discernible effect on ClC-2 pH dependence (not shown). ClC-2-H532N and ClC-2-H532A showed no activity when expressed in HEK-293 cells, despite the fact that HA-tagged mutants were seen to be expressed in the plasma membrane (not shown). However, as shown in Fig. 3B, a robust expression could be measured when mutant ClC-2-H532F was used. Currents mediated by ClC-2-H532F were similar to those of the non-mutated channel but differed in voltage dependence. As seen in Fig. 3C the V0.5 for the mutant measured at 135 mm intracellular Cl− was −115 ± 6 mV with a slope factor of −24 ± 1.6 mV (n= 5). This V0.5 value is some 45 mV more negative than that of WT ClC-2 (Catalán et al. 2004) but there was no change in slope factor (triangles in Fig. 3C). What was remarkable in the mutant channel was its dependence upon extracellular pH, as acidification even to pH 5.5 enhanced the currents mediated by ClC-2-H532F (Fig. 3D). In Fig. 3E, current recorded at −130 mV is plotted as a function of extracellular pH. The currents rose monotonically with acidification and activation took place with a pK1/2 of 7.6. The inhibition normally seen in WT or ClC-2-E207V (shown for comparison as triangles) channels was absent in ClC-2-H532F.
Figure 3. Effect of neutralisation of H532 by mutation on ClC-2 function.

A, topological model of ClC-2 showing the position of histidine residues potentially exposed to the extracellular solution. In grey, regions thought to be involved in the selectivity filter. The voltage dependence of ClC-2-H532F is shown in B as currents elicited by pulses to the indicated voltages from a holding potential of 0 mV and the indicated intra- and extracellular Cl− concentrations. The extracellular pH was 7.4. C, Boltzmann equation fit to voltage dependence of apparent open probability obtained from experiments as in B. Means ±s.e.m., n= 8, circles. Triangles show previously published data for WT ClC-2 under the same conditions (Catalán et al. 2004) for comparison. D, effect of extracellular pH on currents elicited by pulses to −130 mV. E, steady-state currents as a function of extracellular pH observed for ClC-2-H532F (circles) and ClC-2-E207V (triangles) mutant channels. The fits of single Hill equations to the data gave pK1/2 values of 7.57 ± 0.08 for ClC-2-H532F and 6.25 ± 0.10 for ClC-2-E207V. Respective nH values were 0.8 ± 0.15 and 2.04 ± 0.21. Means ±s.e.m., n= 7 for both sets of experiments.
The H532F mutant of ClC-2 allows a better test of whether activation by hyperpolarisation and acidification are additive. We compared the instantaneous tail currents obtained at 40 mV after a saturating test pulse of −200 mV at pHo 7.4 with that obtained by after a pulse at −80 mV, but at pHo of 5.5, as paired experiments in the same cell (Fig. 4A and B). The instantaneous currents measured as tail currents at 40 mV were the same, whether elicited by the saturating −200 mV stimulus at pH 7.4 or by a subsaturating voltage of −80 mV but at pH 5.5. Figure 4C shows average results of pooled experiments that confirmed this view. This experiment indicates that the same conductance, or apparent open probability, can be reached by maximal hyperpolarisation and maximal acidification.
Figure 4. Eliciting maximal currents by hyperpolarisation or acidification.

A, example of the maximal conductance evoked by a saturating −200 mV squared voltage pulse followed by a tail current measured at 40 mV at pHo 7.4. B, currents elicited in the same cell as in A by a pulse to −80 mV followed by a tail-current measuring pulse to 40 mV at pHo 7.4 (grey trace) or pHo 5.5. Notice that there was no difference in the instantaneous tail currents in A (1.36 nA) or in B at pHo 5.5 (1.40 nA). C, summary of tail currents measured as in A and B (means ±s.e.m., n= 4).
Voltage-driven influx of H+ into the selectivity filter gates ClC-2 Cl− channel
As shown above, neutralisation of the pH-sensing histidine in the mutant H532F-ClC-2 abolished inhibition by acidification but preserved proton activation. This affords the possibility to study proton activation within a wide pH range. The experiment shown in Fig. 5A confirms that proton activation of the ClC-2 channels is dependent on the membrane potential. Titration curves for ClC-2 activity measured at 200 mm intracellular chloride and four different membrane potentials are shown. There was a marked displacement of pK1/2 to more alkaline values as the potential was decreased from −50 to −110 mV. At a given membrane potential, proton sensitivity of H532F-ClC-2 was also strongly increased by intracellular chloride. Figure 5B illustrates measurements taken at –110 mV and shows an alkaline displacement of pK1/2 as intracellular Cl− concentration increased.
Figure 5. Modulation of proton activation of ClC-2 by transmembrane voltage and intracellular Cl− concentration.

A and B, examples of extracellular pH dependence curves at different transmembrane potentials (A) and intracellular Cl− concentrations (B). C, a summary of results derived from experiments as in A and B, with pK1/2 for activation values plotted against transmembrane potential at the indicated intracellular Cl− concentrations (n= 5, 7, 1, 5 and 3 for 200, 135, 60, 35 and 10 mm[Cl−]i respectively). The lines are fits of a model based on the Woodhull formalism as explained in Methods (Eq. 2). D and E, parameters derived from the linear fits in C, giving the pK1/2 value in the absence of an applied voltage and the distance down the electrical field of the membrane to the site of proton action, as functions of intracellular Cl− concentration.
Results of experiments at various potentials and intracellular chloride concentrations are shown in Fig. 5C. Here pK1/2 values are plotted against voltage at five different intracellular chloride concentrations. Independently of [Cl−]i, hyperpolarisation increased the apparent pK1/2 value for the activation of H532F-ClC-2 by extracellular acidification. A change in the concentration of H+ at its site of action by voltage would occur if the location of the pHo sensor were within the electric field of the membrane and hyperpolarisation effectively accumulated H+ locally. The straight lines are fits to the data of a model (Woodhull, 1973) originally used to explain voltage-dependent proton block. The fits indicate that the apparent changes in pK1/2 with voltage can be explained by voltage-driven accumulation of protons at a site located some 62% down the electrical field across the membrane. The effect of intracellular Cl− concentration on pK1/2 in the absence of an electrical gradient (0 mV) is plotted in Fig. 5D. The pK1/2 (0 mV) values increased from about 5.6 to 6.1 when increasing [Cl−]i from 10 to 135 mm. As seen in Fig. 5E the distance down the membrane electrical field remained constant.
Molecular simulation predicts a weak influence of Cl− bound at Sint upon E207 pKa
Crystallographic structures of bacterial ClCs (Dutzler et al. 2002, 2003) have revealed three Cl− coordination sites (Sint, Scen and Sext) bridging intra- and extracellular sides of the membrane. A closed state is defined by occupation of Sext by the side chain of a glutamate residue termed Eext (E207 in ClC-2). One possible mechanism for the dependence of pK1/2 for proton activation of ClC-2 on intracellular Cl− could be an effect on the acidity of E207 (see Discussion) side-chain through an electrostatic influence favouring its protonation. To gain some insight into this possibility we have done quantum-chemical calculations to predict the change in E207 pKa as a consequence of Cl− occupying Scen only (ΔpKa1) or after additional occupation of site Sint to give the doubly occupied channel containing chlorides at Scen and Sint (ΔpKa2). The calculations used atoms of the pore structure of the bacterial EcClC (PDB: 1OTS) or from a homology model of ClC-2 (Fig. 6). The data are shown in Table 2, where values for ΔpKa1 and ΔpKa2 for the glutamate side-chain in the closed or open configuration are listed. Considering solely the distances of Cl− at Scen and Sint to the glutamate residue (Fig. 6A), these gave alkaline shifts in pKa of near 1 pH unit for occupation of Scen and of around 0.2 for the additional occupation of Sint. The effect of Cl− at Scen was reduced by ∼50% in the open configuration (Fig. 6B) and this can be attributed to the increased distance of the Cl− to Eext. An alternative calculation corresponds to a model that takes into account the gate glutamate plus its near environment in the selectivity filter (Fig. 6C and D). Under these conditions ΔpKa2 was not markedly changed. It is concluded therefore that the electrostatic effect exerted by Cl− bound at Sint on the acidity of the E207 side-chain is predicted to be weak and significantly smaller than that predicted for Cl− at Scen.
Figure 6. Atomic models used to calculate the pKa shifts (ΔpKa) of the gating glutamate due to the presence of chlorides at Scen and Sint A and B are minimum models of the closed and open configuration respectively, which considers the gating glutamate and chlorides at geometries equivalent to Scen and Sint.

Representative distances between chlorides and glutamate are depicted as dashed lines for reference. C and D are atomic models of the selectivity filter of ClC-2, in the closed and open configuration of the gating glutamate. The amino acids are represented as sticks and the chlorides as spheres (N, blue; O, red; C, light-grey; H, white; S, yellow; Cl, green).
Table 2.
Calculated pKashifts due to chloride occupation in Scenand Sint
| Model | ClC | ΔpKa1 |
ΔpKa2 |
||
|---|---|---|---|---|---|
| Closed | Open | Closed | Open | ||
| Model 1 | EcClC | 1.17 | 0.40 | 0.24 | 0.24 |
| CpClC-2 | 0.87 | 0.41 | 0.25 | 0.21 | |
| SF100 | EcClC | 3.04 | 1.05 | 0.02 | 0.22 |
| CpClC-2 | 2.22 | 0.68 | 0.18 | 0.32 | |
ΔpKa1= pKa(Clcen)− pKa(no Cl), pKa shift due to Cl− occupation of Scen and ΔpKa2= pKa(Clcen,in)− pKa(Clcen), pKa shift after additional occupation of Sint by Cl−. Calculated for the open and closed conformation of the gating glutamate. Model 1 considers Eext and Cl− ions only. SF100 considers in addition 100 atoms of the selectivity filter amino acids.
Discussion
The activity of ClC-2 Cl− channel has a biphasic response to extracellular pH with activation by moderate acidification followed by an abrupt channel closure at pH values lower than ∼7. This behaviour is compatible with the presence of two independent protonatable sites with opposing effects on channel gating (Arreola et al. 2002). Our previous work (Niemeyer et al. 2003) suggested that protonation of the side chain of the selectivity filter E207 accounts for the activation site. We have now explored further ClC-2 gating by extracellular pH using electrophysiological recording of heterologously expressed ClC-2 and site-directed mutagenesis. The two main findings of the present work were (a) that activation by acidification is a voltage-dependent phenomenon compatible with an action of H+ ions in a region of the channel buried within the electrical field of the membrane, and (b) that inhibition at acid pH is mediated by a sensor histidine residue located at the extracellular end of a putative transmembrane helix far from the pore entrance and exposed to the external solution.
Activation of ClC-2 by hyperpolarisation and extracellular acidification
Unlike its other ion channel congeners, ClC-2 Cl− channels are gated open by hyperpolarisation, intracellular Cl− and moderate extracellular acidification (Jordt & Jentsch, 1997; Niemeyer et al. 2003; Zúñiga et al. 2004). Although ClC-2 channel activity is abolished by extracellular acidification to pH 5.5, it is possible to see a fast transient activation of ClC-2 channels with pH 5.5 before the much slower inhibition takes place (Arreola et al. 2002; Niemeyer et al. 2003). Examination of fast activation, which is abolished by E207V neutralising mutation (Niemeyer et al. 2003), led us to the idea that ClC-2 channel opening by hyperpolarisation entails protonation of the selectivity filter E207. In addition we found pK1/2 values for activation by protons increased with hyperpolarisation, without apparent effects of the inhibition seen at very acid pH. As a more detailed study of the activation of ClC-2 by acidification was obscured by the concurrent inhibition, we sought to identify the sensor mediating this last effect in order to disable it.
An extracellular-facing histidine residue mediates the sensing of extracellular H+ to close ClC-2 channels at acid pH
We identified by mutation histidine 532 as responsible for detecting extracellular acidification leading to complete closure of ClC-2 channels. The mutant ClC-2-H532F presented a near-normal activity but lacked inhibition by strong acidification. Interestingly, although it appears they reach the membrane, mutants ClC-2-H532N and ClC-2-H532A had no detectable channel activity. This suggests that an interaction between perhaps neutral H532 and other neighbouring residue(s) is preserved by replacement with phenylalanine, but not by alanine or asparagine, to maintain the channel open.
Examination of a topological model of ClC-2 (see Fig. 3A) shows H532 to be located at the N-terminal end of α-helix Q. A homology model of ClC-2 based on the bacterial ClC EcClC (Dutzler et al. 2002) also indicates that H532 can be accessible from the extracellular solution (see Supplemental Fig. 1). We speculate that when H532 gets protonated, i.e. charged positively, this could modify its interaction with hydrophobic amino acids and consequently the conformational flexibility of ClC-2 at acid pH, through a still unknown mechanism. It is interesting that the intracellular C-terminus end of α-helix Q is connected by a 5-amino acid loop to helix R which has been associated with the gating mechanism in ClC proteins (Bell et al. 2006). Helix R also harbours Y555, which is homologous to Y445 in a bacterial ClC where it forms a crucial part of an intracellular gate (Jayaram et al. 2008). The existence of a similar intracellular gate in ClC-2 could account for the effect mediated by titration of H532. A long range effect on helix R might involve a large conformational change consistent with the slow kinetics of the acidification-induced inhibition.
As observed originally (Arreola et al. 2002), inhibition of ClC-2 by acidification is cooperative and can be described with a Hill coefficient of 2. This is confirmed here using wild-type ClC-2 and the E207V mutant, where this phenomenon occurs without the concurrent activation by extracellular protons (see legends to Figs 2 and 3). This cooperativity could be consistent with an interaction between the H532 sensors present in the separate monomers of the dimeric channel. Another possible explanation would be the involvement of a second site mediating the acidification-induced channel closing, but we have no evidence for this. Further speculation on the mechanism of H532-mediated, pH-dependent gating of ClC-2 is unwarranted here and its understanding will require more experimentation. The unravelling of H532 as a sensor for acidification-dependent inhibition of ClC-2 and its silencing by mutation, however, has allowed us to explore in more detail the activation of the channel by protons.
Gating of ClC-2 Cl− channel by voltage is mediated by a protonation event
Proton activation of the ClC-2 channel, as studied with the H532F-ClC-2 mutant, is dependent on the membrane potential, with a marked alkaline displacement of activation pK1/2 with hyperpolarisation. An increase in [Cl−]i also increases the pK1/2 value for activation, but this effect was not voltage dependent. The data are in fact consistent with a change in H+ concentration at its site of action by voltage, suggesting the presence of a pHo sensor at a site located some 62% down the electrical field across the membrane.
Much of the mechanistic insight into the gating of ClC channels by voltage, Cl− and H+ has been inferred from data for ClC-0, a channel from Torpedo electroplax that was the first member of the family to be analysed functionally (Miller, 1982) and cloned (Jentsch et al. 1990). ClC proteins are now firmly established as homodimeric structures with each monomer containing a separate pore. The two-pore structure had been successfully predicted from functional studies of ClC-0 which revealed the presence of two clearly separated gating mechanisms, a protopore and a common gate that could close the two pores simultaneously (Miller, 1982). The analysis of the gating of ClC-2 has represented a challenge. Although its voltage dependence can be accommodated by a complex model incorporating features of protopore and common gates (de Santiago et al. 2005), the clear separation of gating modes by, for example, temperature dependence (Zúñiga et al. 2004) or mutation (Yusef et al. 2006) has not been possible in ClC-2. Assuming, as has been proposed for ClC-0, that the reactions leading to the neutralisation of E207 constitute the protopore gating process, we have analysed the data using a simplified partial model that explains satisfactorily most features of the activation of ClC-2-H532F mutant by extracellular H+, as modified by intracellular Cl− and voltage. The scheme, shown below, is similar to that used to describe the fast gating of ClC-0, in which all the voltage dependence resides in the protonation/deprotonation step with the opening and closing rates being voltage independent (Traverso et al. 2006). An additional assumption is that intracellular Cl− interacts with a closed channel state and does not in itself lead to channel opening, an assumption that is supported by the fact that there is no channel activity at alkaline pHo independently of voltage or [Cl−]i.
| 4 |
Where, KClH=KClH (0) exp (−zδVF/RT) and C, CCl and CClH are closed states whilst OClH is an open state. Constants designated by K are dissociation constants, and α and β are rate constants.
It must be stressed that this model does not provide a complete description of ClC-2 gating. For instance the apparent gating valence of 1 for the Boltzmann distribution describing the voltage dependence of ClC-2 suggests a single electronic charge crossing fully the membrane field, compared with 62% predicted by the present description. Interestingly, a very recent study of ClC-0 gating suggests that H+-dependent gating might involve full migration of H+ across the membrane (Lisal & Maduke, 2008), a possibility that might be worth exploring in future work with ClC-2. Also, although not revealed in the present experiments, we cannot completely discard an effect from a voltage-dependent Cl− movement that might also contribute to the overall voltage dependence of ClC-2.
If maximal activation reaches an open probability of unity, the model predicts:
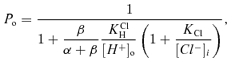 |
(3) |
This expression provides a description of the pHo dependence of K1/2 for proton activation of ClC-2 and its modulation by voltage and [Cl−]i, where 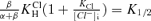 . Fit of this expression to the results in Fig. 5D (continuous line in the graph) gives values of 7.3 × 10−7m for
. Fit of this expression to the results in Fig. 5D (continuous line in the graph) gives values of 7.3 × 10−7m for  , and of 24 × 10−3m for KCl.
, and of 24 × 10−3m for KCl.
Structural studies of bacterial ClCs (Dutzler et al. 2002, 2003) have yielded useful information on which to interpret functional data for ClC channels and transporters. The structure revealed three Cl− coordination sites (Sext, Scen and Sint) that span the protein. A closed state was defined as blockade of Sext by the side chain of a residue termed Eext, and swinging out of this side-chain upon competition with extracellular Cl− promoted by depolarisation was proposed as the opening event (Dutzler et al. 2003). This scheme provided a mechanism for the functional coupling between Cl− ion conduction and gating observed in ClC-0 (and ClC-1) in which, in the absence of a proteinaceous voltage sensor, Cl− itself would act as the gating particle (Pusch et al. 1995). More recently, however, it has been suggested that protonation of Eext might be the event that opens the protopore gate (Miller, 2006), a view that has gained experimental support from the study of a mutant of ClC-0 in which Eext is replaced by an aspartic acid (Traverso et al. 2006). In this last work it is proposed that the activation of the fast gate of ClC-0-E166D is the consequence of voltage-dependent protonation (of D166) from the intracellular side, a process that would require the side chain of D166 extending towards the intracellular mouth of the channel pore. Further work has unsuccessfully attempted to discover an amino acid to act as an intracellular pH sensor in ClC-0 (Zifarelli et al. 2008). These authors propose a model in which a proton is generated by the dissociation of a water molecule within the selectivity filter which is delivered to the gating glutamate leading to channel opening whilst the hydroxyl would become stabilized in Scen.
As the main structural features of the bacterial ClC permeation pathway are highly conserved in the ClC-2, we had speculated that Eext, E207 in ClC-2, acts as a protopore gate with opening requiring hyperpolarisation to remove its carboxylate group from Sext. Intracellular Cl− would favour the open state of the channel by competition for this site (Niemeyer et al. 2003). Our present results, however, could be interpreted to suggest that protonation of E207, promoted by hyperpolarisation and H+ influx into the channel's mouth, is an important component in the voltage-dependent process of channel opening. In this scheme Cl−i would modulate the gating in a voltage-independent manner. Neutralisation of E207, which has a free solution pKa of 4.25, occurs because its local proton concentration becomes higher than that of bulk solution when a negative membrane potential is applied. Our calculations suggest that the protonation site is located 62% down the electrical distance across the membrane. In addition, there is an alkaline shift in pK1/2 promoted by intracellular Cl− interacting with a site with a dissociation constant KCl of 24 mm in a voltage-independent manner. We speculate that this site could correspond to Sint, because recent crystallographic measurements of binding affinity of Cl− for Sint in EcClC give a KD of the same order of magnitude (Lobet & Dutzler, 2006); and secondly, the intracellular Cl− effect is voltage independent, which would be consistent with the water accessible state of the Cl− ion bound to the Sint site (Dutzler et al. 2002) and by its location deduced from Poisson–Boltzmann calculations being barely into the electrical field from the internal side of the membrane (Faraldo-Gomez & Roux, 2004). What could be the nature of the effect of Cl−i? One possibility is that Cl− occupying Sint exerts an electrostatic effect on E207 side-chain to favour its protonation. A solely electrostatic effect is not supported by our ΔpKa calculations, which suggest an alkaline shift of only 0.2–0.3 pH units due to the presence of Cl− at Sint. An additional action, such as an allosteric effect taking place upon Cl− interaction with Sint, might be involved.
The plot of pK1/2(0 mV) vs. [Cl−]i (Fig. 5D) does not provide a direct estimate of KClH that, we could argue, ought to correspond to the Ka of E207 in situ. Instead the figure obtained is modified by the fraction β/α+β. To obtain the observed value of 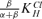 , α would be required to be about 75 times β. Considering that ClC-2 opens maximally to a Po value near unity (Yusef et al. 2006), this does not seem far-fetched.
, α would be required to be about 75 times β. Considering that ClC-2 opens maximally to a Po value near unity (Yusef et al. 2006), this does not seem far-fetched.
Concerning the effect of intracellular Cl−, ΔpKa calculations point to a marked electrostatic effect of Cl− at Scen upon E207, but not of binding at Sint. The presence of an occluded Cl− within Scen in the closed state of the channel would be consistent with functional data obtained with ClC-0 (Richard & Miller, 1990), the high affinity of Scen for Cl− measured for a bacterial ClC (Lobet & Dutzler, 2006), and the fact that examination of the structure of EcClC reveals that in addition to Eext blocking the external aspect of the pore, the exit to the intracellular aspect is also impeded by the highly conserved Ser107 and Tyr445 that constrict the pore between Scen and Sint (Corry et al. 2004; Miloshevsky & Jordan, 2004). Although some data suggest that there is no cytoplasmic-end gate in ClC channels (Lin & Chen, 2003), an additional conformational change at the inner aspect of the pore has been proposed to ensure free transmembrane Cl− passage (Accardi & Pusch, 2003). The presence of an occluded Cl− ion at Scen could also be an inheritance of transporter ancestors of ClC-2. Mutational and anion replacement experiments emptying this site uncouple H+ transport in a ClC exchanger, suggesting that Cl− at Scen might be essential to allow effective H+ capture by Eext (Nguitragool & Miller, 2006; Accardi et al. 2006). It is remarkable that whilst activation of ClC-2 requires hyperpolarisation, intracellular Cl− and extracellular acidification, this is the opposite behaviour to ClC-0 and ClC-1, which are mainly opened by depolarisation, extracellular Cl− and acid intracellular pH. There are no obvious structural determinants to account for these differences as the pore regions are highly conserved between ClC channels. The answer might lie in the way the gating of different channels is controlled by a complex C-terminus of unknown structure and low conservation. Recent work has investigated the role of C-terminus cystathionine β-synthase CBS domains in controlling gating of ClC-0, -1 and -2 and has unravelled diverse and profound effects as yet not well understood (Estévez et al. 2004; Niemeyer et al. 2004; Hebeisen et al. 2004; Bennetts et al. 2005; Garcia-Olivares et al. 2008).
In summary, we postulate a model for ClC-2 protopore gating in which protonation of the selectivity filter E207 is the essential voltage-dependent step in channel opening. Intracellular Cl− increases E207 pKa by binding at Sint. These characteristics of gating appear as partial reactions of a putative exchanger cycle, which is starting to be unravelled by mutagenesis, functional and structural studies, fitting with a scenario where ClC channels have evolved from their exchanger relatives. In addition we identify the molecular sensor for a mechanism that closes ClC-2 channels under strong acidification and which might exert a long-range control of an intracellular, cooperative gating mechanism. Further work will be needed to provide a better understanding of the molecular mechanisms that we have merely glimpsed. The understanding of the function of the complex H+-dependent gating mechanisms of ClC-2 in the tissue, cell and subcellular locations where it is expressed (Bösl et al. 2001; Peña-Münzenmayer et al. 2005) might also help to reveal the essential but so far poorly understood physiological role of this widely expressed Cl− channel.
Acknowledgments
Supported by Fondecyt grants 1070722 and 3080017. The Centro de Estudios Científicos (CECS) is funded by the Chilean Government through the Millennium Science Initiative and the Centers of Excellence Base Financing Program of Conicyt. CECS is also supported by a group of private companies which at present includes Antofagasta Minerals, Arauco, Empresas CMPC, Indura, Naviera Ultragas and Telefónica del Sur. CIN is funded by Conicyt and the Gobierno Regional de Los Ríos. We are grateful to Dr David C. Gadsby for illuminating comments on our results before this paper was written and to Dr L. Felipe Barros for many useful suggestions.
Supplemental material
Online supplemental material for this paper can be accessed at:
http://jp.physoc.org/cgi/content/full/jphysiol.2008.167353/DC1
References
- Accardi A, Lobet S, Williams C, Miller C, Dutzler R. Sinergism between halide binding and proton transport in a CLC-type exchanger. J Mol Biol. 2006;362:691–699. doi: 10.1016/j.jmb.2006.07.081. [DOI] [PubMed] [Google Scholar]
- Accardi A, Miller C. Secondary active transport mediated by a prokaryotic homologue of ClC Cl− channels. Nature. 2004;427:803–807. doi: 10.1038/nature02314. [DOI] [PubMed] [Google Scholar]
- Accardi A, Pusch M. Conformational changes in the pore of CLC-0. J Gen Physiol. 2003;122:277–293. doi: 10.1085/jgp.200308834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Accardi A, Walden M, Nguitragool W, Jayaram H, Williams C, Miller C. Separate ion pathways in a Cl−/H+ exchanger. J Gen Physiol. 2005;126:563–570. doi: 10.1085/jgp.200509417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arreola J, Begenisich T, Melvin JE. Conformation-dependent regulation of inward rectifier chloride channel gating by extracellular protons. J Physiol. 2002;541:103–112. doi: 10.1113/jphysiol.2002.016485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell SP, Curran PK, Choi S, Mindell JA. Site-directed fluorescence studies of a prokaryotic ClC antiporter. Biochemistry. 2006;45:6773–6782. doi: 10.1021/bi0523815. [DOI] [PubMed] [Google Scholar]
- Bennetts B, Rychkov GY, Ng HL, Morton CJ, Stapleton D, Parker MW, Cromer BA. Cytoplasmic ATP-sensing domains regulate gating of skeletal muscle ClC-1 chloride channels. J Biol Chem. 2005;280:32452–32458. doi: 10.1074/jbc.M502890200. [DOI] [PubMed] [Google Scholar]
- Bösl MR, Stein V, Hubner C, Zdebik AA, Jordt SE, Mukhopadhyay AK, Davidoff MS, Holstein AF, Jentsch TJ. Male germ cells and photoreceptors, both dependent on close cell-cell interactions, degenerate upon ClC-2 Cl− channel disruption. EMBO J. 2001;20:1289–1299. doi: 10.1093/emboj/20.6.1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catalán M, Niemeyer MI, Cid LP, Sepúlveda FV. Basolateral ClC-2 chloride channels in surface colon epithelium: regulation by a direct effect of intracellular chloride. Gastroenterology. 2004;126:1104–1114. doi: 10.1053/j.gastro.2004.01.010. [DOI] [PubMed] [Google Scholar]
- Cid LP, Niemeyer MI, Ramírez A, Sepúlveda FV. Splice variants of a ClC-2 chloride channel with differing functional characteristics. Am J Physiol Cell Physiol. 2000;279:C1198–C1210. doi: 10.1152/ajpcell.2000.279.4.C1198. [DOI] [PubMed] [Google Scholar]
- Corry B, O'Mara M, Chung SH. Conduction mechanisms of chloride ions in ClC-type channels. Biophys J. 2004;86:846–860. doi: 10.1016/S0006-3495(04)74160-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Angeli A, Monachello D, Ephritikhine G, Frachisse JM, Thomine S, Gambale F, Barbier-Brygoo H. The nitrate/proton antiporter AtCLCa mediates nitrate accumulation in plant vacuoles. Nature. 2006;442:939–942. doi: 10.1038/nature05013. [DOI] [PubMed] [Google Scholar]
- de Santiago JA, Nehrke K, Arreola J. Quantitattive analysis of the voltage-dependent gating of mouse parotid ClC-2 chloride channel. J Gen Physiol. 2005;126:591–603. doi: 10.1085/jgp.200509310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutzler R, Campbell EB, Cadene M, Chait BT, MacKinnon R. X-ray structure of a ClC chloride channel at 3.0 A reveals the molecular basis of anion selectivity. Nature. 2002;415:287–294. doi: 10.1038/415287a. [DOI] [PubMed] [Google Scholar]
- Dutzler R, Campbell EB, MacKinnon R. Gating the selectivity filter in ClC chloride channels. Science. 2003;300:108–112. doi: 10.1126/science.1082708. [DOI] [PubMed] [Google Scholar]
- Estévez R, Pusch M, Ferrer-Costa C, Orozco M, Jentsch TJ. Functional and structural conservation of CBS domains from CLC chloride channels. J Physiol. 2004;557:363–378. doi: 10.1113/jphysiol.2003.058453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estévez R, Schroeder BC, Accardi A, Jentsch TJ, Pusch M. Conservation of chloride channel structure revealed by an inhibitor binding site in ClC-1. Neuron. 2003;38:47–59. doi: 10.1016/s0896-6273(03)00168-5. [DOI] [PubMed] [Google Scholar]
- Faraldo-Gomez JD, Roux B. Electrostatics of ion stabilization in a ClC chloride channel homologue from Escherichia coli. J Mol Biol. 2004;339:981–1000. doi: 10.1016/j.jmb.2004.04.023. [DOI] [PubMed] [Google Scholar]
- Gadsby DC. Ion transport: spot the difference. Nature. 2004;427:795–797. doi: 10.1038/427795a. [DOI] [PubMed] [Google Scholar]
- Gadsby DC, Vergani P, Csanády L. The ABC protein turned chloride channel whose failure causes cystic fibrosis. Nature. 2006;440:477–483. doi: 10.1038/nature04712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Olivares J, Alekov A, Boroumand MR, Begemann B, Hidalgo P, Fahlke C. Gating of human ClC-2 chloride channels and regulation by carboxy-terminal domains. J Physiol. 2008;586:5325–5336. doi: 10.1113/jphysiol.2008.158097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graves AR, Curran PK, Smith CL, Mindell JA. The Cl−/H+ antiporter ClC-7 is the primary chloride permeation pathway in lysosomes. Nature. 2008;453:788–792. doi: 10.1038/nature06907. [DOI] [PubMed] [Google Scholar]
- Gründer S, Thiemann A, Pusch M, Jentsch TJ. Regions involved in the opening of ClC-2 chloride channel by voltage and cell volume. Nature. 1992;360:759–762. doi: 10.1038/360759a0. [DOI] [PubMed] [Google Scholar]
- Hebeisen S, Biela A, Giese B, Muller-Newen G, Hidalgo P, Fahlke C. The role of the carboxyl terminus in ClC chloride channel function. J Biol Chem. 2004;279:13140–13147. doi: 10.1074/jbc.M312649200. [DOI] [PubMed] [Google Scholar]
- Jayaram H, Accardi A, Wu F, Williams C, Miller C. Ion permeation through a Cl–-selective channel designed from a CLC Cl−/H+ exchanger. Proc Natl Acad Sci U S A. 2008;105:11194–11199. doi: 10.1073/pnas.0804503105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen JH, Li H, Robertson AD, Molina PA. Prediction and rationalization of protein pKa values using QM and QM/MM methods. J Phys Chem A. 2005;109:6634–6643. doi: 10.1021/jp051922x. [DOI] [PubMed] [Google Scholar]
- Jentsch TJ, Poet M, Fuhrmann JC, Zdebik AA. Physiological functions of CLC Cl− channels gleaned from human genetic disease and mouse models. Annu Rev Physiol. 2005;67:779–807. doi: 10.1146/annurev.physiol.67.032003.153245. [DOI] [PubMed] [Google Scholar]
- Jentsch TJ, Steinmeyer K, Schwarz G. Primary structure of Torpedo marmorata chloride channel isolated by expression cloning in Xenopus oocytes. Nature. 1990;348:510–514. doi: 10.1038/348510a0. [DOI] [PubMed] [Google Scholar]
- Jordt SE, Jentsch TJ. Molecular dissection of gating in the ClC-2 chloride channel. EMBO J. 1997;16:1582–1592. doi: 10.1093/emboj/16.7.1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CW, Chen TY. Probing the pore of ClC-0 by substituted cysteine accessibility method using methane thiosulfonate reagents. J Gen Physiol. 2003;122:147–159. doi: 10.1085/jgp.200308845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisal J, Maduke M. The ClC-0 chloride channel is a ‘broken’ Cl−/H+ antiporter. Nat Struct Mol Biol. 2008;15:805–810. doi: 10.1038/nsmb.1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobet S, Dutzler R. Ion-binding properties of the ClC chloride selectivity filter. EMBO J. 2006;25:24–33. doi: 10.1038/sj.emboj.7600909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller C. Open-state substructure of single chloride channels from Torpedo electroplax. Philos Trans R Soc Lond B Biol Sci. 1982;299:401–411. doi: 10.1098/rstb.1982.0140. [DOI] [PubMed] [Google Scholar]
- Miller C. ClC chloride channels viewed through a transporter lens. Nature. 2006;440:484–489. doi: 10.1038/nature04713. [DOI] [PubMed] [Google Scholar]
- Miloshevsky GV, Jordan PC. Anion pathway and potential energy profiles along curvilinear bacterial ClC Cl− pores: electrostatic effects of charged residues. Biophys J. 2004;86:825–835. doi: 10.1016/S0006-3495(04)74158-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguitragool W, Miller C. Uncoupling of a CLC Cl−/H+ exchange transporter by polyatomic anions. J Mol Biol. 2006;29:682–690. doi: 10.1016/j.jmb.2006.07.006. [DOI] [PubMed] [Google Scholar]
- Niemeyer MI, Cid LP, Zúñiga L, Catalán M, Sepúlveda FV. A conserved pore-lining glutamate as a voltage- and chloride-dependent gate in the ClC-2 chloride channel. J Physiol. 2003;553:873–879. doi: 10.1113/jphysiol.2003.055988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niemeyer MI, Yusef YR, Cornejo I, Flores CA, Sepúlveda FV, Cid LP. Functional evaluation of human ClC-2 chloride channel mutations associated with idiopathic generalized epilepsies. Physiol Genomics. 2004;19:74–83. doi: 10.1152/physiolgenomics.00070.2004. [DOI] [PubMed] [Google Scholar]
- Peña-Münzenmayer G, Catalán M, Cornejo I, Figueroa CD, Melvin JE, Niemeyer MI, Cid LP, Sepúlveda FV. Basolateral localization of native ClC-2 chloride channels in absorptive intestinal epithelial cells and basolateral sorting encoded by a CBS-2 domain di-leucine motif. J Cell Sci. 2005;118:4243–4252. doi: 10.1242/jcs.02525. [DOI] [PubMed] [Google Scholar]
- Picollo A, Pusch M. Chloride/proton antiporter activity of mammalian CLC proteins ClC-4 and ClC-5. Nature. 2005;436:420–423. doi: 10.1038/nature03720. [DOI] [PubMed] [Google Scholar]
- Pusch M, Jordt SE, Stein V, Jentsch TJ. Chloride dependence of hyperpolarization-activated chloride channel gates. J Physiol. 1999;515:341–353. doi: 10.1111/j.1469-7793.1999.341ac.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pusch M, Ludewig U, Rehfeldt A, Jentsch TJ. Gating of the voltage-dependent chloride channel ClC-0 by the permeant anion. Nature. 1995;373:527–531. doi: 10.1038/373527a0. [DOI] [PubMed] [Google Scholar]
- Richard EA, Miller C. Steady-state coupling of ion-channel conformations to a transmembrane ion gradient. Science. 1990;247:1208–1210. doi: 10.1126/science.2156338. [DOI] [PubMed] [Google Scholar]
- Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- Scheel O, Zdebik AA, Lourdel S, Jentsch TJ. Voltage-dependent electrogenic chloride/proton exchange by endosomal CLC proteins. Nature. 2005;436:424–427. doi: 10.1038/nature03860. [DOI] [PubMed] [Google Scholar]
- Thiemann A, Gründer S, Pusch M, Jentsch TJ. A chloride channel widely expressed in epithelial and non-epithelial cells. Nature. 1992;356:57–60. doi: 10.1038/356057a0. [DOI] [PubMed] [Google Scholar]
- Traverso S, Zifarelli G, Aiello R, Pusch M. Proton sensing of CLC-0 mutant E166D. J Gen Physiol. 2006;127:51–66. doi: 10.1085/jgp.200509340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodhull AM. Ionic blockage of sodium channels in nerve. J Gen Physiol. 1973;61:687–708. doi: 10.1085/jgp.61.6.687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yusef YR, Zúñiga L, Catalán M, Niemeyer MI, Cid LP, Sepúlveda FV. Removal of gating in voltage-dependent ClC-2 chloride channel by point mutations affecting the pore and C-terminus CBS-2 domain. J Physiol. 2006;572:173–181. doi: 10.1113/jphysiol.2005.102392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zifarelli G, Murgia AR, Soliani P, Pusch M. Intracellular proton regulation of ClC-0. J Gen Physiol. 2008;132:185–198. doi: 10.1085/jgp.200809999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zúñiga L, Niemeyer MI, Varela D, Catalán M, Cid LP, Sepúlveda FV. The voltage-dependent ClC-2 chloride channel has a dual gating mechanism. J Physiol. 2004;555:671–682. doi: 10.1113/jphysiol.2003.060046. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.