

Abstract
Methionine aminopeptidases (MetAPs) represent a unique class of protease that is capable of the hydrolytic removal of an N-terminal methionine residue from nascent polypeptide chains. MetAPs are physiologically important enzymes; hence, there is considerable interest in developing inhibitors that can be used as anti-angiogenic and antimicrobial agents. A detailed kinetic and spectroscopic study has been performed to probe the binding of a triazole-based inhibitor and a bestatin-based inhibitor to both Mn(II)- and Co(II)-loaded type-I (Escherichia coli) and type-II (Pyrococcus furiosus) MetAPs. Both inhibitors were found to be moderate competitive inhibitors. The triazole-type inhibitor was found to interact with both active-site metal ions, while the bestatin-type inhibitor was capable of switching its mode of binding depending on the metal in the active site and the type of MetAP enzyme.
Keywords: Hydrolysis, Manganese, Electron paramagnetic resonance, Antibiotics, Electronic absorption
Introduction
Methionine aminopeptidases (MetAPs) represent a unique class of metalloprotease that is responsible for removing N-terminal methionine residues from polypeptide chains [1, 2]. On the basis of fumagillin affinity chromatography and mass spectrometry, MetAPs were identified as the molecular targets of ovalicin, fumagillin, and TNP-470 [3]. The activity of this sesquiterpiene epoxide containing molecule has been evaluated for the treatment of a variety of cancers such as Kaposi's sarcoma, cervical cancer, brain cancer, and renal cell carcinoma [4]. Furthermore, in vivo studies showed that one of the observed effects of the inhibition of MetAPs by antiangiogenesis agents is the failure to expose glycine residues at the N termini of certain signaling proteins involved in cell cycle regulation, preventing myristolyation [5]. Selective inhibition of tumor cells by targeting a common molecule required for signal transduction of numerous growth stimulators provides an effective approach to treat cancer [6-10]. MetAPs represent just such a target and, therefore, understanding the catalytic mechanism of MetAPs is critically important for the future development of drugs that prevent tumor vasculature formation, growth, and proliferation [11-14]. In addition, sesquiterpiene epoxide containing molecules have been shown to inhibit the in vitro growth of the malaria parasites Plasmodium falciparum and Leishmania donavani; therefore, drug design efforts that target MetAPs may have additional applications in the treatment of malaria and leishmaniasis [15].
MetAPs are organized into two classes (types I and II) on the basis of the absence or presence of an extra 62 amino acid sequence (of unknown function) inserted near the catalytic domain. The type-I MetAPs from Escherichia coli (EcMetAP-I), Staphylococcus aureus, Thermotoga maritime, and Homo sapiens and the type-II MetAPs from H. sapiens (HsMetAP-II) and Pyrococcus furiosus (PfMetAP-II) have been crystallographically characterized [16-21]. All six display a novel “pita-bread” fold with an internal pseudo twofold symmetry that structurally relates the first and second halves of the polypeptide chain to each other. Both domains contribute conserved residues to the metallo-active site. In all six structures, a bis(μ-carboxylato)(μ-aquo/hydroxo)dicobalt core is observed with an additional carboxylate residue at each metal site and a single histidine bound to Co1. An X-ray crystal structure of EcMetAP-I was recently reported with partial occupancy (40%) of a single Mn(II) ion bound in the active site [22]. This structure was obtained by adding the transition-state analog inhibitor l-norleucine phosphonate, to impede divalent metal binding to the second site, and by limiting the amount of metal ion present during crystal growth. This structure provides the first structural verification that MetAPs can form mononuclear active sites and the single divalent metal ion resides on the H171 side of the active site as previously predicted by 1H NMR and extended X-ray absorption fine structure spectroscopy [23, 24].
A current challenge facing the rational design of highly potent inhibitors of MetAPs is that some candidate compounds fail to have similar efficacy between in vitro and in vivo experiments [25]. Such differences between in vivo and in vitro effectiveness have been partially ascribed to the lack of certainty about the identity of the in vivo metal ion for MetAPs and its stoichiometry [25, 26]. The dissimilarity in metal identity and its occupancy number within the active site may have subtle effects on the catalytic mechanism of MetAPs, which manifests itself in significant differences in the efficacy of potential inhibitors. It has been established that MetAPs can be activated by Co(II), Fe(II), and Mn(II), but that Zn(II) will not activate either type-I or type-II enzymes [23, 26-28]. However, a single study on the type-I MetAP from Saccharomyces cerevisiae suggested that Zn(II) ions, in the presence of 5 mM glutathione and 2 mM EDTA under aerobic conditions, provided an active MetAP enzyme, while the Co(II)-containing enzyme was largely inactivated under similar conditions and no activity could be observed in the presence of Fe(II) [29]. Even more recently, the physiologically relevant metal ion for HsMetAP-II was shown to be Mn(II) using Co(II)-specific synthetic inhibitors to probe intracellular HsMetAP-II (Fig. 1) [25]. To understand the mechanism of inhibitor binding to MetAP and to determine why a triazole-containing inhibitor, 3-((2-naphthylmethyl)sulfanyl)-4H-1,2,4-triazole (A-310840), strongly inhibits Co(II)-loaded HsMetAP-II but not Mn(II)-loaded HsMetAP-II, while a bestatin-like inhibitor, ((2RS,3R)-3-amino-2-hydroxy-5-ethylthio)pentanoyl-((S)-(−)-(1-naphthyl)ethyl)amide (A-311263), inhibits each metal-substituted HsMetAP-II enzyme equally well, we have investigated the binding affinities and structural aspects of each of these inhibitor interactions with EcMetAP-I and PfMetAP-II.
Fig. 1.
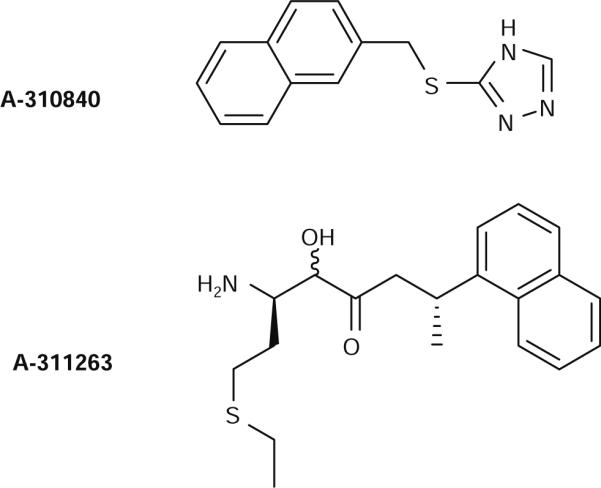
Molecular structures of the triazole-like (A310840) and the bestatin-like (A311263) methionine aminopeptidase (MetAP) inhibitors
Materials and methods
Purification of MetAP enzymes
All chemicals used in this study were purchased commercially and were of the highest quality available. Recombinant EcMetAP-I was expressed and purified as previously described [23, 27, 28]. Purified EcMetAP-I exhibited a single band on sodium dodecyl sulfate polyacrylamide gel electrophoresis. Protein concentrations were obtained spectrophotometrically at 280 nm using an extinction coefficient ε280 nm of 16,445 M−1 cm−1. Apo-EcMetAP-I was washed free of methionine using Chelex-100-treated methionine-free buffer [25 mM N-(2-hydroxyethyl)piperazine-N′-ethanesulfonic acid (HEPES), pH 7.5, 150 mM KCl] and concentrated by microfiltration using a Centricon-10 (Amicon, Beverly, MA, USA) prior to all kinetic assays. Individual aliquots of apo-EcMetAP-I were routinely stored at −80 °C or in liquid nitrogen until needed.
PfMetAP-II was purified as previously reported and exhibited a single band on sodium dodecyl sulfate polyacrylamide gel electrophoresis [28]. Protein concentrations were estimated from the absorbance at 280 nm using an extinction coefficient ε280 nm of 21,650 M−1 cm−1. Metal-free PfMetAP-II was prepared by concentrating purified PfMetAP-II to a volume of approximately 5 mL, after which EDTA was added to a final concentration of 10 mM. The resulting protein solution was dialyzed against 25 mM HEPES buffer (2 L, pH 7.5) containing 10 mM EDTA and 150 mM KCl at 4 °C for 2 days with two buffer changes per day. The protein solution was then dialyzed against Chelex-100-treated 25 mM HEPES buffer (2 L, pH 7.5) containing 150 mM KCl for 3 days against two buffer changes per day. The resulting PfMetAP-II was inactive and found to contain no detectable metal ions via inductively coupled plasma atomic emission spectrometry. This enzyme, deemed “apo-PfMetAP-II,” was stored at −80 °C until needed.
Synthesis of the triazole MetAP inhibitor A-310840
The inhibitor A-310840 was synthesized as previously described [25]. Briefly, to a suspension of 3-mercapto-1,2,4-triazole (0.18 g, 1.8 mmol) and cesium carbonate (0.72 g, 2.2 mmol) in 5 mL of N,N-dimethylformamide was added 2-(bromomethyl)naphthalene (0.38 g, 1.7 mmol). The mixture was heated at 40 °C for 16 h. The volume was reduced by rotary evaporation, and the remaining mixture was shaken with water and methylene chloride and then filtered. The layers of the filtrate were separated, and the organic phase was dried over magnesium sulfate. Filtration and solvent removal gave a white solid (0.143 g). Mass spectrometry (desorption chemical ionization/NH3) m/z 242 (M + H)+, 259 (M + NH4)+; 1H NMR (300 MHz, dimethyl-d6 sulfoxide, δ) 14.08 (s, 1H), 8.57 (bds, 1H), 7.88 (m, 4H), 7.49 (m, 3H), 4.51 (s, 2H); Anal. Calcd for C10H2ON4S: C, 64.70; H, 4.59; N, 17.41. Found: C, 64.93; H, 4.58; N, 17.25.
Synthesis of the bestatin MetAP inhibitor A-311263
The inhibitor A-311263 was synthesized as previously described from d-methionine (Aldrich Chemical Co.) [25]. Mass spectrometry (electrospray ionization + quadrupole 1 MS) m/z 347 (M + H)+, 693 (2 M + H)+; 1H NMR (300 MHz, dimethyl-d6 sulfoxide, δ) 8.75−7.47 (m, 7H), 6.55 (br, 1H), 4.82−4.75 (m, 1H), 4.15 (d, 1H), 3.60−3.33 (br m, 3H), 2.68−2.34 (m, 3.6H), 2.18 (q, 0.4H), 1.87−1.68 (m, 2H), 1.58−1.53 (m, 3H), 1.13 (t, 0.6H), 0.98 (t, 0.4H). Anal. Calcd for C19H27N2O2S·HCl: C, 59.59; H, 7.11; N, 7.32. Found: C, 59.54; H, 7.13; N, 7.16.
Enzymatic assay of MetAPs
All enzymatic assays were performed under strict anaerobic conditions in an inert atmosphere glove box (Coy) with a dry bath incubator to maintain the temperature. Catalytic activities were determined with an error of ±5%. Enzyme activities for both EcMetAP-I and PfMetAP-II were determined in 25 mM HEPES buffer, pH 7.5, containing 150 mM KCl with the tetrapeptide substrate MGMM. The amount of product formation was determined by high-performance liquid chromatography (HPLC; Shimadzu LC-10A class-VP5). A typical assay involved the addition of 4 μL of metal-loaded MetAP to a 16 μL substrate–buffer mixture at 30 °C for 1 min. The reaction was quenched by the addition of 20 μL of 1% trifluoroacetic acid solution. Elution of the product was monitored at 215 nm following separation on a C8 HPLC column (Phenomenex, Luna; 5 μm, 4.6 cm × 25 cm), as previously described [23, 27, 28]. Enzyme activities are expressed as units per milligram, where one unit is defined as the amount of enzyme that releases 1 μmol of product in 1 min at 30 °C. Inhibition studies were performed by the addition of either A-310840 or A-311263 at a concentration of 0.1−2,000 μM to the assay using a substrate concentration of 8 mM.
Isothermal titration calorimetry
Isothermal titration calorimetry (ITC) measurements were carried out with a MicroCal OMEGA ultrasensitive titration calorimeter. The titrants (A-310840 and A-311263) and apo-MetAP solutions were prepared in Chelex-100-treated HEPES buffer at pH 7.5. Stock buffer solutions were thoroughly degassed before each titration. The enzyme solution (50−80 μM) loaded with 2 equiv of metal ions [Co(II) or Mn(II)] was placed in the calorimeter cell and stirred at 200 rpm to ensure rapid mixing. Typically, 4−10 μL of titrant was delivered over 7.6 s with a 6-min interval between injections to allow for complete equilibration. Each titration was continued until 4.5−6 equiv of inhibitor had been added to ensure that no additional complexes were formed in excess titrant. A background titration, consisting of the identical titrant solution but only the buffer solution in the sample cell, was subtracted from each experimental titration to account for heat of dilution. The data were analyzed with a one- or two-site binding model by the Windows-based Origin software package supplied by MicroCal [30].
Spectroscopic measurements
Electronic absorption spectra were recorded using a Shimadzu UV-3101PC spectrophotometer. All apo-MetAP samples used in spectroscopic measurements were made rigorously anaerobic prior to incubation with Co(II) (CoCl2 99.999% or better purity; Strem Chemicals, Newburyport, MA, USA) for approximately 30 min at 25 °C. All Co(II)-containing samples were handled throughout in an anaerobic glove box (Ar/5% H2, 1 ppm or less O2; Coy Laboratories) until frozen. Electronic absorption spectra were normalized for the protein concentration and the absorption due to uncomplexed Co(II) (ε512 nm = 6.0 M−1 cm−1) [23]. Low-temperature EPR spectroscopy was performed using either a Bruker ESP-300E or a Bruker EleXsys E600 spectrometer, each equipped with an ER 4116 DM dual-mode X-band cavity operating at 9.63 GHz (B0⊥B1) or 9.37 GHz (B0∥B1) and an Oxford Instruments ESR-900 helium-flow cryostat. All spectra were recorded using 100-kHz magnetic field modulation. Other EPR running parameters are specified in the figure legends for individual samples. EPR simulations were carried out using matrix diagonalization (XSophe, Bruker Biospin), assuming a spin Hamiltonian H = βgHS + SDS + SAI, with S = 3/2 and D >> βgHS (=hν). Enzyme concentrations for EPR were 1 mM.
Results
Determination of IC50 values
Data on inhibition of MetAP activity by A-311263 and A-310840 (IC50 values) towards the Co(II)- and Mn(II)-loaded PfMetAP-II and EcMetAP-I enzymes are provided in Table 1. A-310840, a triazole-containing inhibitor, binds potently to PfMetAP-II in the presence of Co(II) (IC50 of 15 μM) but is approximately 67-fold less potent for Mn(II)-loaded PfMetAP-II (IC50 of 1,000 μM). A similar trend is observed for EcMetAP-I, except that A-310840 binds to the Co(II)-loaded enzyme well (IC50 of 44 μM) but is approximately 23-fold less potent for Mn(II)-loaded EcMetAP-I (IC50 of 1,000 μM). In comparison, the bestatin-type inhibitor, A-311263, has little or no preference for the type of divalent metal ion that occupies the active site. A-311263 also inhibits PfMetAP-II and EcMetAP-I loaded with either Co(II) or Mn(II) similarly, with IC50 values between 20 and 34 μM.
Table 1.
IC50, inhibition constants (Ki), dissociation constants (Kd), stoichiometry (n), and thermodynamics parameters for 3-((2-naphthylmethyl)sulfanyl)-4H-1,2,4-triazole (A-310840) and ((2RS,3R)-3-amino-2-hydroxy-5-ethylthio)pentanoyl-((S)-(−)-(1-naphthyl)ethyl) amide (A-311263) binding to Co(II)- and Mn(II)-loaded type-II methionine aminopeptidase (MetAP) from Pyrococcus furiosus (PfMetAP-II) and type-I MetAP from Escherichia coli (EcMetAP-I)
| Wild-type enzymes | n | IC50 (μM) | Ki (μM) | Kd (μM) | ΔH (kJ/mol) | TΔS (J/mol) | ΔG (kJ/mol) |
|---|---|---|---|---|---|---|---|
| A-310840 binding to wild-type enzymes | |||||||
| [CoCo(PfMetAP-II)] | 1 | 15 | 3 | 2.0 ± 0.1 | −5.5 ± 2 | 27 × 103 | −32.5 |
| [MnMn(PfMetAP-II)] | 1 | 1,000 | 440 | 666 ± 10 | −0.3 ± 0.1 | 17 × 103 | −18.1 |
| [CoCo(EcMetAP-I)] | 1 | 44 | 8 | 10 ± 0.1 | 8.36 ± 0.3 | 20 × 103 | −28.5 |
| [MnMn(EcMetAP-I)] | 1 | 1,000 | 750 | 714 ± 10 | −24.6 ± 3 | −7 × 103 | −17.9 |
| A-311263 binding to wild-type enzymes | |||||||
| [CoCo(PfMetAP-II)] | 1 | 20 | 2 | 0.45 ± 0.11 | −14.9 ± 2 | 21 × 103 | −36.2 |
| [MnMn(PfMetAP-II)] | 1 | 27 | 20 | 26 ± 5 | −62.7 ± 5 | −36 × 103 | −26.1 |
| [CoCo(EcMetAP-I)] | 1 | 32 | 3 | 2.4 ± 0.1 | 1.57 ± 0.3 | 33 × 103 | −32 |
| [MnMn(EcMetAP-I)] | 1 | 34 | 20 | 50 ± 5 | −19 ± 4 | 6 × 103 | −24.5 |
Kinetic determination of Ki values
Previous work indicated that A-310840 and A-311263 are strong inhibitors of HsMetAP-II on the basis of IC50 values [25]. We have carefully examined the hydrolysis of the tetrapeptide substrate MGMM by both PfMetAP-II and EcMetAP-I in the presence of various concentrations of A-310840 and A-311263. Ki values for A-310840 and A-311263 binding to PfMetAP-II and EcMetAP-I were obtained by fitting the kinetic data to the Michaelis–Menten equation for competitive inhibition (Table 1). A-310840 is a much more potent competitive inhibitor towards Co(II)-loaded PfMetAP-II (Ki = 3 μM) and EcMetAP-I (Ki = 8 μM) compared with the Mn(II)-loaded PfMetAP-II (Ki = 440 μM) and EcMetAP-I (Ki = 750 μM) enzymes. On the other hand, A-311263 inhibits Co(II)-loaded PfMetAP-II (Ki = 2 μM) and EcMetAP-I (Ki = 3 μM) by a magnitude similar to that of Mn(II)-loaded PfMetAP-II (Ki = 27 μM) and EcMetAP-I (Ki = 34 μM).
Isothermal titration calorimetry
The use of ITC to measure the binding of exogenous ligands to biomolecules relies on the fact that such an interaction is accompanied by a heat effect. The heat absorbed (endothermic) or released (exothermic) upon inhibitor–enzyme interaction is used to obtain the dissociation constant, Kd, and the enthalpy of binding, ΔH. ITC measurements for the binding of A-310840 and A-311263 to Co(II)- and Mn(II)-loaded EcMetAP-I and PfMetAP-II were carried out with a MicroCal OMEGA ultrasensitive titration calorimeter at 25.0 ± 0.2 °C. Kd values were obtained by fitting these data, after subtraction of the background heat of dilution, via an interactive process using the Origin software package. This software package uses a nonlinear least-squares algorithm, which allows the concentrations of the titrant and the sample to be fit to the heat flow per injection, to an equilibrium binding equation for either one site or two noninteracting sites. The Ka value, the enzyme–metal stoichiometry (n), and the change in enthalpy (ΔH) were allowed to vary during the fitting process. The relationship between Ka and the dissociation constant is defined as Kd = 1/Ka. The best fits obtained for the binding of A-310840 to Co(II)-loaded PfMetAP-II and EcMetAP-I provided an overall n value of 1 and Kd values of 2.0 and 10 μM, respectively, which indicate approximately 333-fold and approximately 71-fold stronger binding than is observed for A-310840 binding to Mn(II)-loaded PfMetAP-II (Kd = 666 μM) and EcMetAP-I (Kd = 714 μM). On the other hand, the best fits obtained for the binding of A-311263 to Co(II)-loaded PfMetAP-II and EcMetAP-I provided an overall n value of 1 and Kd values of 0.45 and 2.4 μM, respectively. These values indicate approximately 58-fold and approximately 21-fold tighter binding than is observed for Mn(II)-loaded PfMetAP-II and EcMetAP-I, respectively (Table 1).
The heat of reaction, ΔH, measured during an ITC experiment was converted into other thermodynamic parameters using the Gibbs free energy relationship:
| 2 |
The thermodynamic parameters for the binding of A-310840 and A-311263 to Co(II)- and Mn(II)-loaded PfMetAP-II and EcMetAP-I are listed in Table 1. Binding of these inhibitors to both type-I and type-II MetAPs was found to be spontaneous on the basis of the observed negative Gibbs free energy (ΔG). In general, the ΔG values for the binding of both inhibitors to PfMetAP-II are slightly more negative than the ΔG values obtained for EcMetAP-I. The energy change associated with Co(II)-loaded EcMetAP-I with A-311263 is weakly endothermic, with a ΔH value of 1.57 kJ mol−1, while in other cases ΔH was found to be weakly exothermic, with values ranging between −0.3 and −63 kJ mol−1. It is noteworthy that the entropic factor (TΔS) for the binding of each inhibitor to PfMetAP-II and EcMetAP-I varies depending on the enzyme and metal ions present. For example, the TΔS values for the binding of A-310840 to Mn(II)-loaded EcMetAP-I and the binding of A-311263 to Mn(II)-loaded PfMetAP-II are negative, whereas in all other cases the observed TΔS values are positive (Table 1).
Electronic absorption spectroscopy
The electronic absorption spectra of Co(II)-loaded PfMetAP-II and EcMetAP-I were recorded in the absence and presence of A-310840 and A-311263 (Figs. 2, 3) and the absorption due to apoenzyme was subtracted in each case. The absorption spectra of the uncomplexed wild-type Co(II)-loaded PfMetAP-II and EcMetAP-I enzymes were identical to those previously reported [23, 28]. Upon the addition of 1 equiv of A-310840 to both Co(II)-loaded PfMetAP-II and Co(II)-loaded EcMetAP-I, the molar absorptivities increased by approximately140 M−1 cm−1. The observed increase in absorbance at approximately 550 nm is similar in magnitude to that observed at 400 nm, raising the possibility that this increase may be to do to an underlying charge-transfer or π → π* transition with λ < 400 nm, rather than a change in absorptivity of the d–d bands from Co(II). Upon closer inspection, the λmax values of the absorption maxima show small blueshifts compared with those of the wild-type enzymes, indicating that A-310840 directly interacts with the Co(II) active sites of both EcMetAP-I and PfMetAP-II. Interestingly, the addition of 1 equiv of A-311263 to both Co(II)-loaded PfMetAP-II and Co(II)-loaded EcMetAP-I also perturbs the visible absorption spectra of Co(II)-loaded EcMetAP-I and PfMetAP-II, but in a way markedly different from that of A-310840. The maximum molar absorptivity decreases upon the addition of A-311263 to Co(II)-loaded PfMetAP-II by approximately 50 M−1 cm−1, but the observed λmax value remains unaffected. The maximum molar absorptivity does not change upon the addition of A-311263 to Co(II)-loaded EcMetAP-I (approximately 90 M−1 cm−1); however, the λmax value shifts from 580 to 560 nm.
Fig. 2.

UV–vis spectrum of wild-type [CoCo(PfMetAP-II)] (dashed line), A-310840 bound to [CoCo(PfMetAP-II)] (solid line), and A-311263 bound to [CoCo(PfMetAP-II)] (dotted line) in 25 mM N-(2-hydroxyethyl)piperazine-N′-ethanesulfonic acid (HEPES) buffer, pH 7.5, and 150 mM KCl. The absorption due to apoenzyme was subtracted in each case. PfMetAP-II is type-II methionine aminopeptidase (MetAP) from Pyrococcus furiosus
Fig. 3.

UV–vis spectrum of wild-type [CoCo(EcMetAP-I)] (dashed line), A-310840 bound to [CoCo(EcMetAP-I)] (solid line), and A-311263 bound to [CoCo(EcMetAP-I)] (solid line) in 25 mM HEPES buffer, pH 7.5, and 150 mM KCl. The absorption due to apoenzyme was subtracted in each case. EcMetAP-I is type-II MetAP from Escherichia coli
EPR spectra of A-310840 and A-311263 bound to Co(II)- and Mn(II)-loaded PfMetAP-II and EcMetAP-I
EPR spectra for both Co(II)-loaded PfMetAP-II and Co(II)-loaded EcMetAP-I have been reported previously [23]. These spectra contain broad features and exhibit no resolvable 59Co hyperfine splitting, presumably owing to extensive strains in g and/or zero-field-splitting parameters. The spectra observed for Co(II)-loaded PfMetAP-II and EcMetAP-I upon the addition of A-310840 or A-311263 are shown in Fig. 4 along with computer simulations. Spin Hamiltonian parameters derived from these simulations are provided in Table 2. In each case, the EPR spectra of the inhibited forms of Co(II)-loaded PfMetAP-II and EcMetAP-I are very similar to the spectrum from the corresponding unligated enzyme. The observed signals are dominated by an essentially axial component with E/D ∼ 0.09. For Co(II)-loaded EcMetAP-I + A-310840 (Fig. 4, spectrum B) and Co(II)-loaded PfMetAP-II + A-311263 (Fig. 4, spectrum C), a low-field eight-line hyperfine pattern was resolved, indicating the presence of a second, minor species with markedly lower geff strain. This signal may also be present in the spectrum of Co(II)-loaded PfMetAP-II + A-310840 (Fig. 4, spectrum D), but at very low concentrations, and was not observed for Co(II)-loaded EcMetAP-I + A-311263 (Fig. 4, spectrum A). In the EPR spectrum of Co(II)-loaded EcMetAP-I bound by A-310840 (Fig. 4, spectrum B), another feature associated with this second, minor species is observed at an inflection around 1,900 G. On the basis of this feature, gx,y and hence E/D could be reasonably estimated by simulation, resulting in an MS = ±½ species with E/D = 0.22. These parameters are typical for five-coordinate Co(II) centers bound within a rigid coordination sphere [31-33]. For Co(II)-loaded PfMetAP-II + A-311263 (Fig. 4, spectrum C), only the hyperfine pattern could be distinguished, at lower field than that observed for Co(II)-loaded EcMetAP-I + A-310840. The actual values of gx,y and E/D for this minor species are dependent on the identity of the ground-state doublet and therefore cannot be determined from these data.
Fig. 4.

EPR spectra of MetAP species containing Co(II). In A–D, the upper spectrum is the experimental spectrum and the lower spectrum is a computer simulation: [CoCo(EcMetAP-I)] + A-311263 (A); [CoCo(EcMetAP-I)] + A-310840 (B); [CoCo(PfMetAP-II)] + A-311263 (C); [CoCo(PfMetAP-II)] + A-310840 (D). Trace E is of naked [CoCo(PfMetAP-II)]]. Experimental spectra were recorded at 7 K, 0.2 mW (A–C); 9 K, 0.2 mW (D), and 5 K, 0.2 mW (E). Spin Hamiltonian parameters for the simulations are given in Table 2
Table 2.
Spin Hamiltonian parameters for the dominant axial and minor rhombic species obtained from the computer simulation of EPR spectra of Co(II)-loaded EcMetAP-I and PfMetAP-II in the presence of either A-310840 or A-311263
| Wild-type enzymes | Axial |
Rhombic |
||||
|---|---|---|---|---|---|---|
| g(x, y) | g(z) | E/D | g(x, y) [A(y) (10−3 cm−1)] | g(z) | E/D | |
| A-310840 binding to wild-type enzymes | ||||||
| [CoCo(PfMetAP-II)] | 2.21 | 2.09 | 0.09 | ND | ND | ND |
| [CoCo(EcMetAP-I)] | 2.316 | 2.650 | 0.095 | 2.52 [8.6] | 2.56 | 0.22 |
| A-311263 binding to wild-type enzymes | ||||||
| [CoCo(PfMetAP-II)] | 2.21 | 2.09 | 0.09 | 2.5−2.9 [10.6] | ND | ND |
| [CoCo(EcMetAP-I)] | 2.316 | 2.650 | 0.095 | ND | ND | ND |
ND not determined
In addition to the S = 3/2 signals observed for each of the Co(II)-loaded MetAP-inhibited complexes, weak signals were observed between geff = 10 and 12 in parallel mode (not shown) for Co(II)-loaded PfMetAP-II and EcMetAP-I bound by A-311263 and Co(II)-loaded EcMetAP-I + A-310840. These data are consistent with S′ = 2 and/or S′ = 3 transitions from an S = 0, 1, 2, 3 spin ladder due to spin coupling between two S = 3/2 Co(II) ions. While quantification of broad, weak integer spin signals of indeterminate S′ origin and with absorption into zero field is unreliable, integration of the S = 3/2 components based on simulation of the observed spectra suggested that approximately 85 ± 10% of the Co(II) ions are accountable for by the observed S = 3/2 species. The observed integer-spin signals are particularly pronounced for Co(II)-loaded PfMetAP-II + A-311263, and the same integer-spin transitions are likely due to the EPR absorption at very low field (0−500 G) observed in normal-mode EPR (Fig. 4, spectrum C; a small amount of such a signal was included in the simulation but parameters are not given as no attempt was made to fully characterize or quantify this signal). It is unlikely that these signals are dependent upon the inhibitor as similar low-field signals are observed for uncomplexed Co(II)-loaded PfMetAP-II (Fig. 4, spectrum E) and EcMetAP-I enzymes at high pH. Indeed, the lack of such signals from Co(II)-loaded PfMetAP-II + A-310840 may be of more significance.
The EPR spectra of Mn(II)-loaded EcMetAP-I and PfMetAP-II complexed with either A-310840 or A-311263 are presented in Fig. 5. When Mn(II)-loaded EcMetAP-I and PfMetAP-II were incubated with A-310840, both enzymes displayed signals superficially similar to those of the corresponding wild-type enzymes. Characteristic central six-line patterns with approximately 90 G splitting and Curie law temperature dependence are observed, as well as additional features that demonstrate Boltzmann temperature dependence and an approximately 45 G splitting [25, 26]. The central six-line patterns observed for Mn(II)-loaded EcMetAP-I and PfMetAP-II bound by A-310840 are somewhat lower in intensity compared to those of wild-type forms of the enzymes, but the individual lines are better resolved. However, the resonance positions of the transitions in the 3,000−3,800-G region of the spectra are indistinguishable (Fig. 5, spectra F, H). Both sets of spectra provide other features with Boltzmann temperature dependence that exhibit approximately 45 G splitting, characteristic of dinuclear Mn(II) centers (Fig. 5, spectra I, K, L, N).
Fig. 5.

EPR spectra from MetAP containing Mn(II). Traces A–E are 6,000-G (600-mT) experimental scans. Traces F, G, and H and I, J, and K are expanded views of selected regions of A, B, and C, respectively. Traces L, M, and N are derivatives of traces I, J, and K, respectively. The traces correspond to [MnMn(EcMetAP-I)] + A-310840 (A, F, I, L), [MnMn(EcMetAP-I)] + A-311263 (B, G, J, M), [MnMn(Pf(MetAP-II)] + A-310480 (C, H, K, N), and [MnMn (Pf(MetAP-II)] + A-311263 (D). Trace E is a computer simulation of trace D assuming S = 5/2, g = 2.0, AI = 5/2 8.8 × 10−3 cm−1, D = 86 × 10−3 cm−1, σD = 7 × 10−3 cm−1, E/D = 0, and σE/D = 0.14. Experimental spectra were recorded at 30 K, 0.2 mW (A–C, F–H, I–K) or at 40 K, 0.2 mW (D)
In contrast, the addition of A-311263 to Mn(II)-loaded EcMetAP-I and PfMetAP-II provided EPR spectra that are distinctly different from those of wild-type Mn(II)-loaded enzymes. The spectrum of Mn(II)-loaded EcMetAP-I + A-311263 contained a central six-line pattern (Fig. 5, spectrum G) indistinguishable from the patterns of the A-310840 complexes and wild-type enzymes. However, this signal did not exhibit the Boltzmann-temperature-dependent “wings” at 2,200−2,600 G with approximately 45 G splittings that are characteristic of Mn(II)–Mn(II) spin–spin coupling (Fig. 5, spectra J, L). More dramatic differences were seen in the spectrum of Mn(II)-loaded PfMetAP-II + A-311263 (Fig. 5, spectrum D). A very complex spectrum was observed that had intense features across the field range 0−6,000 G. The form of this spectrum was essentially invariant from 5 to 40 K and exhibited Curie law temperature dependence, suggesting that the features are due to isolated S = 5/2 Mn(II) ions. Indeed, this signal could be well simulated assuming a single Mn(II) species with spin Hamiltonian parameters S = 5/2, g = 2.0, AI = 5/2 = 8.8 × 10−3 cm−1, D = 86 × 10−3 cm−1, σD = 7 × 10−3 cm−1, E/D = 0, and σE/D = 0.14 (Fig. 5, spectrum E).
Discussion
Type-II MetAPs have been identified as the molecular target for antiangiogenesis agents such as fumagillin and its derivatives [3, 34], whereas type-I MetAPs have been suggested to be potential antibacterial and/or antifungal targets [35, 36]. Therefore, developing a detailed understanding of how potential small molecule drug candidates interact with the active sites of MetAPs will facilitate the rational design of new, potent MetAP inhibitors with improved in vivo stability and cytotoxicity [37]. As one might expect, most of the information regarding the binding of MetAP inhibitors has come from the inspection of X-ray crystal structures [22, 38, 39]. Although such crystallo-graphic information is extremely valuable, it represents a static view that may not be representative of a dynamic enzyme. Thus, detailed solution characterization of MetAP drug candidates is critically important in the design and development of new drugs. Herein we have investigated the binding affinities and the spectroscopic properties of two inhibitors of PfMetAP-II and EcMetAP-I, namely, a triazole-containing inhibitor 3-((2-naphthylmethyl)sulfanyl)-4H-1,2,4-triazole (A-310840) and a bestatin-like inhibitor ((2RS,3R)-3-amino-2-hydroxy-5-ethylthio)pentanoyl-((S)-(−)-(1-naphthyl)ethyl)amide (A-311263) [25].
Examination of the inhibition patterns of both Co(II)-and Mn(II)-loaded EcMetAP-I and PfMetAP-II in the presence of A-310840 and A-311263 revealed that they are both moderately strong competitive inhibitors. A-310840 preferentially inhibits Co(II)-loaded EcMetAP-I and PfMetAP-II enzymes over the Mn(II)-loaded enzymes, whereas A-311263 shows little or no metal ion selectivity for either type-I or type-II enzymes. The observed Ki values follow the same trend as the IC50 values obtained for EcMetAP-I, PfMetAP-II, and HsMetAP-II except that the IC50 values are substantially weaker for the two bacterial enzymes compared with the human type-II enzyme [25]. For competitive inhibitors, the observed Ki value is equal to the dissociation constant (Kd); therefore, ITC not only provides the opportunity to compare experimentally derived Kd values with Ki values, but also to obtain thermodynamic parameters for inhibitor binding to both the PfMetAP-II and EcMetAP-I enzymes in a single experiment. The Kd values derived from ITC are in reasonable agreement with the kinetically derived Ki values (Table 1). The observed enthalpies (ΔH) for the binding of A-310840 and A-311263 to Mn(II)-loaded PfMetAP-II and EcMetAP-I are negative. However, both A-310840 and A-311263 exhibited positive ΔH values for binding to Co(II)-loaded EcMetAP-I but negative ΔH values for binding to Co(II)-loaded PfMetAP-II. These data exemplify the active-site differences between type-I and type-II MetAPs and indicate that Mn(II)-loaded MetAPs more favorably bind these inhibitors, and possibly substrates. Interesting, the entropic factor (TΔS) follows an opposite trend, with inhibitor binding to the Co(II)-loaded enzymes being more entropically favored than inhibitor binding to Mn(II)-loaded MetAPs. Combination of the observed ΔH and TΔS values for A-310840 and A-311263 binding to both the Co(II)-loaded and the Mn(II)-loaded PfMetAP-II and EcMetAP-I enzymes are all negative, indicating that inhibitor binding is spontaneous. However, the observed ΔG values favor inhibitor binding to the Co(II)-loaded PfMetAP-II and EcMetAP-I enzymes over the Mn(II)-loaded forms by nearly 30%.
To gain some structural insight into how A-310840 and A-311263 bind to both type-I and type-II MetAPs in the presence of either Mn(II) or Co(II), electronic absorption and EPR spectra were recorded in the presence of each inhibitor. The observed changes in electronic absorption spectra of the Co(II)-loaded forms of PfMetAP-II and EcMetAP-I upon A-310840 and A-311263 binding indicate that each ligand interacts with the active-site Co(II) ions. Moreover, the observed molar absorptivities upon A-310840 binding to Co(II)-loaded EcMetAP-I and PfMetAP-II suggest that the active-site Co(II) ions are five-coordinate, while A-311263 binding provides a Co(II) environment that is five- or six-coordinate [40]. Similarly, the observed EPR spectra of the Co(II)-loaded EcMetAP-I and PfMetAP-II enzymes in the presence of A-311263 are due to transitions in the MS = ±½ Kramers doublets and arise from either five- or six-coordinate Co(II) ions The broad, featureless EPR signals from the dominant axial components of the spectra indicate significant structural microheterogeneity in each case; thus, there is a great deal of flexibility in the ligand environment, perhaps related to coordination by water. For Co(II)-loaded EcMetAP-I and PfMetAP-II enzymes in the presence of A-310840 a minor species, in some cases with well-resolved hyperfine splitting and significant rhombic distortion of the axial zero-field splitting, is observed, suggesting an alternative geometry with a rigid coordination environment and likely five-coordinate geometry [41, 42]. Similar species observed in the EPR spectra of Co(II)-substituted forms of the aminopeptidase from Aeromonas proteolytica have been shown to be pH-dependent [41, 42].
For a small percentage (less than 15%) of Co(II) ions bound to either the EcMetAP-I or PfMetAP-II enzymes in the presence of A-311263 and the Co(II)-loaded form of EcMetAP-I bound by A-310840, a weak integer spin signal in the parallel mode is observed. However, for Co(II)-loaded PfMetAP-II bound by A-310840, no integer spin signal is observed. EPR studies on Co(II)-loaded EcMetAP-I revealed that at pH 7.5 there is no significant spin coupling between the two Co(II) ions, though a small proportion (approximately 5%) of the sample exhibited detectable spin–spin interactions at pH values above 9.6 [23]. Therefore, the integer spin signals observed for the EcMetAPI-inhibited enzymes are likely due to inhibitor binding since no spin–spin interactions are observed in the absence of inhibitor. Conversely, the uncomplexed form of Co(II)-loaded PfMetAP-II exhibits detectable (approximately 15%) spin–spin interactions at pH 7.5. Thus, the observed integer spin signal for Co(II)-loaded PfMetAP-II bound by A-311263 is identical to that for the uncomplexed enzyme, suggesting that the binding of A-311263 does not provide a single atom bridge between the two Co(II) ions bound in the active site. However, the lack of an observable integer spin signal when A-310840 is complexed with Co(II)-loaded PfMetAP-II suggests that A-310840 may modulate anti-ferromagnetic coupling via a diazine bridge. Such a coupling interaction is consistent with the limited number of diazine-bridged dinuclear Co(II) model complexes [43-45]. The lack of intense parallel-mode signals is likely due to the weak Kd values observed for the second Co(II) binding event (0.35 and 2.5 mM for PfMetAP-II and EcMetAP-I, respectively), which provides enzyme samples containing a mixture of both mononuclear and dinuclear Co(II) centers with the majority of sites in the mononuclear state [23, 28]. For dinuclear Co(II) species, it appears that the binding of either A-311263 or A-310840 modulates the exchange interaction between the Co(II) ions by altering the precise nature of the electronic wavefunction overlap network (i.e., the bonding network) between these ions.
Both PfMetAP-II and EcMetAP-I exhibit distinct binding constants for two Mn(II) ions, and the higher of these, Kd2, dictates that distinct MetAP species containing either one or two Mn(II) ions will be present in samples prepared even at millimolar concentrations [22, 46, 47]. The dinu-clear forms have been shown to contain two Mn(II) ions in close proximity and, for both PfMetAP-II and EcMetAP-I, features in the EPR spectra have been identified that clearly show spin–spin interaction between the two Mn(II) ions [43-46]. As expected, EPR spectra obtained for the Mn(II)-loaded forms of both PfMetAP-II and EcMetAP-I in the presence of either A-311263 or A-310840 indicate the presence of magnetically isolated S = 5/2 Mn(II) ions coordinated by oxygen- and nitrogen-containing ligands [46]. The S = 5/2 components of the spectra with A-310840 were indistinguishable from those of the corresponding resting enzymes. The EPR spectra of PfMetAP-II and EcMetAP-I incubated with A-310840 exhibited features that are characteristic of spin–spin coupling between the Mn(II) ions; again, these features were very similar to those exhibited by the corresponding resting enzymes. The lack of significant perturbation of the electronic structure of the monomanganese and dimanganese species of these enzymes by A-310840 is likely related to the poor efficacy of this inhibitor for the manganese forms of PfMetAP-II and EcMetAP-I.
Significant differences in the electronic structures of the Mn(II) complements of PfMetAP-II and EcMetAP-I were elicited by incubation with A-311263 in contrast to incubation with A-310840. In the case of Mn(II)-loaded EcMetAP-I + A-311263, no signals or temperature-dependent features corresponding to spin-coupled Mn(II)–Mn(II) were observed between 5 and 50 K. This observation suggested the loss of any spin–spin interaction. The loss of the S′ = 1, 2,..., 5 signals would be expected to result in an increase in the S = 5/2 signal intensity due to the newly separated Mn(II) ions; the loss of the dipolar component of the coupling would, of course, entail physical separation of the two Mn(II) ions. Intriguingly, no significant enhancement of the central six-line pattern signal intensity was observed. Therefore, it is unclear whether no inter-Mn(II) interaction exists or whether the interaction is present but unobservable. An unusually high spin-coupling constant (|J| >> 35 cm−1) would prevent the population of S′ > 0 levels owing to zero-field splitting within the non-Kramers doublets of populated S′ > 0 states (Δ > 0.3 cm−1), which results from broadening by large increases in the values and/or distributions of the individual Mn(II) zero-field-splitting parameters. Alternatively, a relaxation-dependent phenomenon may be in play. Regardless, the inter-Mn(II) interaction in Mn(II)-loaded EcMetAP-I is significantly perturbed, either abolished or greatly enhanced, by A-311286.
The EPR spectrum obtained for Mn(II)-loaded PfMetAP-II + A-311286 looked completely different from any other EPR signal observed for a MetAP species but was very similar to the EPR signal observed for the argE-encoded N-acetyl-l-ornithine deacetylase from E. coli [47]. Despite the complexity of this signal, it was simulated as a single S = 5/2 Mn(II) environment, consistent with its temperature dependence. The lack of an intense central six-line pattern is due to a distribution in E/D; although the system is nominally axially symmetric with a mean E/D = 0 assumed for the sample, the distribution results in a significant proportion of the molecules in the sample exhibiting E/D ≠ 0. Thus, it appears that a rhombic distortion is introduced into the electronic symmetries of the Mn(II) ions bound to PfMetAP-II upon complexation with A-311286. Similar to Mn(II)-loaded EcMetAP-I, no evidence is observed for inter-Mn(II) spin coupling in the EPR spectrum obtained for Mn(II)-loaded PfMetAP-II + A-311286, further supporting the contention that the active site is significantly perturbed by A-311286.
The X-ray crystal structure of A-311263 bound to HsMetAP-II revealed the presence of a μ-alkoxide bridge [48]. In addition, the X-ray crystal structure of a related purported substrate-analog inhibitor (3R)-amino-(2S) hydroxyheptanoyl-l-Ala-l-Leu-l-Val-l-Phe-OMe (SK-V-126) bound to EcMetAP-I has been reported [18]. In this structure, the N-terminal amine coordinated Co2, the keto oxygen (analogous to the carbonyl carbon in a terminal peptide bond) is ligated to Co1, and the (2S)-hydroxyl group displaces the bridging water/hydroxide ion [1, 18]. Taken together with the EPR data, which primarily contain data for mononuclear enzyme species, this suggests the alkoxide moiety displaces the water (hydroxide) oxygen atom and is terminal on M1, while the N-terminal amine nitrogen of the inhibitor is bound by the carboxylate groups making out the purported second metal binding [22]. Inspection of the X-ray crystal structure of the dicobalt(II) form of type-I MetAP from S. aureus complexed with a 1,2,4-triazole-based inhibitor and the crystal structure of a 1,2,3-triazole-based inhibitor bound to the dicobalt(II) form of HsMetAP-II reveals that a common feature for the triazole derivative in enzyme recognition is the interaction of the nitrogen atoms in positions 1 and 2 with the dicobalt(II) center such that their N–N bond is nearly collinear with the metal–metal vector in the dinuclear site [49-51].
Combination of EPR and crystallographic data suggests that when two metal ions are bound to MetAPs, A-310840 binds via a diazine bridge (Fig. 6). On the other hand, when only one Co(II) ion is bound in a MetAP active site, binding of A-310840 clearly results in a more rigid five-coordinate environment. Conversely, the addition of A-310840 does not markedly perturb the EPR spectra of either Mn(II)-loaded PfMetAP-II or Mn(II)-loaded EcMetAP-I. These data indicate nonbinding or a very weak binding interaction of the triazole ligand with the Mn(II) ions bound in the MetAP active sites. Therefore, the specificity of A-310840 for Co(II) over Mn(II) is likely due to Co(II)'s preference for softer donor ligands (nitrogen) over harder donor ligands (oxygen) [52, 53]. However, this explanation does not tell the entire story. Since MetAPs are likely a mixture of mononuclear and dinuclear enzymes under physiological conditions [54, 55] and the putative partially empty, second metal binding site is carboxylate-rich, the lone pair of electrons on the N2 atom of the triazole ligand will be repelled by the negative charge associated with the carboxylate groups in this site. Alternatively, the N-terminal amine moiety of A-311263 will form a zwitterionic interaction with this carboylate site, resulting in an additional binding interaction over the A-310840 molecule (Fig. 7). This hypothesis is consistent with the observed thermodynamic parameters for both A-310840 and A-311263 binding to Co(II)- and Mn(II)-loaded PfMetAP-II or EcMetAP-I, particularly in the decrease in ΔG observed for A-310840 binding to Mn(II)-loaded MetAPs compared with Co(II)-loaded enzymes.
Fig. 6.

Proposed scheme for binding of A-310840 to the active sites of both Co(II)- and Mn(II)-loaded EcMetAP-I (residue numbering) and PfMetAP-II
Fig. 7.

Proposed scheme for binding of inhibitor A-311263 to the active site of both Co(II)- and Mn(II)-loaded EcMetAP-I (residue numbering) and PfMetAP-II
Acknowledgments
This work was supported by the National Science Foundation (CHE-0652981, R.C.H.) and the National Institutes of Health (AI056231, B.B.). The Bruker Elexsys spectrometer was purchased by the Medical College of Wisconsin and is supported with funds from the National Institutes of Health (EB001980, B.B.).
Footnotes
Present Address: S. Mitra Department of Chemistry, Boston University, Boston, MA 02215−2521, USA
Contributor Information
Brian Bennett, Department of Biophysics, The National Biomedical EPR Center, Medical College of Wisconsin, Milwaukee, WI 53226−0509, USA.
Richard C. Holz, Department of Chemistry, Loyola University-Chicago, 1068 W. Sheridan Rd, Chicago, IL 60626, USA
References
- 1.Lowther TW, Zhang Y, Sampson PB, Honek JF, Matthews BW. Insights into the mechanism of E. coli methionine aminopeptidase from the structural analysis of reaction products and phosphorous-based transition state analogs. Biochemistry. 1999;38:14810–14819. doi: 10.1021/bi991711g. [DOI] [PubMed] [Google Scholar]
- 2.Lowther WT, Matthews BW. Metalloaminopeptidases: common functional themes in disparate structural surroundings. Chem Rev. 2002;102:4581–4607. doi: 10.1021/cr0101757. [DOI] [PubMed] [Google Scholar]
- 3.Griffith EC, Su Z, Turk BE, Chen S, Chang Y-H, Wu Z, Biemann K, Liu JO. Methionine aminopeptidase (type 2) is the common target for angiogenesis inhibitors AGM-1470 and ovalicin. Chem Biol. 1997;4:461–471. doi: 10.1016/s1074-5521(97)90198-8. [DOI] [PubMed] [Google Scholar]
- 4.Selvakumar P, Lakshmikuttyamma A, Kanthan R, Kanthan SC, Dimmock JR, Sharma RK. High expression of methionine aminopeptidase 2 in human colorectal adenocarcinomas. Clin Cancer Res. 2004;10:2771–2775. doi: 10.1158/1078-0432.ccr-03-0218. [DOI] [PubMed] [Google Scholar]
- 5.Selvakumar P, Lakshmikuttyamma A, Lawman Z, Bonham K, Dimmock JR, Sharma RK. Expression of methionine aminopeptidase 2, N-myristoyltransferase, and N-myristoyltransferase inhibitor protein 71 in HT29. Biochem Biophys Res Commun. 2004;322:1012–1017. doi: 10.1016/j.bbrc.2004.08.021. [DOI] [PubMed] [Google Scholar]
- 6.Bradshaw RA. Protein translocation and turnover in eukaryotic cells. Trends Biochem Sci. 1989;14:276–279. doi: 10.1016/0968-0004(89)90063-7. [DOI] [PubMed] [Google Scholar]
- 7.Meinnel T, Mechulam Y, Blanquet S. Methionine as translation start signal—a review of the enzymes of the pathway in Escherichia coli. Biochimie. 1993;75:1061–1075. doi: 10.1016/0300-9084(93)90005-d. [DOI] [PubMed] [Google Scholar]
- 8.Bradshaw RA, Brickey WW, Walker KW. N-terminal processing: The methionine aminopeptidase and Na-acetyl transferase families. Trends Biochem Sci. 1998;23:263–267. doi: 10.1016/s0968-0004(98)01227-4. [DOI] [PubMed] [Google Scholar]
- 9.Arfin SM, Bradshaw RA. Cotranslational processing and protein turnover in eukaryotic cells. Biochemistry. 1988;27(21):7979–7984. doi: 10.1021/bi00421a001. [DOI] [PubMed] [Google Scholar]
- 10.Taunton J. How to starve a tumor. Chem Biol. 1997;4:493–496. doi: 10.1016/s1074-5521(97)90320-3. [DOI] [PubMed] [Google Scholar]
- 11.Wang J, Tucker LA, Stavropoulos J, Zhang Q, Wang Y-C, Bukofzer G, Niquette A, Meulbroek JA, Barnes DM, Shen J, Bouska J, Donawho C, Sheppard GS, Bell RL. Correlation of tumor growth suppression and methionine aminopetidase-2 activity blockade using an orally active inhibitor. Proc Natl Acad Sci USA. 2008;105:1838–1843. doi: 10.1073/pnas.0708766105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tucker LA, Zhang Q, Sheppard GS, Lou P, Jiang F, McKeegan E, Lesniewski R, Davidsen SK, Bell RL, Wang J. Ectopic expression of methionine aminopeptidase-2 causes cell transformation and stimulates proliferation. Oncogene. 2008;27:3967–3976. doi: 10.1038/onc.2008.14. [DOI] [PubMed] [Google Scholar]
- 13.Benny O, Fainaru O, Adini A, Cassiola F, Bazinet L, Adini I, Pravda E, Nahmias Y, Koirala S, Corfas G, D'Amato RJ, Folkman J. An orally delivered small-molecule formulation with anti-angiogenic and anticancer activity. Nat Biotechnol. 2008;26:799–807. doi: 10.1038/nbt1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Satchi-Fainaro R, Mamluk R, Wang L, Short SM, Nagy JA, Feng D, Dvorak AM, Dvorak HF, Puder M, Mukhopadhyay D, Folkman J. Inhibition of vessel permeability by TNP-470 and its polymer conjugate, caplostatin. Cancer Cell. 2005;7:251–261. doi: 10.1016/j.ccr.2005.02.007. [DOI] [PubMed] [Google Scholar]
- 15.Zhang P, Nicholson DE, Bujnicki JM, Su X, Brendle JJ, Ferdig M, Kyle DE, Milhous WK, Chiang PK. Angiogenesis inhibitors specific for methionine aminopeptidase 2 as drugs for malaria and leishmaniasis. J Biomed Sci. 2002;9:34–40. doi: 10.1007/BF02256576. [DOI] [PubMed] [Google Scholar]
- 16.Douangamath A, Dale GE, D'Arcy A, Almstetter M, Eckl R, Frutos-Hoener A, Henkel B, Illgen K, Nerdinger S, Schulz H, MacSweeney A, Thormann M, Treml A, Pierau S, Wadman S, Oefner C. Crystal structures of Staphylococcus aureus methionine aminopeptidase complexed with keto heterocycle and aminoketone inhibitors reveal the formation of a tetrahedral intermediate. J Med Chem. 2004;47:1325–1328. doi: 10.1021/jm034188j. [DOI] [PubMed] [Google Scholar]
- 17.Tahirov TH, Oki H, Tsukihara T, Ogasahara K, Yutani K, Ogata K, Izu Y, Tsunasawa S, Kato I. Crystal structure of the methionine aminopeptidase from the hyperthermophile, Pyrococcus furiosus. J Mol Biol. 1998;284:101–124. doi: 10.1006/jmbi.1998.2146. [DOI] [PubMed] [Google Scholar]
- 18.Lowther WT, Orville AM, Madden DT, Lim S, Rich DH, Matthews BW. Escherichia coli methionine aminopeptidase: implications of crystallographic analyses of the native, mutant and inhibited enzymes for the mechanism of catalysis. Biochemistry. 1999;38:7678–7688. doi: 10.1021/bi990684r. [DOI] [PubMed] [Google Scholar]
- 19.Roderick LS, Matthews BW. Structure of the cobalt-dependent methionine aminopeptidase from Escherichia coli: a new type of proteolytic enzyme. Biochemistry. 1993;32:3907–3912. doi: 10.1021/bi00066a009. [DOI] [PubMed] [Google Scholar]
- 20.Liu S, Widom J, Kemp CW, Crews CM, Clardy J. Structure of the human methionine aminopeptidase-2 complexed with fumagillin. Science. 1998;282:1324–1327. doi: 10.1126/science.282.5392.1324. [DOI] [PubMed] [Google Scholar]
- 21.Spraggon G, Schwarzenbacher R, Kreusch A, McMullan D, Brinen LS, Canaves JM, Dai X, Deacon AM, Elsliger MA, Eshagi S, Floyd R, Godzik A, Grittini C, Grzechnik SK, Jaroszewski L, Karlak C, Klock HE, Koesema E, Kovarik JS, Kuhn P, McPhillips TM, Miller MD, Morse A, Moy K, Ouyang J, Page R, Quijano K, Rezezadeh F, Robb A, Sims E, Stevens RC, van den Bedem H, Velasquez J, Vincent J, von Delft F, Wang X, West B, Wolf G, Xu Q, Hodgson KO, Wooley J, Lesley SA, Wilson IA. Crystal structure of a methionine aminopeptidase (TM1478) from Thermotoga maritima at 1.9Å resolution. Proteins. 2004;56:396–400. doi: 10.1002/prot.20084. [DOI] [PubMed] [Google Scholar]
- 22.Ye QZ, Xie SX, Ma ZQ, Huang M, Hanzlik RP. Structural basis of catalysis by monometalated methionine aminopeptidase. Proc Natl Acad Sci USA. 2006;103:9470–9475. doi: 10.1073/pnas.0602433103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.D'souza VM, Bennett B, Copik AJ, Holz RC. Characterization of the divalent metal binding properties of the methionyl aminopeptidase from Escherichia coli. Biochemistry. 2000;39:3817–3826. doi: 10.1021/bi9925827. [DOI] [PubMed] [Google Scholar]
- 24.Cosper NJ, D'souza V, Scott R, Holz RC. Structural evidence that the methionyl aminopeptidase from Escherichia coli is a mononuclear metalloprotease. Biochemistry. 2001;40:13302–13309. doi: 10.1021/bi010837m. [DOI] [PubMed] [Google Scholar]
- 25.Wang J, Sheppard GS, Lou P, Kawai M, Park C, Egan DA, Schneider A, Bouska J, Lesniewski R, Henkin J. Physiologically relevant metal cofactor for methionine aminopeptidase-2 is manganese. Biochemistry. 2003;42:5035–5042. doi: 10.1021/bi020670c. [DOI] [PubMed] [Google Scholar]
- 26.Chai SC, Wang W-L, Ye Q-Z. Fe(II) is the native cofactor for Escherichia coli methionine aminopeptidase. J Biol Chem. 2008;283 doi: 10.1074/jbc.M804345200. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.D'souza VM, Holz RC. The methionyl aminopeptidase from Escherichia coli is an iron(II) containing enzyme. Biochemistry. 1999;38:11079–11085. doi: 10.1021/bi990872h. [DOI] [PubMed] [Google Scholar]
- 28.Meng L, Ruebush S, D'souza VM, Copik AJ, Tsunasawa S, Holz RC. Overexpression and divalent metal binding studies for the methionyl aminopeptidase from Pyrococcus furiosus. Biochemistry. 2002;41:7199–7208. doi: 10.1021/bi020138p. [DOI] [PubMed] [Google Scholar]
- 29.Walker KW, Bradshaw RA. Yeast methionine aminopeptidase I can utilize either Zn(II) or Co(II) as a cofactor: A case of mistaken identity. Protein Sci. 1998;7:2684–2687. doi: 10.1002/pro.5560071224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Copik AJ, Swierczek SI, Lowther WT, D'souza V, Matthews BW, Holz RC. Kinetic and spectroscopic characterization of the H178A mutant of the methionyl aminopeptidase from Escherichia coli. Biochemistry. 2003;42:6283–6292. doi: 10.1021/bi027327s. [DOI] [PubMed] [Google Scholar]
- 31.Stamper C, Bennett B, Edwards T, Holz RC, Ringe D, Petsko G. Inhibition of the aminopeptidase from Aeromonas proteolytica by l-leucinephosphonic acid. Spectroscopic and crystallographic characterization of the transition state of peptide hydrolysis. Biochemistry. 2001;40:7034–7046. doi: 10.1021/bi0100891. [DOI] [PubMed] [Google Scholar]
- 32.Stamper C, Bienvenue D, Moulin A, Bennett B, Ringe D, Petsko G, Holz RC. Spectroscopic and X-ray crystallographic characterization of the bestatin bound form of the aminopeptidase from Aeromonas proteolytica. Biochemistry. 2004;43:9620–9628. doi: 10.1021/bi049126p. [DOI] [PubMed] [Google Scholar]
- 33.Kumar A, Periyannan GR, Narayanan B, Kittell AW, Kim J-J, Bennett B. Experimental evidence for a metallohydrolase mechanism in which the neucleophile is not delivered by a metal ion: EPR spectrokinetic and structural studies of aminopeptidase from Vibrio proteolyticus. Biochem J. 2007;403:527–536. doi: 10.1042/BJ20061591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sin N, Meng L, Wang MQW, Wen JJ, Bornmann WG, Crews CM. The anti-angiogenic agent fumagillin covalently binds and inhibits the methionine aminopeptidase, MetAP-2. Proc Natl Acad Sci USA. 1997;94:6099–6103. doi: 10.1073/pnas.94.12.6099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Swierczek K, Copik AJ, Swierczek SI, Holz RC. Molecular discrimination of type-I over type-II methionyl aminopeptidases. Biochemistry. 2005;44:12049–12056. doi: 10.1021/bi047752g. [DOI] [PubMed] [Google Scholar]
- 36.Luo Q-L, Li J-Y, Liu Z-Y, Chen L-L, Li J, Qian Z, Shen Q, Li Y, Lushington GH, Ye Q-Z, Nan F-J. J Med Chem. 2003;46:2631–2640. doi: 10.1021/jm0300532. [DOI] [PubMed] [Google Scholar]
- 37.Bradshaw R, Yi E. Methionine aminopeptidases and angiogenesis. Essays Biol Med. 2002;38:65–78. doi: 10.1042/bse0380065. [DOI] [PubMed] [Google Scholar]
- 38.Lowther WT, Matthews BW. Structure and function of the methionine aminopeptidases. Biochim Biophys Acta. 2000;1477:157–167. doi: 10.1016/s0167-4838(99)00271-x. [DOI] [PubMed] [Google Scholar]
- 39.Addlagatta A, Hu X, Liu JO, Matthews BW. Structural basis for the functional differences between type I and type II human methionine aminopeptidases. Biochemistry. 2005;44:14741–14749. doi: 10.1021/bi051691k. [DOI] [PubMed] [Google Scholar]
- 40.Bertini I, Luchinat C. High-spin cobalt(II) as a probe for the investigation of metalloproteins. Adv Inorg Biochem. 1984;6:71–111. [PubMed] [Google Scholar]
- 41.Bennett B, Holz RC. EPR studies on the mono- and dicobalt(II)-substituted forms of the aminopeptidase from Aeromonas proteolytica. Insight into the catalytic mechanism of dinuclear hydrolases. J Am Chem Soc. 1997;119:1923–1933. [Google Scholar]
- 42.Bennett B, Holz RC. Spectroscopically distinct cobalt(II) sites in heterodimetallic forms of the aminopeptidase from Aeromonas proteolytica: characterization of substrate binding. Biochemistry. 1997;36:9837–9846. doi: 10.1021/bi970735p. [DOI] [PubMed] [Google Scholar]
- 43.Gavrilova AL, Bosnich B. Principles of mononucleating and bionucleating ligand design. Chem Rev. 2004;104:349–383. doi: 10.1021/cr020604g. [DOI] [PubMed] [Google Scholar]
- 44.Escuer A, Vicente R, Mernari B, El Gueddi A, Pierrot M. Syntheses, structure, and magnetic behavior of two new nickel(II) and cobalt(II) dinuclear complexes with 1, 4-dicarboxylatopyridazine. MO calculations of the superexchange pathway through the pyridazine bridge. Inorg Chem. 1997;36:2511–2516. [Google Scholar]
- 45.Yoo HS, Lim JH, Kang JS, Koh EK, Hong CS. Triazole-bridged magnetic M(II) assemblies (M = Co, Ni) capped with the end-on terephthalate dianion involving multi-intermolecular contacts. Polyhedron. 2007;26:4383–4388. [Google Scholar]
- 46.Reed GH, Markham GD. Biol Magn Reson. 1984;6:73–142. [Google Scholar]
- 47.McGregor WC, Swierczek SI, Bennett B, Holz RC. Characterization of the catalytically active Mn(II)-loaded argE-encoded N-acetyl-l-ornithine deacetylase from Escherichia coli. J Biol Inorg Chem. 2007;12:603–613. doi: 10.1007/s00775-007-0211-4. [DOI] [PubMed] [Google Scholar]
- 48.Sheppard GS, Wang J, Kawai M, BaMaung NY, Craig RA, Erickson SA, Lynch L, Patel J, Yang F, Searle XB, Lou P, Park C, Kim KH, Henkin J, Lesniewski R. Bioorg Med Chem Lett. 2004;14:865–868. doi: 10.1016/j.bmcl.2003.12.031. [DOI] [PubMed] [Google Scholar]
- 49.Kallander LS, Lu Q, Chen W, Tomaszek T, Yang G, Tew D, Meek TD, Hofmann GA, Schulz-Pritchard CK, Smith WW, Janson CA, Ryan MD, Zhang G-F, Johanson KO, Kirkpatrick RB, Ho TF, Fisher PW, Mattern MR, Johnson RK, Hansbury MJ, Winkler JD, Ward KW, Veber DF, Thompson SK. 4-Aryl-1, 2, 3-triazole: a novel template for a reversible methionine aminopeptidase 2 inhibitor, optimized to inhibit angiogenesis in vivo. J Med Chem. 2005;48:5644–5647. doi: 10.1021/jm050408c. [DOI] [PubMed] [Google Scholar]
- 50.Oefner C, Douangamath A, D'Arcy AHS, Mareque D, Mac Sweeney A, Padilla J, Pierau S, Schulz H, Thormann M, Wadman S, Dale GE. J Mol Biol. 2003;332:13–21. doi: 10.1016/s0022-2836(03)00862-3. [DOI] [PubMed] [Google Scholar]
- 51.Marino JP, Fisher PW, Hofmann GA, Kirkpatrick RB, Janson CA, Johnson RK, Ma C, Mattern M, Meek TD, Ryan MD, Schulz C, Smith WW, Tew DG, Tomazek TA, Veber DF, Xiong WC, Yamamoto Y, Yamashita K, Yang G, Thompson SK. Highly potent inhibitors of methionine aminopeptidase-2 based on a 1, 2, 4-triazole pharmacophore. J Med Chem. 2007;50:3777–3785. doi: 10.1021/jm061182w. [DOI] [PubMed] [Google Scholar]
- 52.Xie S-X, Huang W-J, Ma Z-Q, Huang M, Hanzlik RP, Ye Q-Z. Acta Crystallogr D. 2006;62:425–432. doi: 10.1107/S0907444906003878. [DOI] [PubMed] [Google Scholar]
- 53.Sigel H, McCormick DB. Acc Chem Res. 1970;3:201–208. [Google Scholar]
- 54.Larrabee JA, Leung CH, Moore R, Thamrong-nawasawat T, Wessler BH. Magnetic circular dichroism and cobalt(II) binding equilibrium studies of Escherichia coli methionyl aminopeptidase. J Am Chem Soc. 2004;126:12316–12324. doi: 10.1021/ja0485006. [DOI] [PubMed] [Google Scholar]
- 55.Hu XV, Chen X, Han KC, Mildvan AS, Liu JO. Kinetic and mutational studies of the number of interacting divalent cations required by bacterial and human methionine aminopeptidases. Biochemistry. 2007;46:12833–12843. doi: 10.1021/bi701127x. [DOI] [PubMed] [Google Scholar]