

Abstract
Inorganic arsenic shows great promise in human cancer chemotherapy, although hepatotoxicity is a major limiting side‐effect. O2‐Vinyl 1‐[2‐(Carboxylato)pyrrolidin‐1‐yl]diazen‐1‐ium‐1,2‐diolate (V‐PROLI/NO) [Correction added after publication 19 December 2008: 1‐[2‐(Carboxylato)pyrrolidin‐1‐yl]diazen‐1‐ium‐1,2‐diolate (V‐PROLI/NO) was corrected to O2‐Vinyl 1‐[2‐(Carboxylato)pyrrolidin‐1‐yl]diazen‐1‐ium‐1,2‐diolate (V‐PROLI/NO)] is a nitric oxide (NO) donor prodrug that is metabolized by liver cytochromes P450 to release NO. Other NO‐releasing agents have been shown to mitigate arsenic toxicity. Thus, the effects of V‐PROLI/NO pretreatment on the toxicity of inorganic arsenic (as NaAsO2) were studied in vitro in a human liver (HepG2) cell line. HepG2 cells acted upon the prodrug to release NO, as assessed by nitrite levels, in a dose‐ and time‐dependent fashion to maximal levels of 57‐fold above control levels. In cells pretreated with V‐PROLI/NO (200 µM, 24 h) then exposed to arsenic for an additional 24 h, arsenic was much less toxic (LC50 = 151.9 ± 5.9 µM) than in control cells (LC50 = 90.5 ± 6.5 µM) and the reduced cytolethality was directly related to the level of NO produced. V‐PROLI/NO also increased CYP2E1 transcriptional expression in a dose‐dependent manner and CYP2E1 expression was directly related to the level of NO produced and the reduction in arsenic cytotoxicity. V‐PROLI/NO pretreatment markedly reduced arsenic‐induced apoptosis as measured by DNA fragmentation. Pretreatment with V‐PROLI/NO suppressed phosphorylation of JNK1/2 after arsenic exposure. Arsenic increased metallothionein, a metal‐binding protein important in arsenic tolerance, and V‐PROLI/NO pretreatment caused additional increases in metallothionein levels. Thus, the prodrug, V‐PROLI/NO, protects against arsenic toxicity in cultured human liver cells, reducing cytolethality, apoptosis and dysregulation of mitogen‐activated protein kinases, through generation of NO formed after metabolism by liver cell enzymes, possibly including CYP2E1. (Cancer Sci 2009; 100: 382–388)
Arsenicals have shown great promise in the chemotherapy of certain types of human cancers.( 1 , 2 ) In particular, arsenic trioxide treatment has proven to be curative of acute promyelocytic leukemia (APL) and markedly improves the clinical outcome of even refractory or relapsed APL.( 3 ) Arsenicals are now being tested against a variety of other tumors, including solid tumors.( 4 ) However, arsenicals can be quite toxic and concern about serious side‐effects has been raised, such as the potential for fatal hepatotoxicity in patients undergoing arsenical chemotherapy for cancer.( 5 ) The liver is a major organ for arsenic toxicity and metabolism.( 6 ) Individual variation in susceptibility to arsenic‐induced toxicity clearly occurs and is probably related to genetic polymorphisms associated with altered capacity for methylation of inorganic arsenic.( 5 ) Thus, adjuvant pharmacological agents that effectively and specifically limit arsenical hepatotoxicity could potentially increase arsenical therapeutic utility.
Nitric oxide (NO) is a signaling mediator produced by cells involved in numerous critical functions.( 7 ) Recently, several novel NO‐donating compounds have shown promising features related to their pharmacological use in cancer chemotherapy.( 7 ) Indeed, the production of site‐specific and/or novel NO‐donating agents is an important trend in the development of agents with therapeutic and chemo‐preventive potential.( 8 ) For instance, our work indicates that O2‐vinyl 1‐(pyrrolidin‐1‐yl)diazen‐1‐ium‐1,2 diolate (V‐PYRRO/NO) mitigates the hepatotoxicity of various compounds both in vivo and in vitro, including the potentially hepatotoxic chemotherapeutic metalloid, arsenic.( 9 , 10 , 11 ) Another NO prodrug, O2‐Vinyl 1‐[2‐(carboxylato)pyrrolidin‐1‐yl]diazen‐1‐ium‐1,2‐diolate (V‐PROLI/NO) [Correction added after publication 19 December 2008: 1‐[2‐(Carboxylato)pyrrolidin‐1‐yl]diazen‐1‐ium‐1,2‐diolate (V‐PROLI/NO) was corrected to O2‐Vinyl 1‐[2‐(Carboxylato)pyrrolidin‐1‐yl]diazen‐1‐ium‐1,2‐diolate (V‐PROLI/NO)] is structurally similar to V‐PYRRO/NO, but has an additional carboxylic acid and appears to have liver specificity, as it is metabolized to release NO by cytochrome P450,( 12 ) the most important of which appears to be CYP2E1.( 13 ) The major limiting effects of arsenical chemotherapy would be hepatotoxicity.( 14 ) However, whether V‐PROLI/NO is protective against arsenic toxicity in liver or liver cells has not been defined.
Metallothionein (MT) is a small, soluble, cysteine‐rich, metal‐binding protein that helps detoxicate various metals and metalloids.( 15 ) In fact, a recent study showed that human populations poorly expressing MT may be more sensitive to chronic arsenic intoxication.( 16 ) Furthermore, arsenic induces MT expression both in vitro and in vivo,( 17 , 18 , 19 ) and arsenic or its methylated metabolites interacted with MT in a stoichiometric fashion.( 17 ) Taken together, these data suggest that MT can decrease arsenic toxicity. In this regard, V‐PYRRO/NO pretreatment greatly enhances arsenic induction of MT probably as part of its mechanism in reducing arsenic toxicity.( 11 ) NO released from NO‐donating compounds increases MT levels, at least indirectly.( 20 )
Mitogen‐activated protein kinases (MAPK) are a family of serine/threonine phosphorylating proteins which can mediate signal transduction pathways from a variety of extracellular signals to regulate the expression of specific genes. The major MAPK, the extracellular signal‐regulated kinases (ERK), transduce growth factor signals inducing cell proliferation or differentiation. In contrast, stress signals like cytokines activate the c‐Jun NH2‐terminal kinases (JNK), causing stress responses, growth arrest and/or apoptosis.( 21 ) The levels of phosphorylated JNK1/2 and JNK kinase activity are markedly decreased in cells chronically exposed to arsenic.( 21 ) Activation of the JNK pathway can be critical to apoptosis and pretreatment with V‐PYRRO/NO suppresses arsenic‐induced JNK activation in liver cells,( 11 ) indicating that NO has the capacity to alter the adverse effects of arsenic with regard to apoptotic signaling.
Thus, the purpose of this study was to define if V‐PROLI/NO acts as a potential liver NO‐prodrug by blocking arsenic‐induced toxicity and apoptosis in a human liver cell line as a potential prelude to its use as an adjuvant in arsenical chemotherapy of cancer. Because CYP2E1 appears to play a critical role in livermetabolism and other NO‐releasing prodrugs,( 11 ) its potential role with V‐PROLI/NO was also studied in detail.
Materials and Methods
Chemicals. Sodium arsenite was purchased from Sigma Chemical Company (St Louis, MO, USA). V‐PROLI/NO was synthesized as previously described.( 12 ) The chemical structure of V‐PROLI/NO and its metabolism are shown in Figure 1. Anti‐phospho‐JNK, antiphospho‐ERK, antiphospho‐p38 and anti‐JNK1/2 antibodies were purchased from New England Biolabs (Beverly, MA, USA).
Figure 1.
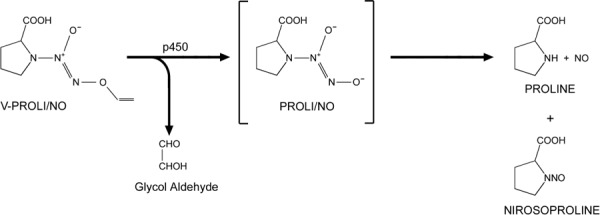
Chemical structure of V‐PROLI/NO and its metabolic pathway.
Cell culture. The HepG2 human liver cells were purchased from the American Type Culture Collection (Manassas, VA, USA). The cells were maintained at 37°C in a humidified 5% CO2 atmosphere and cultured as monolayers in modified Eagle's minimal essential medium (DMEM; pH 7.4), supplemented with 1X nonessential amino acids, 2 mM glutamine, 100 µg/mL streptomycin sulfate, 100 units/mL penicillin G and 10% (v/v) fetal bovine serum.
Metabolic integrity assay. The Promega Cell Titer 96 Non‐Radioactive Cell Proliferation Assay kit (Promega, Madison, WI, USA) was used to determine acute cytotoxicity of arsenic in cells as defined by metabolic integrity. This assay measures the amount of formazan produced by metabolic conversion of Owen's reagent (3‐[4,5‐dimethylthiazol‐2‐yl]‐5‐[3‐carboxymethoxyphenyl]‐2‐[4‐sulfophenyl]‐2H‐tetrazolium, inner salt, MTS) by dehydrogenase enzymes found in the mitochondria of metabolically active cells. The quantity of formazan product, as measured by absorbance at 490 nm, is directly proportional to the number of living cells. A minimum of four replicates of 10 000 cells/well were plated in 96‐well plates and allowed to adhere to the plate for 24 h, at which time the media were removed and replaced with fresh media with or without V‐PROLI/NO (100 or 200 µM). These levels of V‐PROLI/NO were selected because a preliminary study showed them to be non‐toxic. At the end of this period arsenic was added in fresh media. Cells were then incubated for an additional 24 h and cell viability was determined. The LC50 values were determined from analysis of the linear portion of four separately derived metabolic integrity curves.
Nitrite measurement. A Griess reagent‐based system (Promega) was used to determine nitrite concentration in cell culture medium as an indication of NO generated from V‐PROLI/NO. A minimum of four replicates of 10 000 cells/well were plated in 96‐well plates and allowed to adhere to the plate for 24 h. Cells were pretreated with V‐PROLI/NO for 24 h. Extracellular media were collected and nitrite was measured.
Quantification of apoptosis. DNA fragmentation, as an indication of apoptotic cell death, was assessed by determination of cytoplasmic histone‐DNA fragments using the Cell Death Detection ELISA kit (Roche, Indianapolis, IN, USA). In all cases, cells were seeded in 96‐well plates at 10 000 cells/well in 200 µL medium and treatments were initiated 24 h after plating. To examine the effects of V‐PROLI/NO on arsenite‐induced apoptosis, cells were pretreated with V‐PROLI/NO for 24 h and then incubated with arsenic for an additional 24 h. Apoptosis was evaluated in both floating and adherent cells.
CYP2E1 expression. Total RNA was isolated from HepG2 cells using TRIzol (Gibco/BRL Life Technologies) followed by the cleanup using RNeasy Mini kit (Qiagen, Valencia, CA, USA). The resultant DNA‐free RNA was quantitated by ultraviolet spectroscopy at 260 nm and stored in RNase‐free H2O at –70°C. Quantitative real‐time reverse transcription polymerase chain reaction (RT‐PCR) was conducted. Briefly, total RNA from each sample was reverse transcribed with MuLV reverse transcriptase (Applied Biosystems, Foster City, CA, USA) and Oligo d(T) primers. The SYBR Green PCR master mix (Applied Biosystems) was used for quantitative real‐time RT‐PCR analysis. The human primers were designed using Primer Express software (Applied Biosystems) and listed here: CYP2E1, forward 5′‐ACA GTG CAG AGC GCT TGT ACA C‐3′ and reverse, 5′‐GTC TCT GTC CCC GCA AAG AA‐3′; and β‐actin, forward 5′‐GTC CAC CTT CCA GCA GAT GTG‐3′ and reverse, 5′‐GCA TTT GCG GTG GAC GAT‐3′. Relative differences in gene expression between groups were expressed using cycle time (Ct) values; these values were first normalized with that of β‐actin in the same sample and expressed as arbitrary units. Real‐time fluorescence detection was carried out using a MyiQ Single‐Color Real‐Time PCR Detection System (Bio‐Rad, Hercules, CA, USA).
Western blot analysis. Protein samples (30 µg) derived from the various cell preparations were subjected to sodium dodecylsulfate polyacrylamide gel electrophoresis and transferred onto nitrocellulose membranes. The membranes were blocked with 5% non‐fat dry milk in Tris‐buffered saline containing 0.05% Tween 20 (TBST) and probed with phospho‐specific antibodies against JNK1/2, ERK1/2 and p38 MAPK. After incubation with secondary antibodies, immunoblots were visualized with the LumiGlo detection method (New England Biolabs).
MT quantitation. Cellular MT concentrations were measured by the Cd‐hemoglobin radioassay method.( 22 ) Values were adjusted to cell number and are expressed as nanograms of MT per 106 cells.
Statistical analysis. Data are expressed as mean ± SEM. Student's t‐test or ANOVA with subsequent Dunnett's test were used as appropriate. Linear (Pearson's) correlations were used to determine statistical significance of correlations between V‐PROLI/NO concentration or NO production and LC50 for arsenic; correlations between V‐PROLI/NO concentration or NO production and CYP2E1 transcript; correlations between CYP2E1 transcript and LC50 for arsenic. Values are derived from three or more replications. Differences were considered significant at a level of P < 0.05.
Results
Nitrite formation after V‐PROLI/NO exposure in HepG2 cells. To show that V‐PROLI/NO was acted upon by liver HepG2 cells to release NO, cells were exposed to V‐PROLI/NO at various concentrations for 24 h (Fig. 2A) or at various time points (Fig. 2B) with the same concentration (200 µM) and nitrite was measured in extracellular fluid as an indirect measurement of NO production. HepG2 cells clearly acted upon the prodrug to release NO, producing nitrite in a concentration and time‐dependent manner. Addition of V‐PROLI/NO to medium in the absence of cells did not generate nitrite (data not shown).
Figure 2.
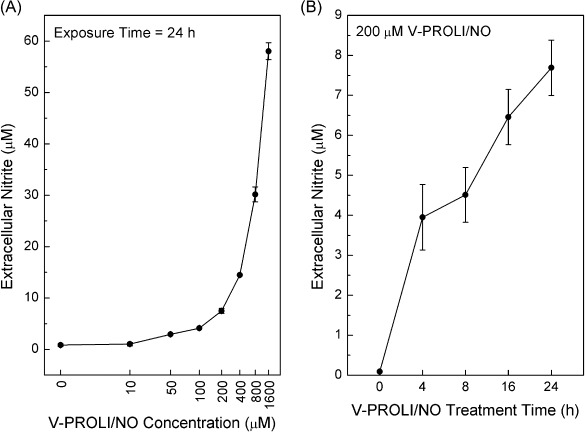
Concentration and time‐dependent nitrite formation after V‐PROLI/NO exposure. Cells were exposed to V‐PROLI/NO for 24 h at the levels indicated for the concentration‐response curve (A) or at 200 µM for the time course (B). Extracellular medium was collected and nitrite was measured by Griess assay as an indirect index of nitric oxide production. Results are presented as the mean ± standard error of the mean of four separate determinations. Note log scale in (A).
V‐PROLI/NO‐induced arsenic tolerance. HepG2 cells were pretreated with V‐PROLI/NO for 24 h and then exposed to arsenic for an additional 24 h at which point LC50 values were determined from cytotoxicity curves. The V‐PROLI/NO pretreatment significantly reduced arsenic‐induced cytotoxicity (Fig. 3). The LC50 value for arsenic in 200 µM V‐PROLI/NO pretreated cells was approximately 70% higher than in control cells. The V‐PROLI/NO pretreatment alone was not cytotoxic at the levels used (not shown).
Figure 3.
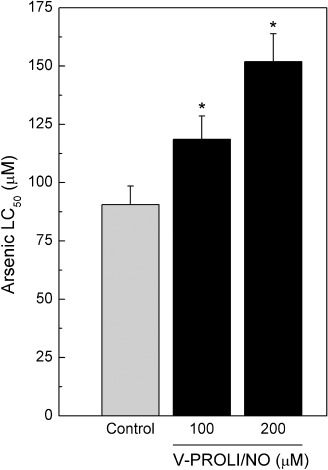
V‐PROLI/NO‐induced tolerance to arsenic‐induced cytotoxicity. HepG2 cells were pretreated with V‐PROLI/NO for 24 h or left untreated. Cells were then incubated with various levels of arsenic for an additional 24 h and LC50 values were measured by the MTS assay. Results are presented as the mean ± standard error of the mean of four separate determinations. *Significantly different (P < 0.05) from untreated control.
Correlation between V‐PROLI/NO concentration or nitrite production and LC50 for arsenic. Cells were first treated with various levels of V‐PROLI/NO for 24 h followed by arsenic for 24 h. In cells pretreated with V‐PROLI/NO, analysis revealed a highly significant correlation between increasing LC50 for arsenic and the pretreatment level of V‐PROLI/NO (Fig. 4A). The relationship between reduced arsenic cytolethality and NO production after V‐PROLI/NO treatment was also assessed by measurement of nitrite products. The results showed again that increasing NO production was also highly correlated with increases in LC50 for arsenic (Fig. 4B). These strong correlations suggest mechanistic significance indicating that when exposed to V‐PROLI/NO, HepG2 cells produce NO which in turn protects against arsenic‐induced cytolethality.
Figure 4.
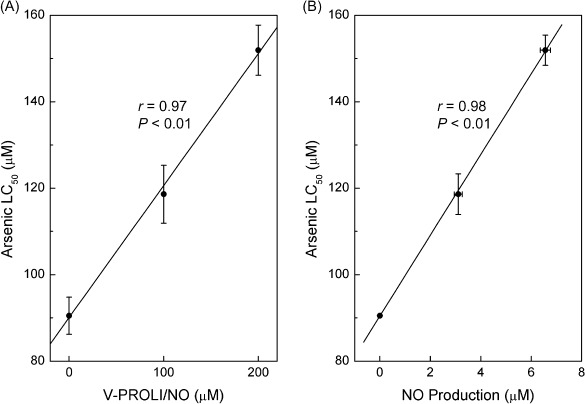
Correlations between V‐PROLI/NO concentrations or nitrite production and LC50 for arsenic. Cells were first treated with various levels of V‐PROLI/NO for 24 h followed by arsenic for 24 h. LC50 values were determined and correlated with V‐PROLI/NO concentrations (A) or nitric oxide (NO) production (B) using Pearson's r correlation. Results are presented as the mean ± standard error of the mean of four separate determinations. Arsenic LC50 values were significantly correlated with V‐PYRRO/NO concentration or NO production.
Correlations between V‐PROLI/NO concentrations, nitrite production, CYP2E1 transcript levels and arsenite cytotoxicity. HepG2 cells were treated with various levels of V‐PROLI/NO for 24 h to determine CYP2E1 levels and NO production, or additionally treated with arsenic for 24 h to determine cytotoxicity. CYP2E1 transcript levels were clearly correlated with V‐PROLI/NO concentration (Fig. 5A) and nitrite production (as an indication of NO production; Fig. 5B). A very strong correlation was observed between CYP2E1 transcript levels and increasing LC50 for arsenic (Fig. 5C). These strong correlations suggest that V‐PROLI/NO is metabolized to release NO by CYP2E1 in HepG2 cells which then protects against arsenic. Thus, it appears V‐PROLI/NO is metabolized by, at least in part, and induces, CYP2E1.
Figure 5.
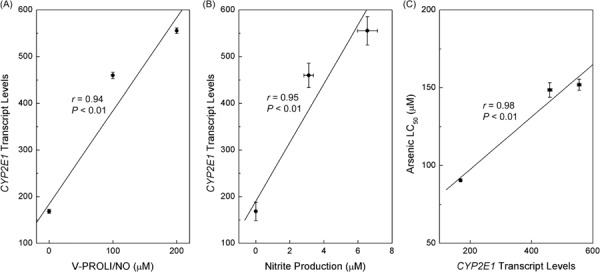
Correlations between V‐PROLI/NO concentrations, nitrite production, CYP2E1 transcript levels and arsenite cytotoxicity. Cells were treated with various levels of V‐PROLI/NO for 24 h. CYP2E1 transcript levels were then determined by real‐time polymerase chain reaction. Nitrite produced after V‐PROLI/NO treatment as an indication of NO production was measured. CYP2E1 transcript levels were then correlated with V‐PROLI/NO concentration (A) or nitrite production (B) or arsenic LC50 (C) using Pearson's r correlation. Results are presented as the mean ± standard error of the mean of four separate determinations. CYP2E1 transcript levels were significantly correlated with V‐PYRRO/NO concentration, nitric oxide production or LC50.
Effect of V‐PROLI/NO pretreatment on arsenic‐induced apoptosis and apoptotic signaling. Cells were pretreated with V‐PROLI/NO for 24 h then arsenic‐induced apoptosis at the cellular level was measured (Fig. 6). V‐PROLI/NO was able to block arsenic‐induced apoptosis in a fashion related to V‐PROLI/NO pretreatment concentration. The levels of phosphorylated JNK1/2 or total JNK1/2, key factors in apoptosis, were also determined (Fig. 7). V‐PROLI/NO blocked the activation of JNK1/2 by phosphorylation induced by arsenic. This is likely an important factor in reducing apoptotic cell death (Fig. 6). The levels of both phosphorylated ERK1/2 and p38 did not show any significant differences induced by arsenic or V‐PROLI/NO treatment (data not shown).
Figure 6.
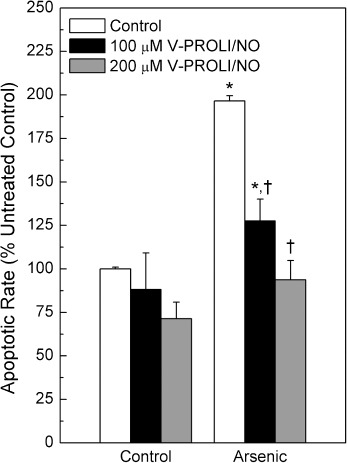
Effect of V‐PROLI/NO pretreatment on arsenic‐induced apoptosis. Cells were pretreated with V‐PROLI/NO (24 h) then arsenic (70 µM; 24 h) and apoptosis was measured by enzyme‐linked immunosorbent assay. Data are expressed as a percent of control (no V‐PROLI/NO or arsenic) ± standard error of the mean of four separate determinations. *Significantly different (P < 0.05) from untreated (no arsenic and no V‐PROLI/NO) control. †Significantly different (P < 0.05) from the arsenic‐alone control (no V‐PROLI/NO).
Figure 7.
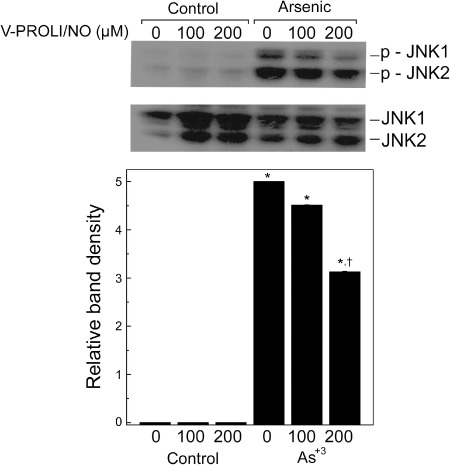
Effect of V‐PROLI/NO on level of phosphorylated JNK1/2 induced by arsenic. Cells were pretreated with V‐PROLI/NO (24 h) followed by arsenic treatment (70 µM; 24 h). The levels of phosphorylated JNK1/2 were determined by western blot analysis (top). After development, the membranes were stripped and reprobed with regular antibodies against JNK1/2 (middle). Blots represent a typical result of three independent experiments. The phosphorylated JNK1/2 protein immunoblots were analyzed by scanning densitometry (bottom) and values were then standardized to untreated control as 1. Results are presented as the mean ± standard error of the mean of three separate determinations. *Significantly different (P < 0.05) from untreated control (no arsenic and no V‐PROLI/NO). †Significantly different (P < 0.05) from the arsenite‐alone control (no V‐PROLI/NO).
Effect of V‐PROLI/NO on MT levels. HepG2 cells were treated with various levels of V‐PROLI/NO for 24 h followed by arsenic treatment and MT levels were measured (Fig. 8). Arsenic alone increased MT, while the higher pretreatment concentration of V‐PROLI/NO facilitated an additional increase in MT levels after subsequent arsenic exposure. Although significant, the additional increase in MT brought about by 200 µM V‐PROLI/NO pretreatment prior to arsenic was relatively small (~28%) compared to arsenic alone.
Figure 8.
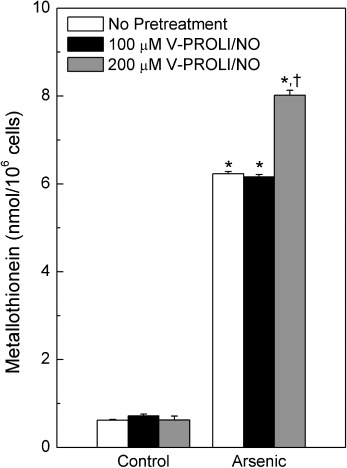
Effect of V‐PROLI/NO pretreatment on metallothionein (MT) levels. Cells were treated with V‐PROLI/NO for 24 h, followed by arsenic (70 µM; 24 h). MT levels were measured by the cadmium‐hemoglobin assay. Results are presented as the mean ± standard error of the mean of four separate determinations. *Significantly different (P < 0.05) from untreated control (no V‐PROLI/NO and no arsenic). †Significantly different (P < 0.05) from arsenic alone control (no V‐PROLI/NO).
Discussion
Arsenic trioxide is a relatively new, yet highly successful, therapy for APL( 23 ) which has now been recommended as a first‐line treatment, even in patients with multiple relapses.( 1 , 2 , 4 , 5 ) Although arsenic has shown its greatest efficacy against APL, and shows promise for a variety of other cancers,( 4 ) the concerns of potentially fatal hepatotoxicity in patients undergoing arsenical cancer chemotherapy have been raised.( 5 , 6 ) Because liver is a key target tissue of arsenic toxicity, this may limit its chemotherapeutic efficacy. Thus, it is very important to develop agents to limit arsenical hepatotoxicity without altering its chemotherapeutic effects. NO is one of the simplest biological molecules in nature and is now accepted as a fundamental signaling molecule in a wide variety of critical cellular functions.( 24 ) For instance, NO affects tumor angiogenesis, blood flow, immune surveillance, apoptosis, cell cycle, invasion and metastasis.( 24 ) Many NO donor prodrugs have been designed that have pharmacological potential.( 8 ) V‐PROLI/NO is a relatively new NO donor prodrug whose p450‐induced NO‐donating metabolite PROLI/NO has vasodilatory and antithrombotic effects.( 25 , 26 , 27 ) These can be localized to the pulmonary vasculature or the site of a vascular surgery procedure without inducing systemic hypotension.( 28 ) The present study investigated the effects of V‐PROLI/NO pretreatment on arsenic‐induced toxicity in human liver cells in vitro. Results indicate that V‐PROLI/NO releases NO apparently, at least in part, via metabolism by CYP2E1 in human liver cells, confirming prior work with metabolism of V‐PROLI/NO by microsomes enriched in human CYP2E1.( 12 ) V‐PROLI/NO‐induced tolerance to arsenic was clearly related to NO release, likely by the metabolic action of the liver cells on the NO prodrug. V‐PROLI/NO protected against the adverse effects of arsenic directly within HepG2 cells, including cytotoxicity, apoptosis and JNK pathway downregulation. Thus, at least in vitro, V‐PROLI/NO acts to reduce arsenic toxicity in a key cell site for limiting side‐effects. Although the release of NO from V‐PROLI/NO would produce other metabolites (Fig. 1), these would probably not be the primary cause of arsenic tolerance because, in several cases, other NO‐releasing prodrugs induce metal tolerance, including tolerance to arsenic toxicity, both in vivo and in vitro, but would produce dissimilar metabolites other than NO,( 11 , 29 , 30 ) indicating that NO is the primary active compound.
Mitogen‐activated protein kinases comprise a family of serine/threonine phosphorylating proteins that mediate a variety of signal transduction pathways.( 21 , 31 ) Activation of JNK is associated with the induction of apoptosis.( 27 ) Arsenic can induce activity of JNKs in various cells and activation of JNK by arsenic contributes to apoptosis.( 32 , 33 ) Interestingly, we previously demonstrated that arsenic‐transformed cells become highly resistant to arsenic‐induced apoptosis by perturbation of JNK1/2 activity.( 21 ) Recently, we showed that V‐PYRRO/NO, a liver‐selective NO‐producing prodrug, protects against arsenic‐induced cytotoxicity and apoptosis at the cellular level in cultured rat liver cells.( 11 ) This protection is apparently through generation of NO and the concurrent blockade of arsenic‐activation of the apoptosis‐related JNK pathway. The present results indicate that V‐PROLI/NO generates NO within human liver cells and downregulates levels of phosphorylated JNK1/2 induced by arsenic, an event likely related to reduction of arsenic‐induced apoptosis. Thus, the release of NO from both V‐PYRRO/NO and V‐PROLI/NO appears to be related to a reduction in arsenic‐induced apoptosis by reducing the rate approximately 50%, potentially via perturbed signaling events. It has been reported that NO can function as an intracellular signaling regulator( 34 ) and, in particular, can impact JNK signaling.( 35 , 36 ) NO is a thiol‐reactive molecule and JNK has a cysteine residue that is sensitive to thiol‐modifying agents and endogenously‐produced NO negatively regulates the JNK pathway by means of a thiol‐redox mechanism.( 34 )
Metallothionein can be induced by several inorganics and arsenic has been shown to induce MT both in vitro and in vivo,( 18 , 19 , 37 ) probably through sequestrational binding, and arsenic binds readily to thiols within MT.( 38 ) The binding stoichiometry indicates that each MT molecule binds with up to six inorganic arsenic molecules.( 17 ) MT‐I/II double knock‐out (MT‐null) mice are more sensitive than wild‐type mice to chronic arsenic‐induced hepatotoxicity.( 39 ) Humans that poorly express MT are also most sensitive to arsenic.( 16 ) The present study showed that arsenic alone increased MT levels directly in human liver cells, while only the highest concentration of V‐PROLI/NO caused additional increases in MT levels after subsequent arsenic exposure. Thus, the protective effect of V‐PROLI/NO might in part be a result of increased MT levels facilitating sequestration of arsenic, although this appeared only at the highest dose of V‐PROLI/NO and was limited.
In summary, the present work shows that exposure of human liver cells to the NO‐releasing prodrug, V‐PROLI/NO, protects against the adverse effects of arsenic including cytotoxicity, apoptosis and JNK pathway activation, apparently by generation of NO possibly via CYP2E1 as well as other enzymes. Because hepatotoxicity is a limiting side‐effect of arsenical chemotherapy, the potential of V‐PROLI/NO as an adjuvant in arsenic chemotherapy should be explored in vivo.
Acknowledgments
The authors thank Drs Erik Tokar and Chikara Kojima for critical review of this manuscript and Mr Matthew Bell for the great assistance in preparation of the graphics. This research was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research and by NCI contract NO1‐C012400 with SAIC Frederick.
References
- 1. Chen GQ, Zhu J, Shi XG et al . In vitro studies on cellular and molecular mechanisms of arsenic trioxide (As2O3) in the treatment of acute promyelocytic leukemia: As2O3 induces NB4 cell apoptosis with downregulation of Bcl‐2 expression and modulation of PML‐RAR alpha/PML proteins. Blood 1996; 88: 1052–61. [PubMed] [Google Scholar]
- 2. Shen ZX, Chen GQ, Ni JH et al . Use of arsenic trioxide (As2O3) in the treatment of acute promyelocytic leukemia (APL). II. Clinical efficacy and pharmacokinetics in relapsed patients. Blood 1997; 89: 3354–60. [PubMed] [Google Scholar]
- 3. Wang ZY, Chen Z. Acute promyelocytic leukemia: from highly fatal to highly curable. Blood 2008; 111: 2505–15. [DOI] [PubMed] [Google Scholar]
- 4. Hede K. Chinese folk treatment reveals power of arsenic to treat cancer, new studies under way. J Natl Cancer Inst 2007; 99: 667–8. [DOI] [PubMed] [Google Scholar]
- 5. Mathews V, Desire S, George B et al . Hepatotoxicity profile of single agent arsenic trioxide in the treatment of newly diagnosed acute promyelocytic leukemia, its impact on clinical outcome and the effect of genetic polymorphisms on the incidence of hepatotoxicity. Leukemia 2006; 20: 881–3. [DOI] [PubMed] [Google Scholar]
- 6. Liu J, Waalkes MP. Liver is a target of arsenic carcinogenesis. Toxicol Sci 2008; 105: 24–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kozoni V, Rosenberg T, Rigas B. Development of novel agents based on nitric oxide for the control of colon cancer. Acta Pharmacologica Sinica 2007; 28: 1429–33. [DOI] [PubMed] [Google Scholar]
- 8. Keefer LK. Progress toward clinical application of the nitric oxide‐releasing diazeniumdiolates. Annu Rev Pharmacol Toxicol 2003; 43: 585–607. [DOI] [PubMed] [Google Scholar]
- 9. Saavedra JE, Billiar TR, Williams DL, Kim YM, Watkins SC, Keefer LK. Targeting nitric oxide (NO) delivery in vivo. Design of a liver‐selective NO donor prodrug that blocks tumor necrosis factor‐alpha‐induced apoptosis and toxicity in the liver. J Med Chem 1997; 40: 1947–54. [DOI] [PubMed] [Google Scholar]
- 10. Liu J, Li C, Waalkes MP et al . The nitric oxide donor, V‐PYRRO/NO, protects against acetaminophen‐induced hepatotoxicity in mice. Hepatology 2003; 37: 324–33. [DOI] [PubMed] [Google Scholar]
- 11. Qu W, Liu J, Fuquay R, Saavedra JE, Keefer LK, Waalkes MP. The nitric oxide prodrug, V‐PYRRO/NO, mitigates arsenic‐induced liver cell toxicity and apoptosis. Cancer Lett 2007; 256: 238–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chakrapani H, Showalter BM, Kong L, Keefer LK, Saavedra JE. V‐PROLI/NO, a prodrug of the nitric oxide donor, PROLI/NO. Org Lett 2007; 9: 3409–12. [DOI] [PubMed] [Google Scholar]
- 13. Inami K, Nims RW, Srinivasan A et al . Metabolism of a liver‐selective nitric oxide‐releasing agent, V‐PYRRO/NO, by human microsomal cytochromes P450. Nitric Oxide 2006; 14: 309–15. [DOI] [PubMed] [Google Scholar]
- 14. Douer D, Tallman MS. Arsenic trioxide: new clinical experience with an old medication in hematologic malignancies. J Clin Oncol 2005; 23: 2396–410. [DOI] [PubMed] [Google Scholar]
- 15. Klaassen CD, Liu J, Choudhuri S. Metallothionein: an intracellular protein to protect against cadmium toxicity. Ann Rev Pharmacol Toxicol 1999; 39: 267–94. [DOI] [PubMed] [Google Scholar]
- 16. Liu J, Cheng ML, Yang Q et al . Blood metallothionein transcript as a biomarker for metal sensitivity: low blood metallothionein transcripts in arsenicosis patients from Guizhou, China. Environ Health Perspect 2007; 115: 1101–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jiang G, Gong Z, Li XF, Cullen WR, Le XC. Interaction of trivalent arsenicals with metallothionein. Chem Res Toxicol 2003; 16: 873–80. [DOI] [PubMed] [Google Scholar]
- 18. Kreppel H, Bauman JW, Liu J, McKim JM Jr, Klaassen CD. Induction of metallothionein by arsenicals in mice. Fundam Appl Toxicol 1993; 20: 184–9. [DOI] [PubMed] [Google Scholar]
- 19. Falnoga I, Stibilj E, Tusek‐Znidaric M et al . Effect of arsenic trioxide on metallothionein and its conversion to different arsenic metabolites in hen liver. Biol Trace Elem Res 2001; 78: 241–54. [DOI] [PubMed] [Google Scholar]
- 20. Katakai K, Liu J, Nakajima K, Keefer LK, Waalkes MP. Nitric oxide induces metallothionein (MT) gene expression apparently by displacing zinc bound to MT. Toxicol Lett 2001; 119: 103–8. [DOI] [PubMed] [Google Scholar]
- 21. Qu W, Bortner CD, Sakurai T, Hobson MJ, Waalkes MP. Acquisition of apoptotic resistance in arsenic‐induced malignant transformation: role of the JNK signal transduction pathway. Carcinogenesis 2002; 23: 151–9. [DOI] [PubMed] [Google Scholar]
- 22. Eaton DL, Toal BF. Evaluation of the Cd/hemoglobin affinity assay for the rapid determination of metallothionein in biological tissues. Toxicol Appl Pharmacol 1982; 66: 134–42. [DOI] [PubMed] [Google Scholar]
- 23. Levy M, Wofford MM, Powell BL, McLean TW. Hyperleukocytosis from arsenic trioxide. Pediatr Blood Cancer 2008; 50: 1265–7. [DOI] [PubMed] [Google Scholar]
- 24. Kozoni V, Rosenberg T, Rigas B. Development of novel agents based on nitric oxide for the control of colon cancer. Acta Pharmacol Sin 2007; 28: 1429–33. [DOI] [PubMed] [Google Scholar]
- 25. Waterhouse DJ, Saavedra JE, Davies KM et al . Injectable formulation of disodium 1‐[2‐(carboxylato) pyrrolidin‐1‐yl]diazen‐1‐ium‐1,2‐diolate (PROLI/NO), an ultrafast nitric oxide donor prodrug. J Pharm Sci 2006; 95: 108–15. [DOI] [PubMed] [Google Scholar]
- 26. Saavedra JE, Southan GJ, Davies KM et al . Localizing antithrombotic and vasodilatory activity with a novel, ultrafast nitric oxide donor. J Med Chem 1996; 39: 4361–5. [DOI] [PubMed] [Google Scholar]
- 27. Adrie C, Hirani WM, Holzmann A, Keefer L, Zapol WM, Hurford WE. Selective pulmonary vasodilation by intravenous infusion of an ultrashort half‐life nucleophile/nitric oxide adduct. Anesthesiology 1998; 88: 190–5. [DOI] [PubMed] [Google Scholar]
- 28. Chen C, Hanson SR, Keefer LK et al . Boundary layer infusion of nitric oxide reduces early smooth muscle cell proliferation in the endarterectomized canine artery. J Surg Res 1997; 67: 26–32. [DOI] [PubMed] [Google Scholar]
- 29. Qu W, Liu J, Fuquay R et al . The nitric oxide prodrug, V‐PYRRO/NO, protects against cadmium toxicity and apoptosis at the cellular level. Nitric Oxide 2005; 12: 114–20. [DOI] [PubMed] [Google Scholar]
- 30. Liu J, Qu W, Saavedra JE, Waalkes MP. The nitric oxide donor, O2‐vinyl 1‐(pyrrolidin‐1‐yl) diazen‐1‐ium‐1,2‐diolate (V‐PYRRO/NO), protects against cadmium‐induced hepatotoxicity in mice. J Pharmacol Exp Ther 2004; 310: 18–24. [DOI] [PubMed] [Google Scholar]
- 31. Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Opposing effects of ERK and JNK‐p38 MAP kinases on apoptosis. Science 1995; 270: 1326–31. [DOI] [PubMed] [Google Scholar]
- 32. Samet JM, Graves LM, Quay J et al . Activation of MAPKs in human bronchial epithelial cells exposed to metals. Am J Physiol 1998; 275: L551–8. [DOI] [PubMed] [Google Scholar]
- 33. Huang C, Ma WY, Li J, Dong Z. Arsenic induces apoptosis through a c‐Jun NH2‐terminal kinase‐dependent, p53‐independent pathway. Cancer Res 1999; 59: 3053–8. [PubMed] [Google Scholar]
- 34. Park HS, Huh SH, Kim MS, Lee SH, Choi EJ. Nitric oxide negatively regulates c‐Jun N‐terminal kinase/stress‐activated protein kinase by means of S‐nitrosylation. Proc Natl Acad Sci USA 2000; 97: 14382–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lo YY, Wong JM, Cruz TF. Reactive oxygen species mediate cytokine activation of c‐Jun NH2‐terminal kinases. J Biol Chem 1996; 271: 15703–7. [DOI] [PubMed] [Google Scholar]
- 36. Wang D, Yu X, Brecher P. Nitric oxide and N‐acetylcysteine inhibit the activation of mitogen‐activated protein kinases by angiotensin II in rat cardiac fibroblasts. J Biol Chem 1998; 273: 33027–34. [DOI] [PubMed] [Google Scholar]
- 37. Flora SJ, Tripathi N. Hepatic and renal metallothionein induction following single oral administration of gallium arsenide in rats. Biochem Mol Biol Int 1998; 45: 1121–7. [DOI] [PubMed] [Google Scholar]
- 38. Ngu TT, Stillman MJ. Arsenic binding to human metallothionein. J Am Chem Soc 2006; 128: 12473–83. [DOI] [PubMed] [Google Scholar]
- 39. Liu J, Liu Y, Goyer RA, Achanzar W, Waalkes MP. Metallothionein‐I/II null mice are more sensitive than wild‐type mice to the hepatotoxic and nephrotoxic effects of chronic oral or injected inorganic arsenicals. Toxicol Sci 2000; 55: 460–7. [DOI] [PubMed] [Google Scholar]