

Abstract
Large conductance Ca2+-activated K+ (BK) channels modulate many physiological processes including neuronal excitability, synaptic transmission and regulation of myogenic tone. A gain-of-function (E/D) mutation in the pore-forming α subunit (Slo1) of the BK channel was recently identified and is linked to human neurological diseases of coexistent generalized epilepsy and paroxysmal dyskinesia. Here we performed macroscopic current recordings to examine the effects of the E/D mutation on the gating kinetics, and voltage and Ca2+ dependence of the BK channel activation in the presence of four different β subunits (β1–4). These β subunits are expressed in a tissue-specific pattern and modulate BK channel function differently, providing diversity and specificity for BK channels in various physiological processes. Our results show that in human (h) Slo1-only channels, the E/D mutation increased the rate of opening and decreased the rate of closing, allowing a greater number of channels to open at more negative potentials both in the presence and absence of Ca2+ due to increased Ca2+ affinity and enhanced activation compared with the wild-type channels. Even in the presence of β subunits, the E/D mutation exhibited these changes with the exception of β3b, where Ca2+ sensitivity changed little. However, quantitative examination of these changes shows the diversity of each β subunit and the differential modulation of these subunits by the E/D mutation. For example, in the presence of the β1 subunit the E/D mutation increased Ca2+ sensitivity less but enhanced channel activation in the absence of Ca2+ more than in hSlo1-only channels, while in the presence of the β2 subunit the E/D mutation also altered inactivation properties. These findings suggest that depending on the distribution of the various β subunits in the brain, the E/D mutation can modulate BK channels differently to contribute to the pathophysiology of epilepsy and dyskinesia. Additionally, these results also have implications on physiological processes in tissues other than the brain where BK channels play an important role.
BK channels have a large unitary conductance (∼150–300 pS) and are activated by membrane depolarization and increasing intracellular Ca2+ concentration ([Ca2+]i). The opening of BK channels would effectively repolarize the membrane and reduce Ca2+ entry through voltage-dependent Ca2+ channels. Through this negative feedback mechanism, BK channels regulate smooth muscle tone in arteries (Brayden & Nelson, 1992; Knot et al. 1998; Ledoux et al. 2006), trachea (Semenov et al. 2006) and urinary bladder (Herrera et al. 2000,2005; Petkov et al. 2001), neuronal excitability (Lancaster & Nicoll, 1987; Hu et al. 2001; Faber & Sah, 2003; Gu et al. 2007) and transmitter release (Roberts et al. 1990; Robitaille & Charlton, 1992; Hu et al. 2001; Faber & Sah, 2003), endocrine secretion (Findlay et al. 1985; Orio et al. 2002), and electrical tuning in the inner hair cells (Fettiplace & Fuchs, 1999). The pore-forming α subunit of the BK channel is encoded by a single Slo1 gene (Atkinson et al. 1991; Adelman et al. 1992). In addition, four types of β subunits (β1–4) have been identified. Each β subunit has a tissue-specific expression and modulates channel function uniquely (Orio et al. 2002), which provides a major mechanism for diverse BK channel phenotypes in various tissues.
A recent study found a mutation in the Slo1 subunit that was linked to generalized epilepsy and paroxysmal dyskinesia (GEPD) (Du et al. 2005). This epilepsy/dyskinesia (E/D) mutation enhances channel activity at the same voltage and [Ca2+]i, so that more E/D channels are open than the wild-type (WT) channels at physiological conditions. The gain of function in BK channels is believed to be responsible for the GEPD syndrome (Du et al. 2005). The original study on the E/D mutation was done in the absence of β subunits, but there are multiple β subunits localized in the brain (Behrens et al. 2000; Brenner et al. 2000a; Uebele et al. 2000). Therefore, in order to understand the molecular and cellular mechanisms of how the mutation is associated with GEPD, it is essential to first identify changes induced by the E/D mutation to the properties of the BK channels containing various β subunits. In addition, since only a single gene expresses the Slo1 subunit, the E/D mutation is likely to be found in places outside the brain where BK channels normally exist. Thus, to understand the impact of the E/D mutation on the physiological function of various tissues, it is imperative to examine the changes as a result of the E/D mutation with every β subunit.
A previous study focusing on the β4 subunit used single channel recordings to study the effect of the mutation on the Ca2+-dependent open probabilities (Diez-Sampedro et al. 2006). In the present study, we performed macroscopic recordings to determine the changes to the activation and deactivation kinetics, and Ca2+ sensitivity of channel activation by the E/D mutation in the presence of various β subunits (β1, β2, β3b and β4). A possible mechanism for the change in Ca2+ sensitivity was further explored using an allosteric gating model (MWC model) for BK channels (Cox et al. 1997). Our results show that in the hSlo1-only channels, the E/D mutation increased Ca2+ affinity and enhanced channel activation in the absence of Ca2+. Thus, more channels open under physiological conditions and remain open for a longer time than in the WT channels. Furthermore, even in the presence of the β subunits, the E/D mutation modulates the channel's kinetics and Ca2+-dependent properties with the same trend as in the hSlo1-only channels with the exception of the β3b subunit. However, quantitative examination of the channel's voltage- and Ca2+-dependent gating highlights the diverse properties of each β subunit as modified by the E/D mutation. This differential modulation of BK channels containing β subunits suggests that regions in the brain expressing different combinations of β subunits may play different roles in the aetiology of epilepsy and dyskinesia.
Methods
Mutagenesis and expression
Human β1 (KCNMB1, GenBank Accession No. U25138), β2 (KCNMB2, GenBank Accession No. AF209747), β3b (KCNMB3, GenBank Accession No. AF214561) and β4 (KNCMB4, GenBank Accession No. AF207992) cDNAs were subcloned into pcDNA3.1(+). WT and E/D mutant (D434G) human Slo1 (hSlo1, GenBank Accession No. NM_002247) were coexpressed with the β1, β3b and β4 subunits. WT and E/D mutant (D369G) hSlo1 (GenBank Accession No. U11058) were coexpressed with the β2 and β2ND subunits. All hSlo1 cDNAs were subcloned into pcDNA3.1(+). The β2 with NH2-terminus deleted (β2ND) subunit was created by removing amino acids from positions 2 to 20. Subcloning of β1, β2, β3b and β4 constructs and mutations of hSlo1 and β2ND were made using PCR and Pfu polymerase (Stratagene, La Jolla, CA, USA). The PCR-amplified regions for all constructs were verified by sequencing. mRNA was transcribed in vitro using T7 polymerase (Ambion, Austin, TX, USA).
Oocyte harvesting and mRNA injection
The use of Xenopus laevis for harvesting oocytes was approved by Washington University's Animal Studies Committee. The frogs were anaesthetized using 0.13% ethyl 3-aminobenzoate methanesulfonate (MS-222), in sodium bicarbonate-buffered tap water treated to remove chlorine and chloramine, until there was no response to pinching of the toe (∼10–15 min). Next, a 1–2 cm incision was made in the abdomen to excise the ovarian tissue. The incision was closed with sutures and the frog was placed in a recovery tank for ∼1–2 days. This procedure was performed for a total of six times for each frog, three incisions per side. At the end of the 6th surgery, the frogs were killed by excising the heart. The ovarian tissue was manually teased apart and placed in Ca2+-free OR-2 solution (in mm: 82.5 NaCl, 2.5 KCl, 1 MgCl2, 5 Hepes, pH 7.6). Using collagenase, type 1A (Sigma-Aldrich, St Louis, MO, USA), the follicle cells were removed and stage IV–V oocytes were selected for injection. hSlo1 mRNA, 0.05–20 ng, or a mixture of 5–15 ng hSlo1 and 25–50 ng β subunit mRNAs were injected into each oocyte and incubated in ND96 solution (in mm: 96 NaCl, 2 KCl, 1.8 CaCl2, 1 MgCl2, 5 Hepes, pH 7.6) at 18°C for 2–4 days before recording.
Electrophysiology
Macroscopic currents were recorded from inside-out patches formed with borosilicate pipettes of ∼0.9–1.5 MΩ resistance. The data were acquired using an Axopatch 200-B patch-clamp amplifier (Axon Instruments, Union City, CA, USA) and Pulse acquisition software (HEKA Electronik, Lambrecht/Pfalz, Germany). Recordings were digitized at 20 μs intervals and low-pass filtered at 10 kHz with the 4-pole Bessel filter built into the amplifier. Capacitive transients and leak currents were subtracted using a P/5 protocol. Experiments were conducted at room temperature (20–22°C). The pipette solution contained (in mm) 140 KMeSO3, 20 Hepes, 2 KCl and 2 MgCl2, pH 7.2. The internal solution contained (in mm) 140 KMeSO3, 20 Hepes, 2 KCl, and 1 N-(2-hydroxyethyl)ethylenediamine-N,N,N-triacetic acid (HEDTA), pH 7.2. CaCl2 was added to the internal solution to give the appropriate free [Ca2+]i, which was measured with a calcium-sensitive electrode (Orion Research, Cambridge, MA, USA). 18-Crown-6-tetracarboxylic acid (50 μm; Sigma-Aldrich) was added to internal solutions to chelate Ba2+. For nominal 0 μm[Ca2+]i, the same internal solution was used except that HEDTA was replaced by 5 mm EGTA and no CaCl2 was added. The free [Ca2+] in nominal 0 μm[Ca2+]i solution is 0.5 nm.
Data analysis
The relative conductance was determined by measuring tail current amplitudes at indicated voltages for WT and E/D channels with and without β subunits. The conductance–voltage (G–V) relationships for the WT and E/D channels with and without β subunits were fitted with the Boltzmann equation:  , where G/Gmax is the ratio of conductance to maximum conductance, z is the number of equivalent charges, V1/2 is the voltage at which the channel is 50% activated, e is the elementary charge, k is Boltzmann's constant, and T is the absolute temperature. Curve fittings were done with Igor Pro software (WaveMetrics, Inc., Lake Oswego, OR, USA) using the Levenberg–Marquardt algorithm to perform non-linear least squares fits. The means of the data were obtained by averaging from 3–47 patches and error bars represent standard error of the mean (s.e.m.). Statistics were performed using Origin 6.1 (OriginLab Corp., Northampton, MA, USA); independent/unpaired t tests were performed. A P value of < 0.05 was considered significant.
, where G/Gmax is the ratio of conductance to maximum conductance, z is the number of equivalent charges, V1/2 is the voltage at which the channel is 50% activated, e is the elementary charge, k is Boltzmann's constant, and T is the absolute temperature. Curve fittings were done with Igor Pro software (WaveMetrics, Inc., Lake Oswego, OR, USA) using the Levenberg–Marquardt algorithm to perform non-linear least squares fits. The means of the data were obtained by averaging from 3–47 patches and error bars represent standard error of the mean (s.e.m.). Statistics were performed using Origin 6.1 (OriginLab Corp., Northampton, MA, USA); independent/unpaired t tests were performed. A P value of < 0.05 was considered significant.
Monod–Wyman–Changeux (MWC) model
MWC model fits were performed using the following equation:
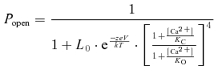 |
where Popen is the channel's open probability, L0 is the steady-state equilibrium constant from open to closed channels ([C0]/[O0]) in the absence of Ca2+ binding at 0 mV, z, e, k and T are the same as in the Boltzmann equation (see above), KC and KO are the dissociation constants of Ca2+ in the closed and open states, respectively. The MWC model code was written and executed in MATLAB v7.4 (The MathWorks, Inc.).
Since BK channels are activated by both voltage and Ca2+, the ideal conditions for measuring Ca2+ sensitivity of channel activation would be in the absence of voltage sensor movements. Such measurements have been done at very negative voltages (< −130 mV) where the voltage sensor of BK channels is kept at the resting state (Horrigan & Aldrich, 2002). Ca2+-dependent activation of BK channels under such conditions could be fitted by the MWC model, and the results show that the parameters KC and KO have very similar values to those obtained by the MWC model fitting to the G–V relations (Cox et al. 1997; Horrigan & Aldrich, 2002), which is the method we used in this study. The lack of a large influence of voltage on Ca2+ affinity measurement is not surprising because it has been shown that voltage and Ca2+ activate the channel through distinct mechanisms that have little interaction (Cui & Aldrich, 2000; Horrigan & Aldrich, 2002). In the MWC model used in this study, each hSlo1 subunit is assumed to contain a single Ca2+ binding site although previous studies have proposed two putative Ca2+ binding sites in each hSlo1 subunit (Schreiber & Salkoff, 1997; Xia et al. 2002). A model composed of two Ca2+ binding sites would not provide any additional information since the effect of the E/D mutation contributed by each site remains to be determined. Notwithstanding such an approximation, the MWC model used in this study provides an appropriate account of the overall changes to the Ca2+-dependent properties of BK channels due to the β subunits and E/D mutation.
Results
The epilepsy/dyskinesia mutation modifies gating properties of hSlo1
Characterization of the E/D mutant hSlo1 channels identified changes to three major gating properties. First, the rate of channel opening was increased by the mutation; the activation time constant of the macroscopic current (τAct) was shortened at all voltages tested (Fig. 1A and B), consistent with previous observations (Du et al. 2005). Second, the rate of channel closing upon repolarization was slower in the mutant channels; the deactivation time constant (τDact) was prolonged (Fig. 1A and C). Thus, the E/D mutation rendered the channel to open faster and close slower than the WT hSlo1. Collectively, these changes prolonged the time for K+ conduction. Third, the steady-state open probability of the channel was increased by the mutation. In the near absence of Ca2+, the conductance–voltage (G–V) relations of the mutant channels shows a small but significant shift to more negative voltages compared to that of the WT channel; the voltage at half-activation, V1/2, for the WT and E/D mutant hSlo1 is 180.1 ± 1.6 mV and 173.5 ± 2.3 mV, respectively with P < 0.05 (Fig. 1D and E). In the presence of intracellular Ca2+, the difference in V1/2 between the E/D mutant and WT channels was even larger (Fig. 1D and E). Thus, at a given voltage and [Ca2+]i, the E/D mutant channels have a higher open probability than the WT channels. Because the G–V shift resulting from the E/D mutation depends on [Ca2+]i, the Ca2+ sensitivity, measured as the difference in V1/2 values between nominal 0 and the saturating [Ca2+]i, is larger in the E/D mutant than in the WT hSlo1 channels (ΔV1/2,0−100Ca= 183.6 ± 3.2 mV and 199.7 ± 3.5 mV in the WT and E/D mutant hSlo1, respectively, P < 0.05). In the intermediate [Ca2+]i (∼2–10 μm), the difference in V1/2 between the WT and the E/D channels reaches a maximum of ∼50 mV (Fig. 1E), which is similar to previous reports (Du et al. 2005).
Figure 1. Functional differences between the WT and E/D mutant hSlo1 channels.

A, macroscopic current traces of WT and E/D mutation in ∼2 μm[Ca2+]i. Voltage ranges from −80 to +200 mV with 10 mV increments and the repolarizing potential at −50 mV. B, activation kinetics of WT and E/D mutation in ∼2 μm[Ca2+]i. τAct in this and other figures was obtained by fitting current traces with a single exponential function. C, deactivation kinetics of WT and E/D mutation in ∼2 μm[Ca2+]i. In this and other figures, tail currents at various voltages after 20 ms prepulse were fitted with a single exponential function to obtain τDact. Prepulse potential =+200 mV. D, mean G–V relationship of WT and E/D mutant in ∼0, 2 and 100 μm[Ca2+]i fitted with Boltzmann equation. E, V1/2vs.[Ca2+]i plot of WT and E/D mutant hSlo1. F, mean G–V relationship of WT and E/D mutant in different [Ca2+]i are fitted using the MWC model. The specific [Ca2+]i for each symbol is shown. In this and other figures, error bars represent s.e.m.
To further illustrate the changes to Ca2+ sensitivity, we used a 10-state Monod–Wyman–Changeux (MWC) model to fit the G–V relations of WT and E/D mutant hSlo1 channels at seven different [Ca2+]i values (Fig. 1F, Table 1) and identified two major differences exhibited by the E/D mutation. The dissociation constants for Ca2+ binding in the open and closed states (KO and KC, respectively) are reduced (Table 1), which suggests that the E/D mutation alters the Ca2+ binding site to increase Ca2+ affinity. This result is consistent with the above observations that in the presence of Ca2+, the mutation increases the activation rate, decreases the deactivation rate, and increases the Ca2+ sensitivity of the steady-state activation (Fig. 1A–E) due to an increase in Ca2+ binding. The MWC model fitting also showed that the E/D mutant channels have a reduced L0, the equilibrium constant of the channel from open to closed states in the absence of Ca2+ and at 0 mV (Table 1). Since Z is similar in the two channels, this change in L0 is consistent with the negative shift in V1/2 at nominal 0 μm Ca2+ and suggests that the mutation changes channel gating even in the absence of Ca2+ binding. Taken together, our results suggest that the E/D mutation enhances BK channel activation both in the absence and presence of Ca2+ binding leading to more channel openings and longer open times under the same physiological conditions which would repolarize the membrane potential and shut Ca2+ entry more effectively.
Table 1.
Parameters for MWC model fits
| L0 | Z | KC (μm) | KO (μm) | ||
|---|---|---|---|---|---|
| α | WT | 7147 | 1.25 | 7.69 | 0.82 |
| E/D | 6907 | 1.27 | 6.48 | 0.46 | |
| α′ | WT | 7259 | 1.29 | 7.13 | 0.77 |
| E/D | 5780 | 1.25 | 5.85 | 0.49 | |
| α+β1 | WT | 839 | 0.76 | 6.20 | 0.65 |
| E/D | 790 | 0.90 | 7.20 | 0.40 | |
| α′+β2ND | WT | 8838 | 1.18 | 21.01 | 0.47 |
| E/D | 1089 | 0.96 | 9.63 | 0.37 | |
| α+β3b | WT | 9418 | 1.35 | 6.65 | 0.55 |
| E/D | 1200 | 1.07 | 5.39 | 0.72 | |
| α+β4 | WT | 3290 | 1.00 | 10.68 | 1.09 |
| E/D | 4147 | 1.10 | 7.32 | 0.59 |
The epilepsy/dyskinesia mutation modulates channel kinetics in the presence of β subunits
We coexpressed the WT and E/D mutant hSlo1 with β subunits and compared their gating properties. Each of the β subunits expressed robust currents with the E/D mutant, similar to those with the WT hSlo1 (Fig. 2). The currents with the β1, β3b and β4 subunits are shown in ∼2 μm[Ca2+]i. The currents with the β2 subunit exhibited time-dependent inactivation and are shown in ∼10 μm[Ca2+]i to highlight the inactivation properties (Brenner et al. 2000a; Ding & Lingle, 2002; Wang et al. 2002; Benzinger et al. 2006). We found that the mutation altered the time course of both activation and deactivation of the currents in the presence of each of the β subunits.
Figure 2. Macroscopic currents of WT and E/D mutant hSlo1 with β subunits.

All measurements were made in ∼2 μm[Ca2+]i except for those with the β2 subunit, which were in ∼10 μm[Ca2+]i. The voltage protocols were: for hSlo1 +β1: −80 to +200 mV with 10 mV increments and the repolarization potential at −120 mV; for hSlo1 +β2: −100 to +140 mV with 20 mV increments and the repolarization potential at −80 mV; for hSlo1 +β3b: −80 to +200 mV with 10 mV increments and the repolarization potential at −50 mV; and for hSlo1 +β4: −80 to +200 mV with 10 mV increments and the repolarization potential at −80 mV. In this and other figures, hSlo1 splice variant U11058 was coexpressed with the β2/β2ND subunits.
In the left panels of Fig. 3A–D, we normalized the peak amplitudes of the mutant and WT activating currents with each of the β subunits, and compared the time course of the currents to reach maximum amplitude. The τAct was measured by fitting the activating current traces with a single exponential function, and plotted against voltage (right panels, Fig. 3A–D). The E/D mutation increased the activation rate in the presence of β1, β2 and β4 subunits, similar to the results from the hSlo1-only channels. However, in the presence of the β3b subunit, the E/D mutation prolonged the activation time constant. Therefore, the E/D mutant BK channels associated with the β3b subunit would not respond to the stimulus as quickly as the WT +β3b channels.
Figure 3. Activation kinetics of WT and E/D mutant hSlo1 with β subunits.

All measurements were made in ∼2 μm[Ca2+]i except for those with the β2 subunit, which were in ∼10 μm[Ca2+]i. Left column: overlay of normalized current traces for hSlo1 +β1 at +40 mV (A), for hSlo1 +β2 at +40 mV (B), for hSlo1 +β3b at +80 mV (C), and for hSlo1 +β4 at +100 mV (D). Right column: τAct–V relations. The descriptions of the symbols are shown at the bottom.
Equally important to BK channel function are the deactivation kinetics, which measures the rate of channel closing during repolarization. In the left panels of Fig. 4A–D, we normalized the E/D mutant and WT deactivating currents with each β subunit to the amplitude at the beginning of repolarization at −70 mV, and compared the time course of the current relaxation. The τDact was measured by fitting the current traces with a single exponential function, and plotted versus voltage (right panels, Fig. 4A–D). Similar to the results from the hSlo1-only channels, the E/D mutation decreased the deactivation rate in the presence of β1, β3b and β4 subunits. However, the mutation did not significantly change the deactivation rate in the presence of the β2 subunit (P > 0.05 for potentials −180 to −60 mV).
Figure 4. Deactivation kinetics of WT and E/D mutant hSlo1 with β subunits in ∼2 μm[Ca2+]i.

Left column: overlay of normalized current traces at −70 mV. Prepulse potential =+200 mV for hSlo1 +β1 (A), β3b (C), and β4 (D). Prepulse potential =+140 mV for hSlo1 +β2 (B). Right column: τDact–V relations. The descriptions of the symbols are shown at the bottom.
The association of the β2 subunit renders inactivation to BK channels (Fig. 2), because the NH2-terminus of the β2 subunit acts as a peptide ball to block the open channel pore (Wallner et al. 1999; Xia et al. 1999,2003). To examine the effect of the β2 subunit on activation gating of BK channels without the interference of inactivation, we studied the coexpression of the WT and E/D mutant hSlo1 with the β2ND subunit (β2 subunit with NH2-terminal deletion to remove inactivation, see Methods) (Fig. 5) (Orio et al. 2002). A similar strategy has been employed previously by other investigators (Wallner et al. 1999; Brenner et al. 2000a; Xia et al. 2003). The E/D mutation shortened the activation time course in the presence of either the β2 or β2ND subunit (Figs 3B and 5B). However, the E/D mutation prolonged the deactivation time course in association with the β2ND subunit (Fig. 5C), which is in contrast to the effect of the β2 subunit that did not modify the deactivation time course (Fig. 4B). This result supports the idea that the N-terminus of the β2 subunit not only renders inactivation to BK channels, but also affects the activation gating of the channel (Benzinger et al. 2006).
Figure 5. Activation gating of WT and E/D mutant hSlo1 with the β2ND subunit in ∼2 μm[Ca2+]i.

A, macroscopic currents: voltage ranges from −150 to +200 mV for WT +β2ND and −200 to +200 mV for E/D +β2ND with the repolarization potential at −120 mV. B, activation and C, deactivation kinetics: left panels, overlay of normalized current traces at +60 mV (B) and −70 mV (C); right panels, τAct–V (B) and τDact–V (C) relations. The descriptions of the symbols are shown at the bottom.
The epilepsy/dyskinesia mutation modulates channel gating with and without Ca2+ binding in the presence of β subunits
Previous studies have shown that β subunits change the way the BK channel responds to intracellular Ca2+ and voltage dependence (Meera et al. 1996; Wallner et al. 1999; Xia et al. 1999; Behrens et al. 2000; Brenner et al. 2000a; Cox & Aldrich, 2000; Orio et al. 2002; Orio & Latorre, 2005). We examined how the E/D mutation affects voltage dependence and Ca2+ sensitivity of BK channels in the presence of β subunits (Fig. 6).
Figure 6. V1/2/Z vs.[Ca2+]i of WT and E/D mutant hSlo1 with and without β subunits.

Left column shows V1/2vs.[Ca2+]i plots and right column shows Z vs.[Ca2+]i plots for β1, β2ND, β3b and β4 subunits. The descriptions of symbols are shown at the bottom.
As shown in Fig. 1, increasing [Ca2+]i shifts the G–V relation of the E/D mutation to more negative voltages with little change to the slope. The same property holds true in the presence of β1, β2ND and β4 subunits, where the G–V relation with the E/D mutation shifts to more negative voltages with increasing [Ca2+]i with only a minor change to the slope of the G–V relation represented by Z (Z vs.[Ca2+]i plots in Fig. 6). However, with the β3b subunit, the E/D mutation decreased the voltage dependence more prominently, which is reflected in a smaller value of Z (Fig. 6) compared with WT hSlo1 channels at the same [Ca2+]i.
In the presence of the β1 subunit, the Ca2+ sensitivity measured as the difference in V1/2 between nominal 0 and the saturating [Ca2+]i showed little difference between the WT and E/D mutant hSlo1 (WT +β1: ΔV1/2,0−100Ca= 300.4 ± 14.9 mV; E/D +β1: ΔV1/2,0−100Ca= 315.8 ± 5.2 mV, P > 0.05). In other words, the β1 subunit reduced the effect of the E/D mutation in enhancing Ca2+ sensitivity. Consistent with this observation, the MWC model fitting of the G–V relations of WT and E/D mutant hSlo1 with the β1 subunit (Fig. 7) shows that the E/D mutation causes a smaller change in KC and KO than for the hSlo1-only channels (Table 1). However, the mutation caused a negative shift in the G–V relation of a similar amount at each [Ca2+]i tested in the presence of the β1 subunit. Even at nominal 0 μm[Ca2+]i, a significant leftward shift in V1/2 in the E/D mutant channel was observed (WT +β1: 228.0 ± 4.4 mV; E/D +β1: 186.8 ± 2.6 mV, P < 0.05). The magnitude of this change with the β1 subunit in the absence of Ca2+ differs from the effect of the mutation on the hSlo1-only channels (Fig. 6, grey symbols), in which the E/D mutation produced a large negative shift of the G–V relation only in the presence of Ca2+, and thereby enhancing Ca2+ sensitivity. However, in association with the β1 subunit, the E/D mutation caused a parallel shift of the G–V relation at all [Ca2+]i values, with a smaller effect on Ca2+ sensitivity. The increased G–V shift at 0 [Ca2+]i produced by the E/D mutation in the presence of the β1 subunit demonstrates more clearly that besides increasing Ca2+ sensitivity, the mutation alters the gating mechanism even in the absence of Ca2+ binding.
Figure 7. MWC model fits of WT and E/D mutant hSlo1 with β subunits in different [Ca2+]i values.

Mean G–V relationships for WT and E/D mutant are fitted using the MWC model. The specific [Ca2+]i for each symbol is shown.
To examine the impact of the β2 subunit on the modulation of Ca2+ sensitivity by the E/D mutation, we studied the channels that contained β2ND to avoid the interference from inactivation. The modification to the inactivation by the E/D mutation will be described in a later section. With the β2ND subunit, the mutation enhanced Ca2+ sensitivity (WT +β2ND: ΔV1/2,0−100Ca= 295.0 ± 6.7 mV; E/D +β2ND: ΔV1/2,0−100Ca= 325.8 ± 6.1 mV, P < 0.05). The changes in KC and KO from the MWC model fitting are consistent with this result (Fig. 7, Table 1). In addition, the mutation caused a small, but significant shift of the G–V relation in the near absence of Ca2+ (WT +β2ND: 189.6 ± 2.7; E/D +β2ND: 179.7 ± 2.3 mV, respectively, with P < 0.05) (Fig. 6). Thus, in the presence of the β2ND subunit, the epilepsy/dyskinesia mutation caused a more negative G–V shift than in hSlo1-only channels at all [Ca2+]i values.
In the presence of the β3b subunit, the V1/2vs.[Ca2+]i curves for the WT and E/D mutant hSlo1 are virtually overlapping (Fig. 6). Thus, the epilepsy/dyskinesia mutation no longer affects the steady-state activation of BK channels appreciably at any [Ca2+]i with the β3b subunit. This behaviour deviates from the channels composed of hSlo1-only. Likewise, the effect of the E/D mutation on the activation kinetics is also different for the hSlo1-only and hSlo1 +β3b channels (Fig. 3C). These results suggest that the interaction between the β3b subunit and hSlo1 alters the effect of the E/D mutation, which may differ from the interaction of other β subunits with hSlo1. On the other hand, the apparent Ca2+ sensitivity is determined by many functional properties. For example, based on the parameters from the MWC model fits, the E/D +β3b exhibits a reduced L0, Z and KC, but an increased KO (Fig. 7, Table 1). The increase in KO and decrease in KC result in a lower Ca2+ sensitivity because the ratio KO/KC measures the allosteric coupling between Ca2+ binding and channel opening (Cox et al. 1997). However, a reduction in Z makes the G–V shift more sensitive to increases in [Ca2+]i (Cui & Aldrich, 2000). Thus, these changes to the parameters by the E/D mutation may collectively contribute to a lack of Ca2+ sensitivity increase with the association of the β3b subunit (Figs 6 and 7). Similar changes in L0, Z, KC and KO produced by the E/D mutation are also seen in the presence of other β subunits although the combination of these changes differs depending on β subunits. Therefore, it is possible that all the β subunits altered the effects of the E/D mutation with similar mechanisms, while the differences in the effects of the E/D mutation may derive from different details of the interactions of the β subunits with hSlo1.
For the WT hSlo1, its association with the β4 subunit shifted the G–V relation to more positive voltages at low [Ca2+]i (< ∼5 μm), but to more negative voltages at high [Ca2+]i (> ∼5 μm). Thus, the V1/2vs.[Ca2+]i curves for WT hSlo1 and WT +β4 cross over at ∼5 μm[Ca2+]i (Fig. 6). For the E/D mutant hSlo1, the association with the β4 subunit did not affect steady-state channel activation as much as for the WT hSlo1, and the V1/2vs.[Ca2+]i curves for the E/D hSlo1 and E/D +β4 are nearly superimposed. This result does not suggest that the β4 subunit no longer modulates BK channel gating since both the activation and deactivation time course of the E/D mutant channel were modified by the β4 subunit (Figs 3D and 4D). Instead, it shows that for the BK channels containing the β4 subunit, the E/D mutation does not enhance channel activation uniformly; it shifts V1/2 by ≥∼40 mV in 1.2 and 2.3 μm[Ca2+]i, but by ≤∼25 mV in ∼10, 30 and 100 μm[Ca2+]i. The MWC model fitting of the G–V relations with the β4 subunit produced similar changes to the KC and KO values with the E/D mutation comparing to those of the hSlo1-only channels (Fig. 7, Table 1), resulting in increased Ca2+ affinity in both the closed and open conformations.
The epilepsy/dyskinesia mutation modifies inactivation in the presence of the β2 subunit
As noted earlier, the β2 subunit confers inactivation on BK channels. We determined the effects of the E/D mutation on the inactivation properties. Figure 8A (left panel) shows the currents of the WT and E/D mutant hSlo1 +β2 in response to a series of depolarizing voltage pulses. The E/D mutant channel inactivates at the same rate as the WT channel as shown in Fig. 8A (right panel) (P > 0.05 at all potentials, except −80 mV, P= 0.045). The inactivation time constant (τInact) was obtained by fitting the current decay from the peak activation to the steady-state current with a single exponential function for each test pulse. The channels with the β2 subunit do not inactivate completely (Brenner et al. 2000a), and the mutation increased the fraction of channels that do not inactivate (Fig. 8A and B). To quantify the amount of current that remained activated, we calculated the ratio of the currents at steady state to the peak amplitude (ISS/IP), which gave rise to the fraction of non-inactivating current (Fig. 8B). It is apparent that at all potentials tested, the mutant channels have a larger fraction of remaining currents.
Figure 8. Inactivation properties of WT and E/D mutant hSlo1 with β2 in ∼10 μm[Ca2+]i.

A, inactivation kinetics. Left: voltage protocol (upper) and macroscopic currents (lower two). Right: voltage dependence of the inactivation time. τInact was measured by fitting the inactivating current traces with a single exponential function from the peak amplitude to steady-state value. B, inactivation is not complete. Left: normalized current traces at +40 mV. Right: the fraction of non-inactivating current as the ratio of steady-state current (ISS) to peak current (IP) at different voltages. C, steady-state inactivation. Left: voltage protocol (upper) and macroscopic currents (lower two). Right: inactivation was measured as the ratio of the peak current at the +140 mV test pulse with different prepulses (I) versus the peak current at the +140 mV test pulse with the prepulse of −210 mV (I0). The plot was fitted using a sigmoidal function: 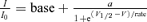 where base is the minimum value of I/I0, a is the I/I0 (max) −I/I0 (min), V1/2 is the half I/I0 voltage and rate is slope of the curve.
where base is the minimum value of I/I0, a is the I/I0 (max) −I/I0 (min), V1/2 is the half I/I0 voltage and rate is slope of the curve.
We then investigated the mutation-induced changes to the voltage dependence of steady-state inactivation. We used a voltage protocol which varied the prepulse voltage from −210 to 0 mV for 300 ms, followed by a test pulse to +140 mV for 400 ms as outlined in Fig. 8C (only part of the prepulse is presented to show the full-length activation pulse). The current traces for the WT and E/D mutant hSlo1 +β2 channels are also shown below the voltage protocol in Fig. 8C. The maximum current during the test pulse with each prepulse voltage was normalized to that with the prepulse voltage of −210 mV (I/I0) to obtain the fractional availability of channels for activation and plotted as the steady-state inactivation curve (right panel, Fig. 8C). The inactivation curve for the E/D mutant channel is shallower than that for the WT, and the V1/2 of the inactivation curves for the WT and E/D mutant hSlo1 +β2 was −118.3 ± 0.9 mV and −136.9 ± 0.9 mV, respectively (Fig. 8C).
The recovery from inactivation was also affected by the E/D mutation. To quantify the time it takes for the channels to recover from inactivation, we used a paired-pulse protocol (Fig. 9, top panel). The channels were activated and inactivated by a voltage pulse to +140 mV from a holding potential −180 mV, then allowed to recover from inactivation at −180 mV for various times before a second voltage pulse to +140 mV was applied to test the amount of the recovered current (Fig. 9, middle panels). We took a ratio of the peak current during the second depolarizing pulse to that during the first depolarizing pulse (I/Imax), and plotted the ratio versus the time interval between the first and second pulses to obtain the recovery curve (Fig. 9, bottom panel). Apparently, the E/D mutant hSlo1 +β2 channels took a longer time to recover from inactivation than the WT hSlo1 +β2 channels. The recovery curves were fitted with a single exponential function and the recovery time was ∼2.5 times slower in the E/D mutant channels (28.8 ms) as compared to the WT channels (11.7 ms).
Figure 9. Recovery from inactivation in WT and E/D mutant hSlo1 with the β2 subunit at ∼10 μm[Ca2+]i.

Top three panels show the paired-pulse recovery protocol (upper) and current traces (middle two). Channels inactivated during the +140 mV prepulse (400 ms), were allowed to recover from inactivation during various intervals (0.4, 0.8, 1.6, 3.2, 6.4, 12.8, 25.6, 51.2, 102.4 and 204.8 ms) at −180 mV, and then were activated again by a test pulse to +140 mV (200 ms). Bottom graph plots the ratio of the peak current at each of the test pulses (I) to the peak current at the test pulse following the longest interval between the prepulse and the test pulse (Imax). The continuous and dashed lines are fits using a single exponential function with the time constants of 11.7 ms for WT +β2 and 28.8 ms for E/D +β2.
Discussion
Since the Slo1 subunit of BK channels is broadly distributed, the E/D mutation is likely to follow the same pattern of expression. However, the auxiliary β subunits have a tissue-specific distribution to fine tune BK channel properties. This study reports the changes to the kinetics, voltage dependence, Ca2+ sensitivity and inactivation properties of BK channels composed of different auxiliary β subunits in the presence of the E/D mutation. Our results show that the mutation affects channel properties uniquely with each β subunit. These results provide a basis for further examining how the changes caused by the E/D mutation at the molecular level can present themselves at the system level to be associated with GEPD and to affect the function of organs other than the brain. Additionally, by studying the E/D mutation in the presence of β subunits, we provide novel insights into the molecular mechanism of how the mutation affects the channel function.
Wild-type BK channels containing β1 subunits show an increased Ca2+ sensitivity with a leftward shift in the G–V relation in the presence of Ca2+ (Meera et al. 1996; Nimigean & Magleby, 1999; Behrens et al. 2000; Brenner et al. 2000a; Cox & Aldrich, 2000; Orio & Latorre, 2005) (Fig. 6), thereby small increases in [Ca2+]i can induce channel opening. The β1 subunit also slows the rate of channel opening and closing, and decreases the voltage dependence compared to hSlo1-only channels (Meera et al. 1996; Brenner et al. 2000a; Cox & Aldrich, 2000; Bao & Cox, 2005; Orio & Latorre, 2005; Yang et al. 2008) (Figs 3A, 4A and 6). In the presence of the E/D mutation, these channels opened faster (Fig. 3A) and closed slower (Fig. 4A) with ion conduction occurring at a more negative voltage in the presence and absence of Ca2+ than in WT +β1 channels (Fig. 6). It is known that the β1 subunit alters intrinsic gating (Orio & Latorre, 2005; Wang & Brenner, 2006). The changes observed with the E/D mutation in the presence of the β1 subunit may have been facilitated by a change in the intrinsic gating equilibrium to promote channel opening and a reduction in KO (Fig. 7, Table 1). The increased activity at all [Ca2+]i values and in the length of time that the channels remain open contribute to a faster repolarization of the membrane and thus a decreased Ca2+ entry through voltage-dependent Ca2+ channels. The β1 subunits of BK channels are predominantly expressed in smooth muscles of urinary bladder (Markwardt & Isenberg, 1992; Behrens et al. 2000), the vascular (Brayden & Nelson, 1992; Behrens et al. 2000; Ledoux et al. 2006) and the respiratory systems (Savaria et al. 1992; Behrens et al. 2000) to regulate myogenic tone in these organs (Petkov et al. 2001; Ledoux et al. 2006; Semenov et al. 2006). Previous studies showed that the loss of function of the β1 subunit or Slo1, which decreases BK channel Ca2+ sensitivity or removes the channels in smooth muscle of these organs, resulted in hypertension (Brenner et al. 2000b), increased tracheal constriction (Semenov et al. 2006), urinary incontinence (Meredith et al. 2004) and overactive bladder (Meredith et al. 2004; Thorneloe et al. 2005) due to increased contractile tone. Conversely, a gain of function of the β1 subunit by a polymorphism was correlated to low incidence of hypertension (Fernandez-Fernandez et al. 2004; Senti et al. 2005). Since the epilepsy/dyskinesia mutation causes a gain of function in BK channels containing the β1 subunit, it remains to be determined if the mutation has a destructive or beneficial effect on these organs.
Similar to the β1 subunit, the β2 subunit of BK channels in the absence of inactivation increases Ca2+ sensitivity by a leftward shift of the G–V plot in the presence of Ca2+ and slows the rate of channel opening and closing (Brenner et al. 2000a; Orio & Latorre, 2005) (Figs 3–6). Additionally, the β2 subunit confers fast inactivation (Wallner et al. 1999; Xia et al. 1999) (Figs 2, 8 and 9). These β2 subunits are highly expressed in kidney, pancreas and ovary, and weakly distributed in testes, small intestine, adrenal chromaffin cells and brain (Xia et al. 1999; Behrens et al. 2000; Brenner et al. 2000a; Uebele et al. 2000). The role of the β2 subunit in these cell types is not clear, but in chromaffin cells and pancreas, the BK channels are hypothesized to participate in secretion (Petersen & Findlay, 1987; Solaro et al. 1995). The epilepsy/dyskinesia mutation in the BK channels containing β2 subunits increases Ca2+ sensitivity of channel activation (Fig. 6) by a mechanism similar to hSlo1-only channels (Fig. 7, Table 1) and the activation rate (Fig. 3B), which enhances the function of BK channels. The changes to the inactivation by the E/D mutation in general are also to enhance the function of BK channels (Figs 8 and 9). The rate of inactivation between the E/D mutant and WT hSlo1 channels is not significantly different, particularly at potentials closer to physiological conditions (at +40 mV, P > 0.05) (Fig. 8A). However, the steady-state inactivation is less in E/D mutant channels when the holding potential is at −50 to −100 mV, suggesting that in physiological conditions, the BK channels with β2 are mostly inactivated, but the E/D mutation increases the availability of these channels. Due to the presence of the β2 subunit in the brain and the exhibited changes to the function of the hSlo1 +β2 channels by the E/D mutation, the BK channels containing the β2 subunit may play an important role in the pathology of GEPD. This hypothesis is based on the channel's behaviour in chromaffin cells where the inhibitory currents lead to repetitive firing of action potentials (Solaro et al. 1995). However, in the presence of the E/D +β2 channels, we expect a further increase in the firing frequency primarily due to persistent BK current and greater channel availability than the WT channels resulting in a faster repolarization of action potentials. Since BK channels are localized at the axons and excitatory synapses (Hu et al. 2001; Sailer et al. 2006; Sausbier et al. 2006), it is plausible that the E/D mutation containing β2 subunits may lead to hyperexcitability associated with epilepsy in a mechanism similar to the one shown in the β4 knockout (KO) study (Brenner et al. 2005).
The β3b subunit increases channel activity by a similar leftward shift of the G–V relation at all [Ca2+]i including nominal 0 μm (Fig. 6), as well as by an increase in the rate of channel opening (Brenner et al. 2000a). The E/D mutation slowed the rates of both channel opening and closing in the presence of the β3b subunit (Figs 3C and 4C) and had a pronounced reduction in the voltage dependence, while keeping the Ca2+ sensitivity virtually unchanged (Fig. 6). These results are unexpected since with the β1, β2 and β4 subunits, the E/D mutation caused a small change to the voltage dependence and a distinct leftward G–V shift, similar to that in the hSlo1-only channels. The β3b subunit has a broad tissue expression pattern, including in different regions of the brain (Behrens et al. 2000; Brenner et al. 2000a; Uebele et al. 2000). Little work has been done on this subunit to identify its role in the tissues. Nevertheless, a mutation on the β3 gene is linked to idiopathic epilepsy (Lorenz et al. 2007), thus supporting the idea that changes in the function of BK channels containing the β3b subunit can be detrimental. It is not known how the β3 mutation modulates the BK channel function. Since the epilepsy/dyskinesia mutation slows down the activation rate of the BK channels with the β3b subunit, the function of the channel during an action potential would be reduced. It will be interesting to compare this functional change with that of the β3 mutation in order to gain insights on how the function of the BK channels containing the β3 subunit affects physiology at system level.
The effect of the β4 subunit on the BK channel function is complex: it increases channel activity at high [Ca2+]i (≥∼5 μm), but decreases channel activity at low [Ca2+]i (Brenner et al. 2000a; Orio et al. 2002) (Fig. 6). The β4 subunit also slows the rate of channel opening and closing (Brenner et al. 2000a). The E/D mutation enhances channel activity at all [Ca2+]i values and increases opening rate (Figs 3D and 6) in the presence of the β4 subunit. A previous study using single channel recordings also found that the E/D mutation increased channel function in association with the β4 subunit in hSlo1 (Diez-Sampedro et al. 2006). However, the present study shows that the increased channel activity by the E/D mutation is not as uniform as observed in hSlo1-only channels. The β4 subunit is known as the neuronal subunit because it is primarily found in various regions of the brain (Behrens et al. 2000; Brenner et al. 2000a). The BK channels in these regions regulate neurotransmitter release (Robitaille & Charlton, 1992; Hu et al. 2001; Faber & Sah, 2003) and neuronal excitability through spike frequency adaptation (Lancaster & Nicoll, 1987; Hu et al. 2001; Faber & Sah, 2003; Gu et al. 2007). Thus, it is plausible that the changes in BK channel property by the E/D mutation may interfere with these physiological processes and contribute to GEPD. An example of such a possibility has been reported by Brenner et al. who generated a β4 KO mouse and observed seizures emanating from the temporal lobe of the brain (Brenner et al. 2005). Based on the V1/2vs.[Ca2+]i plot of the hSlo1 +β4 channels (Fig. 6), the KO of the β4 subunit would result in a gain of BK channel function at physiological [Ca2+]i values, which increases excitability of the granule cells in the KO mouse (Brenner et al. 2005).
The study of the hSlo1-only channels revealed that the E/D mutation increases Ca2+ sensitivity as well as enhancing channel activation in the absence of Ca2+ binding (Fig. 1). In the presence of the β1 subunit, the enhancement of channel activation in the absence of Ca2+ binding produced by the E/D mutation became more pronounced (Fig. 6), thus clearly demonstrating that the E/D mutation activates the channel not merely by increasing Ca2+ sensitivity. The increase in Ca2+ sensitivity produced by the E/D mutation can be approximated by changes in KC and KO of the MWC model (Figs 1 and 7; Table 1). Ca2+ activates BK channels in two major steps: Ca2+ binds to the channel protein, which then changes its conformation to open the activation gate (Cui et al. 2008). Thus in the context of the MWC model, KC and KO can be changed by at least two different mechanisms. First, the E/D mutation may directly alter the Ca2+ binding site and change Ca2+ affinity. Second, the MWC model assumes that channel opening can alter the Ca2+ binding site through an allosteric coupling, thus the KO differs from KC. Therefore, the E/D mutation may also change the allosteric coupling to alter Ca2+ affinity. The first mechanism is an attractive hypothesis because the E/D mutation (D434G) is located close to one of the putative Ca2+ binding sites, D432 (Xia et al. 2002). The E/D mutation could directly alter the conformation of the Ca2+ binding site and change Ca2+ affinity. However, such a direct change in the Ca2+ binding site is expected to either increase or decrease Ca2+ affinity in both the closed and open conformations. Nevertheless, while in the hSlo1-only channel the E/D mutation reduced both KC and KO, in the presence of the β1 and β3b subunits the E/D mutation changed KC and KO in opposite directions. Therefore, it is more likely that the E/D mutation alters Ca2+ sensitivity by changing the allosteric coupling because the allosteric interaction between the Ca2+ binding site and the activation gate depends on the open and closed states. This mechanism is also consistent with the results that the E/D mutation can enhance channel activation both in the absence and presence of Ca2+ binding. If the E/D mutation merely changes the Ca2+ binding site, it would not have changed the channel activation in the absence of Ca2+ binding.
Acknowledgments
The wild-type hSlo1 (GenBank Accession No. U11058), β1, β2, β3b and β4 clones were kindly provided by Robert Brenner (University of Texas Health Science Center, San Antonio). Qing Wang (Cleveland Clinic, Cleveland) kindly provided wild-type and D434G mutant hSlo1 clones (GenBank Accession No. NM_002247). We thank Huanghe Yang and Di Wu for comments on the manuscript. This work was supported by Epilepsy Foundation (U.S.L.) and National Institutes of Health Grant R01-HL70393 (J.C.). J.C. is Associate Professor of Biomedical Engineering on the Spencer T. Olin Endowment.
Glossary
Abbreviations
- BK
large conductance Ca2+-activated K+
- E/D
epilepsy/dyskinesia
- GEDP
generalized epilepsy and paroxysmal dyskinesia
- G–V
conductance–voltage
- hSlo1
human Slo1
- KO
Ca2+ dissociation constant in the open state
- KC
Ca2+ dissociation constant in the closed state
- KO
knockout
- MWC
Monod–Wyman–Changeux
- WT
wild-type
Author contributions
U.S.L. and J.C. designed the research; U.S.L. performed the research and analysed the data; and U.S.L. and J.C. wrote the paper.
References
- Adelman JP, Shen KZ, Kavanaugh MP, Warren RA, Wu YN, Lagrutta A, Bond CT, North RA. Calcium-activated potassium channels expressed from cloned complementary DNAs. Neuron. 1992;9:209–216. doi: 10.1016/0896-6273(92)90160-f. [DOI] [PubMed] [Google Scholar]
- Atkinson NS, Robertson GA, Ganetzky B. A component of calcium-activated potassium channels encoded by the Drosophila slo locus. Science. 1991;253:551–555. doi: 10.1126/science.1857984. [DOI] [PubMed] [Google Scholar]
- Bao L, Cox DH. Gating and ionic currents reveal how the BKCa channel's Ca2+ sensitivity is enhanced by its β1 subunit. J Gen Physiol. 2005;126:393–412. doi: 10.1085/jgp.200509346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrens R, Nolting A, Reimann F, Schwarz M, Waldschutz R, Pongs O. hKCNMB3 and hKCNMB4, cloning and characterization of two members of the large-conductance calcium-activated potassium channel β subunit family. FEBS Lett. 2000;474:99–106. doi: 10.1016/s0014-5793(00)01584-2. [DOI] [PubMed] [Google Scholar]
- Benzinger GR, Xia XM, Lingle CJ. Direct observation of a preinactivated, open state in BK channels with β2 subunits. J Gen Physiol. 2006;127:119–131. doi: 10.1085/jgp.200509425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brayden JE, Nelson MT. Regulation of arterial tone by activation of calcium-dependent potassium channels. Science. 1992;256:532–535. doi: 10.1126/science.1373909. [DOI] [PubMed] [Google Scholar]
- Brenner R, Chen QH, Vilaythong A, Toney GM, Noebels JL, Aldrich RW. BK channel β4 subunit reduces dentate gyrus excitability and protects against temporal lobe seizures. Nat Neurosci. 2005;8:1752–1759. doi: 10.1038/nn1573. [DOI] [PubMed] [Google Scholar]
- Brenner R, Jegla TJ, Wickenden A, Liu Y, Aldrich RW. Cloning and functional characterization of novel large conductance calcium-activated potassium channel β subunits, hKCNMB3 and hKCNMB4. J Biol Chem. 2000a;275:6453–6461. doi: 10.1074/jbc.275.9.6453. [DOI] [PubMed] [Google Scholar]
- Brenner R, Perez GJ, Bonev AD, Eckman DM, Kosek JC, Wiler SW, Patterson AJ, Nelson MT, Aldrich RW. Vasoregulation by the β1 subunit of the calcium-activated potassium channel. Nature. 2000b;407:870–876. doi: 10.1038/35038011. [DOI] [PubMed] [Google Scholar]
- Cox DH, Aldrich RW. Role of the β1 subunit in large-conductance Ca2+-activated K+ channel gating energetics. Mechanisms of enhanced Ca2+ sensitivity. J Gen Physiol. 2000;116:411–432. doi: 10.1085/jgp.116.3.411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox DH, Cui J, Aldrich RW. Allosteric gating of a large conductance Ca-activated K+ channel. J Gen Physiol. 1997;110:257–281. doi: 10.1085/jgp.110.3.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui J, Aldrich RW. Allosteric linkage between voltage and Ca2+-dependent activation of BK-type mslo1 K+ channels. Biochemistry. 2000;39:15612–15619. doi: 10.1021/bi001509+. [DOI] [PubMed] [Google Scholar]
- Cui J, Yang H, Lee US. Molecular mechanisms of BK channel activation. Cell Mol Life Sci. 2008 doi: 10.1007/s00018-008-8609-x. DOI 10.1007/s00018--008-8609-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diez-Sampedro A, Silverman WR, Bautista JF, Richerson GB. Mechanism of increased open probability by a mutation of the BK channel. J Neurophysiol. 2006;96:1507–1516. doi: 10.1152/jn.00461.2006. [DOI] [PubMed] [Google Scholar]
- Ding JP, Lingle CJ. Steady-state and closed-state inactivation properties of inactivating BK channels. Biophys J. 2002;82:2448–2465. doi: 10.1016/S0006-3495(02)75588-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du W, Bautista JF, Yang H, Diez-Sampedro A, You SA, Wang L, Kotagal P, Luders HO, Shi J, Cui J, Richerson GB, Wang QK. Calcium-sensitive potassium channelopathy in human epilepsy and paroxysmal movement disorder. Nat Genet. 2005;37:733–738. doi: 10.1038/ng1585. [DOI] [PubMed] [Google Scholar]
- Faber ES, Sah P. Calcium-activated potassium channels: multiple contributions to neuronal function. Neuroscientist. 2003;9:181–194. doi: 10.1177/1073858403009003011. [DOI] [PubMed] [Google Scholar]
- Fernandez-Fernandez JM, Tomas M, Vazquez E, Orio P, Latorre R, Senti M, Marrugat J, Valverde MA. Gain-of-function mutation in the KCNMB1 potassium channel subunit is associated with low prevalence of diastolic hypertension. J Clin Invest. 2004;113:1032–1039. doi: 10.1172/JCI20347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fettiplace R, Fuchs PA. Mechanisms of hair cell tuning. Annu Rev Physiol. 1999;61:809–834. doi: 10.1146/annurev.physiol.61.1.809. [DOI] [PubMed] [Google Scholar]
- Findlay I, Dunne MJ, Petersen OH. High-conductance K+ channel in pancreatic islet cells can be activated and inactivated by internal calcium. J Membr Biol. 1985;83:169–175. doi: 10.1007/BF01868748. [DOI] [PubMed] [Google Scholar]
- Gu N, Vervaeke K, Storm JF. BK potassium channels facilitate high-frequency firing and cause early spike frequency adaptation in rat CA1 hippocampal pyramidal cells. J Physiol. 2007;580:859–882. doi: 10.1113/jphysiol.2006.126367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrera GM, Etherton B, Nausch B, Nelson MT. Negative feedback regulation of nerve-mediated contractions by KCa channels in mouse urinary bladder smooth muscle. Am J Physiol Regul Integr Comp Physiol. 2005;289:R402–R409. doi: 10.1152/ajpregu.00488.2004. [DOI] [PubMed] [Google Scholar]
- Herrera GM, Heppner TJ, Nelson MT. Regulation of urinary bladder smooth muscle contractions by ryanodine receptors and BK and SK channels. Am J Physiol Regul Integr Comp Physiol. 2000;279:R60–68. doi: 10.1152/ajpregu.2000.279.1.R60. [DOI] [PubMed] [Google Scholar]
- Horrigan FT, Aldrich RW. Coupling between voltage sensor activation, Ca2+ binding and channel opening in large conductance (BK) potassium channels. J Gen Physiol. 2002;120:267–305. doi: 10.1085/jgp.20028605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu H, Shao LR, Chavoshy S, Gu N, Trieb M, Behrens R, Laake P, Pongs O, Knaus HG, Ottersen OP, Storm JF. Presynaptic Ca2+-activated K+ channels in glutamatergic hippocampal terminals and their role in spike repolarization and regulation of transmitter release. J Neurosci. 2001;21:9585–9597. doi: 10.1523/JNEUROSCI.21-24-09585.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knot HJ, Standen NB, Nelson MT. Ryanodine receptors regulate arterial diameter and wall [Ca2+] in cerebral arteries of rat via Ca2+-dependent K+ channels. J Physiol. 1998;508:211–221. doi: 10.1111/j.1469-7793.1998.211br.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancaster B, Nicoll RA. Properties of two calcium-activated hyperpolarizations in rat hippocampal neurones. J Physiol. 1987;389:187–203. doi: 10.1113/jphysiol.1987.sp016653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledoux J, Werner ME, Brayden JE, Nelson MT. Calcium-activated potassium channels and the regulation of vascular tone. Physiology. 2006;21:69–78. doi: 10.1152/physiol.00040.2005. [DOI] [PubMed] [Google Scholar]
- Lorenz S, Heils A, Kasper JM, Sander T. Allelic association of a truncation mutation of the KCNMB3 gene with idiopathic generalized epilepsy. Am J Med Genet B Neuropsychiatr Genet. 2007;144B:10–13. doi: 10.1002/ajmg.b.30369. [DOI] [PubMed] [Google Scholar]
- Markwardt F, Isenberg G. Gating of maxi K+ channels studied by Ca2+ concentration jumps in excised inside-out multi-channel patches (myocytes from guinea pig urinary bladder) J Gen Physiol. 1992;99:841–862. doi: 10.1085/jgp.99.6.841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meera P, Wallner M, Jiang Z, Toro L. A calcium switch for the functional coupling between α (hslo) and β subunits (Kv,Caβ) of maxi K channels. FEBS Lett. 1996;382:84–88. doi: 10.1016/0014-5793(96)00151-2. [DOI] [PubMed] [Google Scholar]
- Meredith AL, Thorneloe KS, Werner ME, Nelson MT, Aldrich RW. Overactive bladder and incontinence in the absence of the BK large conductance Ca2+-activated K+ channel. J Biol Chem. 2004;279:36746–36752. doi: 10.1074/jbc.M405621200. [DOI] [PubMed] [Google Scholar]
- Nimigean CM, Magleby KL. The β subunit increases the Ca2+ sensitivity of large conductance Ca2+-activated potassium channels by retaining the gating in the bursting states. J Gen Physiol. 1999;113:425–440. doi: 10.1085/jgp.113.3.425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orio P, Latorre R. Differential effects of β1 and β2 subunits on BK channel activity. J Gen Physiol. 2005;125:395–411. doi: 10.1085/jgp.200409236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orio P, Rojas P, Ferreira G, Latorre R. New disguises for an old channel: MaxiK channel β-subunits. News Physiol Sci. 2002;17:156–161. doi: 10.1152/nips.01387.2002. [DOI] [PubMed] [Google Scholar]
- Petersen OH, Findlay I. Electrophysiology of the pancreas. Physiol Rev. 1987;67:1054–1116. doi: 10.1152/physrev.1987.67.3.1054. [DOI] [PubMed] [Google Scholar]
- Petkov GV, Bonev AD, Heppner TJ, Brenner R, Aldrich RW, Nelson MT. β1-subunit of the Ca2+-activated K+ channel regulates contractile activity of mouse urinary bladder smooth muscle. J Physiol. 2001;537:443–452. doi: 10.1111/j.1469-7793.2001.00443.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts WM, Jacobs RA, Hudspeth AJ. Colocalization of ion channels involved in frequency selectivity and synaptic transmission at presynaptic active zones of hair cells. J Neurosci. 1990;10:3664–3684. doi: 10.1523/JNEUROSCI.10-11-03664.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robitaille R, Charlton MP. Presynaptic calcium signals and transmitter release are modulated by calcium-activated potassium channels. J Neurosci. 1992;12:297–305. doi: 10.1523/JNEUROSCI.12-01-00297.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sailer CA, Kaufmann WA, Kogler M, Chen L, Sausbier U, Ottersen OP, Ruth P, Shipston MJ, Knaus HG. Immunolocalization of BK channels in hippocampal pyramidal neurons. Eur J Neurosci. 2006;24:442–454. doi: 10.1111/j.1460-9568.2006.04936.x. [DOI] [PubMed] [Google Scholar]
- Sausbier U, Sausbier M, Sailer CA, Arntz C, Knaus HG, Neuhuber W, Ruth P. Ca2+ -activated K+ channels of the BK-type in the mouse brain. Histochem Cell Biol. 2006;125:725–741. doi: 10.1007/s00418-005-0124-7. [DOI] [PubMed] [Google Scholar]
- Savaria D, Lanoue C, Cadieux A, Rousseau E. Large conducting potassium channel reconstituted from airway smooth muscle. Am J Physiol Lung Cell Mol Physiol. 1992;262:L327–L336. doi: 10.1152/ajplung.1992.262.3.L327. [DOI] [PubMed] [Google Scholar]
- Schreiber M, Salkoff L. A novel calcium-sensing domain in the BK channel. Biophys J. 1997;73:1355–1363. doi: 10.1016/S0006-3495(97)78168-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenov I, Wang B, Herlihy JT, Brenner R. BK channel β1-subunit regulation of calcium handling and constriction in tracheal smooth muscle. Am J Physiol Lung Cell Mol Physiol. 2006;291:L802–L810. doi: 10.1152/ajplung.00104.2006. [DOI] [PubMed] [Google Scholar]
- Senti M, Fernandez-Fernandez JM, Tomas M, Vazquez E, Elosua R, Marrugat J, Valverde MA. Protective effect of the KCNMB1 E65K genetic polymorphism against diastolic hypertension in aging women and its relevance to cardiovascular risk. Circ Res. 2005;97:1360–1365. doi: 10.1161/01.RES.0000196557.93717.95. [DOI] [PubMed] [Google Scholar]
- Solaro CR, Prakriya M, Ding JP, Lingle CJ. Inactivating and noninactivating Ca2+- and voltage-dependent K+ current in rat adrenal chromaffin cells. J Neurosci. 1995;15:6110–6123. doi: 10.1523/JNEUROSCI.15-09-06110.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorneloe KS, Meredith AL, Knorn AM, Aldrich RW, Nelson MT. Urodynamic properties and neurotransmitter dependence of urinary bladder contractility in the BK channel deletion model of overactive bladder. Am J Physiol Renal Physiol. 2005;289:F604–610. doi: 10.1152/ajprenal.00060.2005. [DOI] [PubMed] [Google Scholar]
- Uebele VN, Lagrutta A, Wade T, Figueroa DJ, Liu Y, McKenna E, Austin CP, Bennett PB, Swanson R. Cloning and functional expression of two families of β-subunits of the large conductance calcium-activated K+ channel. J Biol Chem. 2000;275:23211–23218. doi: 10.1074/jbc.M910187199. [DOI] [PubMed] [Google Scholar]
- Wallner M, Meera P, Toro L. Molecular basis of fast inactivation in voltage and Ca2+-activated K+ channels: a transmembrane β-subunit homolog. Proc Natl Acad Sci U S A. 1999;96:4137–4142. doi: 10.1073/pnas.96.7.4137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Brenner R. An S6 mutation in BK channels reveals β1 subunit effects on intrinsic and voltage-dependent gating. J Gen Physiol. 2006;128:731–744. doi: 10.1085/jgp.200609596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YW, Ding JP, Xia XM, Lingle CJ. Consequences of the stoichiometry of Slo1 α and auxiliary β subunits on functional properties of large-conductance Ca2+-activated K+ channels. J Neurosci. 2002;22:1550–1561. doi: 10.1523/JNEUROSCI.22-05-01550.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia XM, Ding JP, Lingle CJ. Molecular basis for the inactivation of Ca2+- and voltage-dependent BK channels in adrenal chromaffin cells and rat insulinoma tumor cells. J Neurosci. 1999;19:5255–5264. doi: 10.1523/JNEUROSCI.19-13-05255.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia XM, Ding JP, Lingle CJ. Inactivation of BK channels by the NH2 terminus of the β2 auxiliary subunit: an essential role of a terminal peptide segment of three hydrophobic residues. J Gen Physiol. 2003;121:125–148. doi: 10.1085/jgp.20028667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia XM, Zeng X, Lingle CJ. Multiple regulatory sites in large-conductance calcium-activated potassium channels. Nature. 2002;418:880–884. doi: 10.1038/nature00956. [DOI] [PubMed] [Google Scholar]
- Yang H, Zhang G, Shi J, Lee US, Delaloye K, Cui J. Subunit-specific effect of the voltage sensor domain on Ca2+ sensitivity of BK channels. Biophys J. 2008;94:4678–4687. doi: 10.1529/biophysj.107.121590. [DOI] [PMC free article] [PubMed] [Google Scholar]