

Abstract
The global transcriptional regulator PlcR controls gene expression in Bacillus cereus and Bacillus thuringiensis. Activity of PlcR is regulated by PapR, the product of an open reading frame located immediately downstream of plcR. To be active in B. cereus, PapR must be secreted and then processed to the mature peptide by an unknown protease. This peptide is transported by an oligopeptide permease into the cell where it activates PlcR. In this study, we show that the neutral protease B (NprB) secreted by B. cereus 569 is required for extracellular PapR maturation. Purified recombinant NprB processed the synthetic PapR propeptide to produce a set of peptides derived from the C-terminal domain of PapR. Supplementation of growth media with synthetic PapR-derived C-terminal 5-, 7-, 8-, and 27-amino acid peptides caused activation of intracellular PlcR in a PapR-deficient strain of B. cereus 569 while only the 5- and 7-amino acid peptides activated PlcR in a nprB mutant. The maximum activity was found for the 7-mer peptide. However, even the 7-mer peptide could not activate PlcR with a C-terminal truncation of as few as six amino acids. This indicates that interactions of the C-terminal regions of both PlcR and PapR are important in transcriptional activation of the B. cereus 569 PlcR regulon.
Keywords: Bacillus cereus, Bacillus anthracis, PlcR/PapR regulation, quorum sensing, protease, peptide
Introduction
The Gram-positive spore-forming bacterium Bacillus cereus is considered a predecessor of the B. cereus group that includes Bacillus anthracis, Bacillus mycoides, Bacillus pseudomycoides, Bacillus thuringiensis and Bacillus weihenstephanensis (Helgason et al., 2000; Radnedge et al., 2003). The bacterium is capable of synthesizing a range of membrane-destroying and tissue-degradative enzymes including phospholipases, hemolysins, cereolysin and a putative collagenase (Drobniewski, 1993).
PlcR was first identified as a pleiotropic regulator that transiently induces transcription of various stationary phase B. thuringiensis and B. cereus genes including the phospholipases, enterotoxins and proteases (Lereclus et al., 1996; Okstad et al., 1999). Analysis of the promoter regions of the PlcR-regulated genes revealed the presence of a highly conserved palindromic sequence, TATGNAN4TNCATA (N being any of the four nucleotides) which is presumably the specific recognition target for PlcR binding (Agaisse et al., 1999; Pomerantsev et al., 2004). The PlcR regulation mechanism depends on a 48-amino acid peptide, PapR (Slamti & Lereclus, 2002), encoded by the papR gene that is located immediately downstream of plcR. Disruption of papR diminishes expression of the PlcR regulon, resulting in a large decrease in hemolytic activity. A 21-amino acid signal peptide is predicted to direct secretion of PapR (Okstad et al., 1999). Following extracellular processing, a shortened peptide, presumably a pentapeptide, is imported into the cell via the oligopeptide permease complex Opp (Gominet et al., 2001; Declerck et al., 2007). Once inside the cell, this PapR-derived peptide binds to PlcR and makes it competent for binding to its DNA target (Slamti & Lereclus, 2002).
This system has striking similarities to other peptide-responsive regulatory circuits in Gram-positive bacteria, including the Phr family of extracellular signaling peptides of Bacillus subtilis (Perego, 1997). The peptides of about 40 amino acids in the Phr family contain amino-terminal signal peptides that cause their secretion by the SecA-dependent system. It has been hypothesized that the production of the final carboxy-terminal pentapeptide is the result of two processing events, the first being cleavage to an approximately 20-amino acid propeptide by the type I signal peptidases during export. The final cleavage is done by unidentified extracellular proteases to produce the pentapeptide that is then imported by the oligopeptide permease system (Pottathil & Lazazzera, 2003). Later research demonstrated that the pentapeptide can be produced even if the site thought to be recognized by the signal peptidase is removed, suggesting that cleavage by proteases other than signal peptidases is sufficient to produce the pentapeptide (Stephenson et al., 2003). Lanigan-Gerdes et al. identified three proteases involved in production of the mature Phr signaling peptides (Lanigan-Gerdes et al., 2007). Subtilisin, Epr, and Vpr are members of the subtilisin family of proteases that are widespread in bacteria, suggesting that many bacterial species may be capable of producing Phr signaling peptides (Lanigan-Gerdes et al., 2007).
We recently described an alternative, artificial phenomenon in which PlcR activation can occur through translational fusion of the PapR peptide to PlcR. This tethering of the PapR peptide to PlcR eliminates the need for PapR secretion, extracellular processing, and retrieval into the bacterium (Pomerantsev et al., 2004). Sequence analysis of different bacillus strains has shown that the most variable region of PlcR is the C-terminal portion. We found that 18 sequences in this variable domain fall into two groups. Each of these two sets of PlcR sequences has a matching set of PapR C-terminal peptide sequences that fit a consensus (Pomerantsev et al., 2004). Comparing 29 B. cereus group strains, Slamti and Lereclus (Slamti & Lereclus, 2005) later identified four classes of PlcR-PapR pairs, defining four distinct pherotypes in the B. cereus group. Phylogenetic analysis of the pherotypes along with structural analysis of PlcR-PapR complexes show that PlcR is derived from a common ancestor shared by other Gram-positive peptide sensing systems and thus can be assigned to a group of quorum-sensing proteins termed the RNPP family (for Rap/NprR/PlcR/PrgX) (Declerck et al., 2007).
In this study, we examined the role of neutral protease B (NprB) in PapR maturation. The nprB gene is located adjacent to plcR but has the opposite orientation. The presence of a PlcR recognition palindrome between the genes suggests that both genes are activated by the PlcR protein (in cooperation with PapR) (Okstad et al., 1999). We first disrupted nprB in B. cereus and showed that this eliminated PlcR-dependent gene activation. Regulation by PlcR was restored by complementation with any of three plasmids, two encoding native NprB, and one encoding a translationally fused PlcR-PapR product that bypasses the requirement for NprB action. Also, we found that purified recombinant NprB processed the PapR propeptide to produce a set of C-terminal peptides 11, 8, 7, and 5 amino acids in length. The heptapeptide was found to have the maximum activity, leading to production of PlcR-dependent enzymatic activities demonstrated by the hydrolysis of phosphatidylcholine and red blood cells (RBC).
Materials and Methods
Growth conditions
E. coli strains were grown in Luria-Bertani (LB) broth and used as hosts for cloning. LB agar was used for selection of transformants. B. cereus and B. anthracis strains were grown in brain heart infusion medium (BHI) or in LB medium. Solid media were supplemented with 5% fresh sheep blood or 0.02% L-α-phosphatidylcholine (lecithin, Sigma-Aldrich, St. Louis, MO) for the hemolytic and PC-PLC activity determinations, respectively. Casein agar (Aronson et al., 1971) was used for the differentiation of NprB-deficient and NprB-producing strains of B. anthracis and B. cereus. Antibiotics were purchased from Sigma-Aldrich and added to media when appropriate to give the final concentrations indicated: ampicillin (Amp, 100 μg mL-1 for E. coli), erythromycin (Em, 5 μg mL-1 for B. anthracis/B. cereus and 400 μg mL-1 for E. coli), spectinomycin (Sp, 100 μg mL-1 both for E. coli and B. anthracis/B. cereus), tetracycline (Tc, 5 μg mL-1 for B. anthracis/B. cereus) and kanamycin (Km, 50 μg mL-1 for B. anthracis/B. cereus). SOC medium (Quality Biologicals, Inc., Gaithersburg, MD) was used to grow cells during transformation.
DNA isolation and manipulation
Preparation of plasmid DNA from E. coli, transformation of E. coli, and recombinant DNA techniques were carried out by standard procedures (Sambrook & Russell, 2001). E. coli XL2-blue and SCS110 competent cells were purchased from Stratagene and E. coli TOP10 competent cells from Invitrogen. Recombinant plasmid construction was carried out in E. coli XL2-Blue or TOP10. Plasmid DNA from B. anthracis/B. cereus was isolated according to the QIAGEN Plasmid Protocol: Purification of Plasmid DNA from B. subtilis (QIAGEN Inc., Valencia, CA). Chromosomal DNA from B. anthracis/B. cereus was isolated with the Wizard Genomic Purification Kit (Promega, Madison, WI) in accordance to the protocol for isolation of genomic DNA from Gram-positive bacteria (Promega). B. anthracis/B. cereus was electroporated with unmethylated plasmid DNA isolated from E. coli SCS110. Electroporation-competent cells of B. cereus were prepared using the same method as previously described for B. anthracis (Park & Leppla, 2000).
Restriction enzymes, T4 ligase, T4 DNA polymerase and alkaline phosphatase were purchased from MBI Fermentas or New England Biolabs. Taq polymerase kits were purchased from TaKaRa Shuzo or Invitrogen/Life Technologies. Ready-To-Go PCR Beads (Amersham Biosciences) were used for DNA rearrangement analysis. For routine PCR analysis a single colony was suspended in 200 μl of TE buffer, pH 8.0, heated to 95°C for 60 sec and then cooled to room temperature. Cellular debris was removed by centrifugation at 15,000g for 10 min. One microliter of the lysate contained sufficient template to support a PCR reaction with the PCR beads. The GeneRuler DNA ladder mix (MBI Fermentas) or the 1 kb plus ladder (Invitrogen) was used for determination of DNA fragment length. All constructs were verified by DNA sequencing and/or restriction enzyme digestion. All plasmids used in this study and their relevant characteristics are listed in Table 1. Oligonucleotide primer sequences are listed in Table 2.
TABLE 1.
Plasmids and strains used in this study
| Plasmid or strain | Relevant characteristic(s) | Source or reference |
|---|---|---|
| Plasmid | ||
| pAE5 | Encodes B. cereus 569 plcR under control of B. anthracis protective antigen gene (pagA) promoter; Apr in E. coli; Kmr in B. anthracis | (Pomerantsev et al., 2003) |
| pAE51 | pAE5 with stop codon in plcR truncating six C-terminal amino acids | This work |
| pUTE29-plcR-papR | B. cereus 569 plcR-papR DNA fragment inserted into pUTE29; Apr in E. coli; Tcr in B. anthracis | (Pomerantsev et al., 2004) |
| pFP12 | pUTE29-plcR fusion to papR | (Pomerantsev et al., 2004) |
| pUTE29-papR | B. cereus 569 papR DNA fragment inserted into pUTE29 | (Pomerantsev et al., 2004) |
| p102 | pUTE29-nprB-plcR-papR | This work |
| p182 | pUTE29-nprB-plcR fusion to papR | This work |
| pUTE29-nprB | P182 without BstZ17I-PstI fragment encoding fused PlcR-PapR | This work |
| pJRS312 | pUC18 carrying an Ω element with spectinomycin resistance marker (Ω-sp) | J.R. Scott |
| p102Sp | p102 with Ω-sp cassette flanked by two directly repeated loxP and inserted into BglII-site of nprB | This work |
| p182Sp | p182 with Ω-sp cassette flanked by two directly repeated loxP and inserted into BglII-site of nprB | This work |
| pHY304 | Contains strongly temperature-sensitive replicon for both E. coli and gram-positive bacteria; Emr in both E. coli and B. anthracis | (Pritzlaff et al., 2001) |
| pHU304 | Large PvuII-fragment of pUC18 combined with large Ecl136II-fragment of pHY304 | This work |
| pHU102 | PvuII fragment of p102 containing nprB- plcR-papR gene cluster inserted into BstZ17I-site of pHU304 | This work |
| pHU102Sp | PvuII fragment of p102Sp containing nprB∷(loxP-Ω-sp-loxP) - plcR-papR gene cluster inserted into BstZ17I-site of pHU304 | This work |
| pCrePA | pHY304 with Cre recombinase gene under control of pagA promoter | (Pomerantsev et al., 2006) |
| pDG148-Stu | Gram-Positive-E. coli shuttle vector featuring ligation-independent cloning and inducible expression; Apr in E. coli; Kmr in B. anthracis | (Joseph et al., 2001) |
| pB148 | pDG148-Stu carrying B. cereus 569 nprB gene under control of Pspac promoter | This work |
| pB148Lac- | pB148 without lacI repressor gene | This work |
| Strain | ||
| B. cereus | ||
| 569 | Wild strain, produces extracellular PC-PLC | (Reddy et al., 1987) |
| 541 | 569; intact plcR lacks a promoter; Emr; does not produce extracellular PC-PLC | (Pomerantsev et al., 2003) |
| 537 | 569; nprB knockout, Spr; nprB contains Ω-sp cassette flanked by two directly repeated loxP; demonstrates very low PC-PLC activity | This work |
| 536 | 569; nprB knockout containing one loxP site; demonstrates very low PC-PLC activity | This work |
| 535 | 569; papR replaced by loxP site; demonstrates very low PC-PLC activity | This work |
| B. anthracis | ||
| SdT | Sterne 34F2 cured of pXO1; therefore pXO1- pXO2- | (Ivins et al., 1986) |
| SdT2 | SdT electroporated with pAE5; Kmr; produces intracellular PlcR; does not produce a halo when grown on LB agar with lecithin | (Pomerantsev et al., 2003) |
| SdT251 | SdT electroporated with pAE51; Kmr; produces intracellular PlcR with truncated six C-terminal amino acids; does not produce a halo when grown on LB agar with lecithin | This work |
| SdT12 | SdT electroporated with pFP12; Tcr; produces intracellular PlcR-PapR fused protein; demonstrates does not produce a halo when grown on LB agar with lecithin | (Pomerantsev et al., 2004) |
| Ames 33 | pXO1- pXO2- Ames derivative strain | (Pomerantsev et al., 2003) |
| BH450 | Ames 33 ba_0599 protease knockout, spo0A knockout | (Pomerantsev et al., 2006) |
TABLE 2.
Primers and peptides used in this study
| Primer | Sequencea (5’-3’) | Relevant property | Restriction site |
|---|---|---|---|
| Npr-f | ACTGGTATACCGCACCCGATTCAATTCTGC | Primer pair to amplify nprB-plcR region | BstZ17I |
| Npr-r | ACTGGGATCCTTGCGAAGGCACTTTATCTTTCGCC | BamHI | |
| StuBf | AAGGAGGAAGCAGGTATGAATTATAGAAAACTTGCTAGTGCTTTCG | Primer pair to amplify nprB structural gene | |
| StuBr | GACACGCACGAGGTTTATTGTACCGCTATTACTTTATTTATTTTTTG | ||
| LoxSB | CCCCCCAGATCT CCGATCATATTCAATAACCCT | Primer pair to amplify loxP-Ω-sp-loxP | BglII |
| LoxEB | CCCCCCAGATCTCCAGTACTAGTGAACCTCTTC | BglII | |
| Sp5 | GAGGGTTTGCAACTGCGGG | Ω-sp inner primer for analysis of modifications in nprB from 5’ | |
| Bc-ext | ATTCGTAATGTAATGAAGGG | nprB external primer for analysis of modifications in nprB from 3’ | |
| R3 | CCCGGGATCCTTATTTCTTCATTTTTTTCAAAA | Used to amplify mutated B. cereus 569 plcR gene from 3’ | BamHI |
| BseqF | GAACAACTTTACCACTAACAGT | Primer pair to verify loxP inside nprB | |
| BseqR | TCAAAGTTGAAATTGCTCAG | ||
| PapRF | AAAGCAATTAGAGGCATTAC | Primer pair to verify papR replacement by loxP | |
| PapRR | GCAAACCTGTTTCTAGTAAAGA | ||
| EryF | TTAACGAGTGAAAAAGTACTCAACC | Primer pair to verify ery gene of pHY304 plasmid | |
| EryR | TGACCCATTTTGAAACAAAG | ||
| Peptide | Sequence (N-C) | Relevant property | |
| 5aa | MPFEF | All peptides are derivative of the PapR | |
| 7aa | SDMPFEF | C-terminal region of B. cereus 569 PlcR group II | |
| 8aa | ASDMPFEF | ||
| 27aa | DTALEKSKVISHDNQEVQLASDMPFEF | ||
Restriction enzyme recognition sites are underlined.
DNA cloning and sequencing
The nprB region from B. cereus 569 chromosomal DNA was obtained as a 2.21-kb DNA fragment by PCR using primers Npr-f and Npr-r (Table 2). The PCR fragment restricted with BstZ17I and BamHI was isolated from SeaKem agarose gel (Rockland, ME) and cloned into the corresponding sites of the pUTE29-plcR-papR or pFP12 plasmid (Pomerantsev et al., 2004) using E. coli XL2-Blue. The resulting p102 (pUTE29-nprB-plcR-papR) or p182 (pFP12-nprB-plcR-papR) plasmids were electroporated into B. cereus with selection for tetracycline resistance. The nprB gene was also amplified with primers StuBf and StuBr and introduced into the pDG148-Stu vector under control of the Pspac promoter (Joseph et al., 2001). The resulting pB148 plasmid was electroporated into B. anthracis/B. cereus with selection for kanamycin resistance. The LacI repressor gene in pB148 was eliminated by cleavage with SplI and BamHI, filling of overhangs with T4 DNA polymerase and ligation of the resulting blunt ends with T4 ligase.
The PlcR-expressing plasmid pAE5 containing the B. cereus 569 plcR gene was generated in our previous study by directional cloning of a PCR fragment amplified with primers designated R1 and R2 (Pomerantsev et al., 2003). Here we repeated this cloning using an alternative reverse primer, R3 (Table 2), which deleted a base so as to introduce a terminal codon TGA in place of a methionine codon. The resulting plasmid pAE51 that produces a truncated PlcR protein was introduced into B. anthracis SdT strain to produce B. anthracis SdT251 strain.
Nucleotide sequencing of the cloned fragments and DNA-fragments used for the analysis of mutations was performed by the dideoxy chain-termination technique with a Taq dye primer cycle sequencing kit (Food and Drug Administration Core Facility, National Institutes of Health, Bethesda, MD). The nucleotide sequences encoding the B. cereus 569 nprB gene was previously submitted to Genbank under accession number AY195601 (Pomerantsev et al., 2003). The NCBI BLAST and FASTA programs (http://www.ncbi.nlm.nih.gov/) were used for homology searches in the GenBank and SWISS Protein databases.
Strain construction
Strains, plasmids, and their relevant characteristics are listed in Table 1. All B. cereus strains used here were derived from strain 569. The B. cereus nprB mutant strains 536 and 537 were constructed using Cre/LoxP technology. Briefly, the Ω-spectinomycin (Ω-sp) cassette of the pΩL plasmid (Pomerantsev et al., 2006) was amplified with LoxSB/LoxEB primers to produce a BglII-loxP-Ω-sp-loxP-BglII DNA fragment. The fragment was inserted into the unique BglII-restriction site located in the nprB gene in the p102 plasmid. The PvuII-fragment of the resulting p102Sp plasmid containing the nprB∷sp-plcR-papR – gene cluster was cloned into the BstZ17I-site of the pHU304 plasmid. pHU304 is a recombinant plasmid made by combining the large PvuII-fragment from pUC18 and the Ecl136II-fragment from pHY304 (Pritzlaff et al., 2001). The resulting pHU102Sp plasmid was electroporated into B. cereus 569 with selection for erythromycin resistance. Transformants were selected at 30°C on BHI agar containing erythromycin. Erythromycin-resistant colonies were transferred to agar containing spectinomycin, incubated at 37°C to limit replication from the pHY304 origin, and again transferred to fresh agar containing spectinomycin. After the third passage at 37°C, colonies were selected and screened for spectinomycin-resistance and erythromycin-sensitivity. Isolates in which a double-crossover recombination event had occurred were confirmed by PCR and phenotype on blood or lecithin agar. Primer Sp5, located inside the Ω-sp cassette, was used as an internal primer; Bc-ext, located downstream of the Npr-r primer, was used as an external primer. The resulting nprB mutant was designated B. cereus 537.
For elimination of spectinomycin resistance, plasmid pCrePA (Pomerantsev et al., 2006) was introduced by electroporation followed by selection for erythromycin resistance at 30°C. Erythromycin-resistant colonies were transferred to antibiotic-free agar and incubated at 37°C to eliminate pCrePA. Colonies were then patched to three replicate agar plates containing erythromycin, spectinomycin, or no antibiotic. The presence of a single loxP site within the nprB gene of the resulting fully antibiotic-sensitive B. cereus mutant strain 536 was confirmed by PCR and/or sequence analysis. Primer pair BseqF/BseqR was used for the final verification.
The B. cereus papR mutant strain 535 was constructed by replacing the papR coding sequence with a loxP site using a Cre/LoxP technology similar to that described above. Primer pair PapRF/PapRR was used for the final verification.
For the trans-complementation experiments, the pB148 and pB148Lac- plasmids expressing nprB were used for the transformation of B. cereus 536 (nprB-). Kanamycin-resistant clones that reestablished hemolytic and PC-PLC activities were selected on blood or lecithin-containing agar. The in situ nprB gene replacement was accomplished using plasmid pHU102, which was made by combining the PvuII-fragment of the p102 plasmid containing the nprB-plcR-papR gene cluster and the BstZ17I-fragment of pHU304. The pHU102 plasmid was electroporated into B. cereus 536 (nprB-) with selection for erythromycin resistance. Transformants were selected at 30°C on lecithin agar containing erythromycin. Colonies that were erythromycin-resistant and formed halos were transferred to lecithin agar without antibiotic and incubated at 37°C to limit replication from the pHY304 origin and encourage the insertion of the pHU102 plasmid into the chromosomal DNA of B. cereus 536 as a single-crossover event. Halo forming colonies were again transferred to fresh lecithin agar without antibiotic and incubated at 37°C. The entire process was repeated until erythromycin sensitive halo-forming isolates in which a double-crossover recombination event had occurred were obtained. The restoration of the parental nprB gene sequence in the fully antibiotic-sensitive strain was confirmed by PCR and sequence analysis.
In further trans-complementation experiments, the pUTE29-papR (Pomerantsev et al., 2004) plasmid was used for the transformation of B. cereus 535(papR-). Tetracycline-resistant clones that reestablished PC-PLC activity were selected on lecithin-containing agar.
Identification of extracellular B. cereus 569 NprB and its orthologue in B. anthracis
The extracellular proteomes of PlcR-positive B. cereus 569 and B. anthracis SdT12, as well as for the PlcR-negative variants, were determined during the exponential phase (5 h), stationary phase (10 h), and pre-spore growth phase (24 h). Bacteria were grown in LB broth at 37°C. Aliquots of growing cultures were centrifuged for 15 min at 8500 × g. The supernatants were filtered through 0.22-μm (pore size) Millex-GS membrane (Millipore Corp. Bedford, MA) and the proteins were concentrated using Centriprep YM-10 units (Amicon, Inc., Beverly, MA). Samples containing concentrated proteins were focused on pH 3 to 10 IPG strips and separated on 4-12% Bis-Tris NuPAGE gels. The gels were stained with Coomassie Blue. All visible spots were extracted and proteins were identified by mass spectrometry using MALDI-TOF MS (FDA Core Facility). The National Center for Biotechnology Information (NCBI) BLAST and FASTA programs (http://www.ncbi.nlm.nih.gov/) were used for MS peptide homology searches in the GenBank and SWISS Protein databases. Genomic DNA sequences extending 1000 bp upstream of the identified proteins start codons were examined for possible PlcR-binding sites.
NprB purification
Plasmid pB148Lac- was transformed into the sporulation-negative, protease-deficient B. anthracis BH450 strain (formerly designated MSLL33) (Pomerantsev et al., 2006) with selection for kanamycin resistance. The strain was grown in FA medium (Singh et al., 1989) with kanamycin (50 μg mL-1) at 37°C until the beginning of the stationary phase. The supernatant was filter sterilized using a Pellicon cassette system (Millipore Corp., Bedford, MA) with Durapore 0.22-mm membranes. Ammonium sulfate was added to obtain 40% saturation and the sample was loaded onto a Fast Flow Phenyl–Sepharose column (Amersham Pharmacia, Piscataway, NJ) pre-equilibrated with buffer containing 1.5 M ammonium sulfate, 10 mM Tris and 0.5 mM EDTA (pH 7.5). The column was washed with the same buffer and NprB was eluted with an ammonium sulfate gradient from 1.5 to 0 M. Fractions were analyzed on casein agar and NprB-containing fractions were collected and dialyzed with Slide-A-Lyzer Dialysis Cassettes (Pierce, Rockford, IL) overnight against 10 mM Tris (pH 7.5). Finally, the NprB was concentrated using Centriprep YM-10 units (Amicon, Inc., Beverly, MA) and stored at −80°C until used. Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) was used to analyze the purity of NprB. The protein was heated in 1x sample buffer (6x sample buffer contains 0.35 M Tris-HCl, pH 6.8, 10% sodium dodecyl sulfate [SDS], 36% glycerol,0.6 M DTT, and 0.01% bromphenol blue) at 95°C for 5 min and separated on SDS-polyacrylamide (10-20%) gel using the PhastSystem (Amersham Pharmacia, Piscataway, NJ). The N-terminal sequence of NprB was determined by the Edman degradation method, using a gas-phase sequencer at the Research Technologies Branch, NIAID/NIH Twinbrook I Facility, Rockville, MD.
Determination of NprB/PapR cleavage specificity
The PapR propeptide containing 27 C-terminal amino acids was synthesized at the Peptide Synthesis and Analysis Unit of Research Technologies Branch, NIAID/NIH Twinbrook I Facility, Rockville, MD. PapR was incubated with the purified NprB protease (100:1 by mass) in ammonium carbonate buffer, pH 7.5, for 1, 12, or 24 h at 37°C. The hydrolysis products were separated with a C18 reverse phase column and a 5-60% B/40 min gradient (solvent A: 0.1% trifluoroacetic acid/H2O; solvent B: 0.1% trifluoroacetic acid/20% H2O/80% acetonitrile) using a Shimadzu VP series HPLC system (Peptide Synthesis and Analysis Unit). Peptides were identified by mass spectrometry (MS) using a Voyager RP BioSpectrometry Workstation (Peptide Synthesis and Analysis Unit).
PlcR/PapR supplementation test
Bacillus strains were analyzed for phosphatidylcholine hydrolysis activity in two ways. Plates containing LB agar supplemented with 0.04% L-α-phosphatidylcholine (lecithin, Sigma-Aldrich) and different concentrations of the corresponding peptide (titrated from 200 to 0.2 μM) were prepared for visual analysis of the halo. C-terminal peptides of 5, 7, 8, and 27 amino acids of B. cereus 569 PapR were synthesized at the Peptide Synthesis and Analysis Unit of Research Technologies Branch, NIAID/NIH Twinbrook I Facility, Rockville, MD. Kanamycin and tetracycline were added to media when appropriate to give final concentrations of 50 and 5 μg mL-1, respectively. Suspensions of the B. anthracis strains derived from SdT or B. cereus strains were prepared with an A600 close to 0.25, and 3 μl of each suspension was inoculated on plates which were then incubated overnight at 37°C. For quantitative determination of the enzymatic activities, synthetic peptides (5 μM) were added to a culture of the B. cereus strains 535 and 536 grown in LB broth at 37°C until stationary phase (OD600 1.5 ± 0.2). Aliquots of growing cultures were centrifuged for 15 min at 8500 × g 1 h after peptide addition. The supernatants were filtered through 0.22-μm (pore size) Millex-GS membrane and PC-PLC assays were performed. In the same way, B. anthracis SdT2 cells were grown in LB broth supplemented with kanamycin (50 μg mL-1) at 37°C until stationary phase (OD600 1.3 ± 0.2). The culture was then divided and each synthetic peptide was added to individual fractions to final concentrations of 1, 10 and 100 μM. PC-PLC assays were performed 1 h after peptide addition.
Enzymatic activities
Lecithin hydrolyzing activities of the supernatants were determined with the Amplex Red Phosphatidylcholine-Specific Phospholipase C Assay Kit from Molecular Probes, Inc., Eugene, OR, as previously described (Pomerantsev et al., 2003). The activity of the unknowns was compared to that of the standard enzyme supplied with the kit to calculate mU/mg total protein. Casein protease activities of the supernatants were quantified by the Enz Chek Protease Assay Kit E-6638 (Molecular Probes, Eugene, Oregon). For measurement of the protease activity, each reaction mixture contained 5 μg mL-1 of BODIPY FL casein and 5 μl supernatant in 10 mM Tris-HCl (pH 7.8). Reaction mixtures were incubated in the dark at 37°C for 60 min. Fluorescence was measured with a Wallace 1420 VICTOR 96-well plate reader (Perkin Elmer, Boston, Mass.) with excitation at 485 nm and emission at 530 nm.
Results
Comparison of B. cereus group plcR gene regions
The plcR gene is present in, and probably restricted to, all members of the B. cereus group. However, while the B. cereus and B. thuringiensis PlcR proteins appear to be functionally equivalent, the B. anthracis PlcR protein is truncated and does not operate as a transcriptional activator (Agaisse et al., 1999). The truncation of PlcR has been considered the principal, if not only, reason for the nonhemolytic properties of B. anthracis in comparison with the strongly hemolytic properties of B. cereus and B. thuringiensis. In previous work we showed that complementation of the truncated B. anthracis SdT PlcR in B. anthracis with its intact B. cereus 569 homologue was not sufficient to activate the lecithinase gene that contains the PlcR-binding sites (Pomerantsev et al., 2003). In seeking to understand these results, we noted that the PlcR and PapR gene regions of B. cereus 569 (accession number AY195601), 10987 (accession number NC_003909), 14579 (accession number NC_004722) and E33L (accession number NC_006274) as well as B. anthracis Sterne (accession number NC_005945) and Ames (accession number NC_003997) strains have the same gene arrangement with two exceptions: first, B. cereus 10987, E33L, and B. anthracis strains lack the gene corresponding to the B. cereus 14579 and 569 neutral protease B (NprB) (Okstad et al., 1999), and second, B. anthracis strains additionally have a truncated PlcR (Fig. 1A). Analysis of the nucleotide sequence within the B. cereus 569 nprB-plcR intergenic region (Fig. 1B) revealed that both genes are predicted to use the same palindromic PlcR-box for their transcriptional activation. Thus, the -10 and -35 regions for each gene are located on opposite DNA strands in close proximity to the PlcR palindrome. The same arrangement exists in B. thuringiensis, where it was shown that both the plcR (Lereclus et al., 1996) and nprB (Agaisse et al., 1999) genes are transcribed from the same DNA strands as B. cereus 569, in both cases using adenine as a transcriptional start site. Also, two separate Spo0A-boxes were found bracketing the PlcR-box in the regions located between the -10 and -35 sites of the B. cereus nprB and plcR genes (Fig 1B), almost at the same location as in the B. thuringiensis nprB-plcR intergenic region (Lereclus et al., 2000). The presence of Spo0A-boxes in the same functional regions for both plcR and nprB genes indicates that binding of Spo0A~P to the corresponding sites may negatively regulate expression of both genes. Interestingly, the papR gene carries its own PlcR-box and does not have a Spo0A binding site (Fig. 1C). These sequence features suggest that expression of the plcR and nprB genes are coordinately controlled, implying that nprB gene expression may be involved in the mechanism of PlcR regulation in at least the B. cereus 569 and 14579 strains. To test this hypothesis we inactivated the nprB gene and analyzed the resulting phenotypes.
Figure 1.
PlcR, PapR, and NprB genes. (A) B. anthracis Sterne, B. cereus 10987 and 14579 chromosomal DNA regions in the vicinity of plcR and papR genes. B. subtilis gene orthologues were selected by BLAST searches of the NCBI database: yvfR – ABC transporter (ATP-binding protein); yvfS – ABC transporter integral membrane protein; yvfT – two-component sensor histidine kinase; yvfU – two-component response regulator; nprB – neutral thermostable extracellular zinc metalloprotease; ykoW – protein, unknown function, likely to participate in prokaryotic signaling process. (B) Nucleotide sequence of B. cereus 569 nprB-plcR intergenic region. (C) Nucleotide sequence of B. cereus 569 papR promoter region. The predicted translation start codons for NprB, PlcR and PapR proteins are shown in bold type and indicated by arrows. Transcription start sites for all three genes (Okstad et al., 1999) are shown in bold type and underlined. The consensus binding sites for Spo0A and PlcR (Lereclus et al., 2000) are shaded. The putative – 10 and – 35 regions (Okstad et al., 1999; Agaisse et al., 1999) are indicated in italic and bold. All indicated sequences are the same for both B. cereus 569 and 14579 strains.
Inactivation of B. cereus 569 nprB and papR genes
The nprB gene of B. cereus 569 was disrupted as described in the Materials and Methods section according to the scheme presented in Fig. 2A and verified by PCR in Fig. 2B. The presence of a loxP sequence created a frameshift that terminated translation of NprB in the resulting mutant B. cereus strain 536. The B. cereus nprB mutant strain 536 was examined for phenotypic changes (Fig. 2 B,C,D). Hemolytic activity was completely lost (Fig. 2B, lower panel, lane 2). The Enz Chek Protease Assay Kit and Amplex Red Phosphatidilcholine-Specific Phospholipase C Assay Kit provided convenient ways to assess PlcR/PapR dependent casein protease and PC-PLC activities, respectively. The nprB mutant showed greatly decreased activities both for casein and phosphatidylcholine digestions as compared to its B. cereus 569 parent (Fig. 2C and D, respectively).
Figure 2.
Inactivation of B. cereus 569 nprB gene. (A) Diagram of the genetic organization of the B. cereus 569 mutants, B. cereus 537 and B. cereus 536. Construction of the mutants is described in Materials and Methods. Arrows indicate the position and orientation of the loxP sites. Half-head arrows mark the position and orientation of the primers used for PCR analysis. (B) (Upper panel) The PCR fragments amplified with BseqF/BseqR primers from the chromosomal DNA of B. cereus 569 (lane 1), B. cereus 536 (lane 2), B. cereus 536 with plasmid pB148 (lane 3), B. cereus 536 with plasmid pB148Lac- (lane 4), B. cereus 536 with plasmid pFP12 (lane 5), B. cereus 536 with plasmid pHU102 integrated into chromosomal DNA as a single crossover event (lane 6), B. cereus 569 NprB-revertant obtained as a result of a double crossover event between pHU102 and B. cereus 536 chromosomal DNA (lane 7). Locations of the primers correspond to those shown in panel A. The upper fragment (398 bp) presents nprB-loxP allele; the lower fragment (302 bp) presents nprB allele. (Lower panel) Ability of B. cereus 569 to hydrolyze fresh blood is appreciably depressed in B. cereus 536. Transformation with pB148, pB148Lac-, pFP12 or pHU102 restored ability of B. cereus 536 to hydrolyze fresh blood. The locations of the samples correspond to those of the above PCR. (C and D) Casein proteolytic (C) and PC-PLC (D) activities of B. cereus 569, B. cereus 536, and B. cereus 536 (pB148Lac-) extracellular proteins. The strains were grown at 37°C in LB broth for 10 h with supernatant samples taken every hour. Equal amounts of extracellular protein (2-10 μg for PC- PLC and 10-50 μg for casein proteases analysis) were assayed either by the Red Amplex (PC- PLC) or green-fluorescent BODIPY FL (protease) reagents. Maximum root-mean-square deviations did not exceed 10% for the protease (C) and PC-PLC (D) determinations.
The papR gene of B. cereus 569 was replaced by a loxP site as described in the Materials and Methods according to the scheme presented in Fig. 3A. The resulting B. cereus papR mutant strain 535 showed greatly decreased activity for phosphatidylcholine digestion as compared to its B. cereus 569 parent (Fig. 3B).
Figure 3.
Inactivation of B. cereus 569 papR gene. (A) Diagram of the genetic organization of the B. cereus 569 papR mutant, B. cereus 535. Arrows indicate the position and orientation of plcR, papR and loxP. Half-head arrows mark the position and orientation of the primers used for PCR analysis. (B) (Upper panel) The PCR fragments amplified with PapRF/PapRR primers from the chromosomal DNA of B. cereus 569, B. cereus 535, and B. cereus 535 with plasmid pUTE29-papR. Locations of the primers correspond to those shown in panel A. The upper fragment (624 bp) presents the plcR-papR allele; the lower fragment (550 bp) presents the plcR-loxP allele. (Lower panel) Ability of B. cereus 569 to hydrolyze lecithin is appreciably depressed in B. cereus 535. Transformation with pUTE29-papR restored ability of B. cereus 535 to hydrolyze lecithin. The locations of the samples correspond to those of the above PCR. The strains were grown at 37°C in LB broth for 8 h. Equal amounts of extracellular protein were assayed by Red Amplex (PC-PLC). (C) Complementation of both mutant B. cereus 535 and 536 with plasmid producing NprB (pB148Lac-) restores PC-PLC activity only in B. cereus 536 while supplementation of growth medium with 10 μM of 27-aa C-terminal PapR peptide restores phosphatidylcholine hydrolyzing activity only in B. cereus 535. Bacteria were grown at 37°C in LB broth for 8 h with or without the PapR peptide. Equal amounts of extracellular protein were assayed by Red Amplex (PC-PLC).
We reported previously that the hemolytic and lecithinase activities of a B. cereus 569 plcR mutant were markedly reduced or eliminated as compared to the wild-type parent strain (Pomerantsev et al., 2003). Here we obtained very similar data for the nprB and papR mutants B. cereus 536 and 535, respectively. To confirm that B. cereus 536 phenotypic changes were due to nprB disruption, we complemented the mutation with a native B. cereus 569 nprB gene carried on plasmids pB148 or pB148Lac- and also restored the gene in the chromosome (see below). The plasmid transformants partially regained the ability to express the hemolytic activity characteristic of the parent B. cereus 569 strain (Fig. 2B, lanes 3,4). Casein protease and PC-PLC activity were also restored in the B. cereus (pB148Lac-) strain (Fig. 2C and D). In an alternative approach to correct the phenotypic consequences of nprB disruption, the nprB mutant strain was transformed with plasmid pFP12, which contains a translational fusion of the B. cereus PlcR and PapR proteins. The resulting transformant also partially regained the hemolytic activity of the parent strain (Fig. 2B, lane 5). These data are consistent with the previous demonstration that the PlcR-PapR fusion protein activates transcription of PlcR-dependent genes in B. anthracis. In this experiment we found that the fused PlcR-PapR protein can also activate transcription of PlcR-dependent genes in the B. cereus nprB mutant, bypassing the need for extracellular maturation of the PapR protein that appears to depend on the action of NprB. Finally, the hemolytic activity of B. cereus 536 was completely restored by transformation with pHU102 so as to reinsert an intact nprB gene into the original chromosomal location (Fig. 2B, lane 7).
To confirm that the B. cereus 535 phenotypic changes were due to papR replacement with loxP, we complemented the mutation with a native B. cereus 569 papR gene carried on plasmid pUTE29-papR. PC-PLC activity was restored in B. cereus 535(pUTE29-papR) (Fig. 3B). In contrast, no digestion of phosphatidylcholine was found for the B. cereus 535(pB148Lac-) strain, in which the plasmid expresses nprB, while B. cereus 536(pB148Lac-) clearly digested phosphatidylcholine in the same conditions (Fig. 3C). On the other hand, supplementation of LB broth with the 27-aa C-terminal PapR peptide induced PC-PLC activity in B. cereus 535 but not in B. cereus 536 (Fig. 3C). This experiment indicates that PapR cannot be processed in the nprB mutant to produce active peptide.
B. cereus and B. anthracis protease identification
The data shown in Fig. 2C demonstrated that the casein proteolytic activity of B. cereus 569 found at early phases of growth results mostly from NprB. This finding indicates that the nprB gene is required in B. cereus 569 for expression of normal casein proteolytic activity level at the early phase because the B. cereus 569 nprB mutant had drastically decreased activity in the same time interval. Later the activities of both strains became similar. We also found that the hemolytic B. anthracis SdT12 strain expressing the fused PlcR/PapR protein demonstrated a similar induction of proteolytic activity at early phases of growth, while the parent B. anthracis SdT did not. Similarly to the B. cereus 569 nprB mutant, the B. anthracis SdT strain revealed casein proteolytic activity later, but at a reduced level compared with the B. anthracis SdT12 strain (Fig. 4). Because the B. anthracis Sterne strain does not have nprB in the vicinity of plcR (Fig. 1A), we then asked what protease is induced by PlcR in B. anthracis SdT12 at the early phase of growth.
Figure 4.
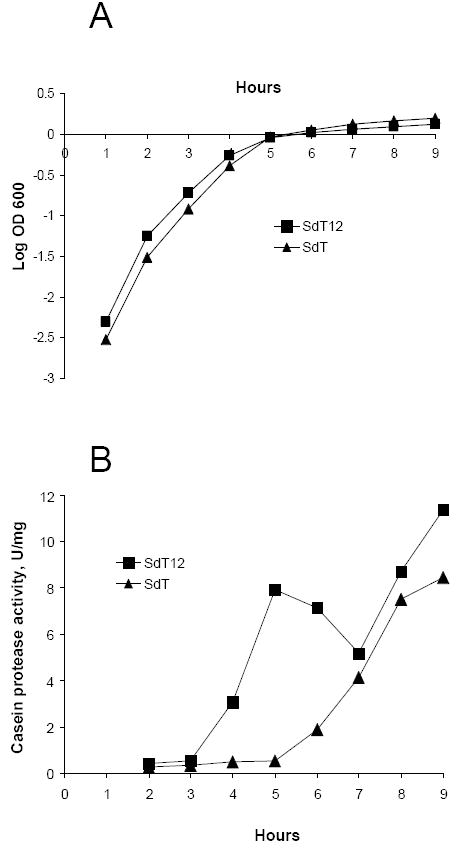
Growth curves (A) and extracellular protein casein proteolytic activity (B) of B. anthracis SdT and B. anthracis SdT12 strains. The growth conditions and activity determinations were performed as was done for B. cereus strains (see description to Fig. 2C).
To identify the proteases produced by the B. cereus and B. anthracis strains being examined here, we used two-dimensional (2D) gel electrophoresis (Fig. 5). In the culture supernatant (secretome) of B. cereus 569 grown for 5 h (corresponding to the peak in Fig. 2C), we identified six proteins, of which only one was a protease - NprB. Expression of four proteins (hemolysin BL, non-hemolytic enterotoxin, NprB, and PC-PLC) was apparently regulated by PlcR because a PlcR-binding site is found upstream of each corresponding gene (data not shown). No PlcR-binding site was found upstream of the genes responsible for the synthesis of either the chitin binding protein or flagellins. Both the chitin binding protein and the flagellin spots were found at the same position on the 2D-gel of B. cereus 536 (Fig. 5, upper right gel). However, spots corresponding to the proteins with PlcR-dependent regulation disappeared, to make way for three proteins with PlcR-independent regulation: N-acetylmuramoyl-L-alanine amidase (BC_5234), hypothetical membrane spanning protein (BC_1726), and cytochrome aa3 quinol oxidasepolypeptide (BC_0698). It is interesting that a protease (BAS2543) of the same M4 neutral protease class as NprB was one of the three proteins found in the 5 h secretome of B. anthracis SdT12 (Fig. 5). Expression of all three proteins containing a PlcR-binding site upstream of the corresponding genes (neutral protease M4, murein endopeptidase and enterotoxin A) was obviously regulated by PlcR because the corresponding secretomes of B. anthracis SdT strain contained only one protein, the S-layer SAP protein (BAS0841), which lacks PlcR regulation. Neither NprB from B. cereus 569 nor neutral protease M4 from B. anthracis SdT12 were found in the secretomes after 10 and 24 h of growth (data not shown).
Figure 5.
Secretome analyses. Two-dimensional electrophoretic separation of proteins secreted by B. cereus 569 and 536 as well as B. anthracis SdT12 and SdT grown in LB broth for 5 h. Mr, molecular mass (in kDa); NL, nonlinear. The numbers mark protein spots which were identified by MALDI-TOF-MS in accordance with the B. cereus 14579 (accession number NC_004722) and B. anthracis Sterne (accession number NC_005945) databases. B. cereus 569: 1 - chitin binding protein (BC_2798); 2, 3 - hemolysin BL lytic and binding components (BC_3104 and BC_3103, respectively); 4, 6, 7 - three isoforms of non-hemolytic enterotoxin corresponding to lytic component L2 (enterotoxin A, BC_1809) and lytic component L1 (enterotoxin B, BC_1810); 5 - neutral protease B (BC_5351); 8 - flagellins A, B, C (BC_1657-1659); 9 -phosphatidylcholine-specific phospholipase C (BC_0670). B. cereus 536: 1- chitin binding protein (BC_2798); 2 - flagellins A, B, C (BC_1657-1659); 3 - cytochrome aa3 quinol oxidasepolypeptide (BC_0698); 4 - hypothetical membrane spanning protein (BC_1726); 5 - N-acetylmuramoyl-L-alanine amidase (BC_5234); B. anthracis SdT12: 1 - murein endopeptidase (BAS1852); 2 - neutral protease M4 (BAS2543); 3,4 - enterotoxin A (BAS1749) ; B. anthracis SdT: 1, 2 - S-layer protein, Sap (BAS0841).
NprB characteristics
NprB of B. cereus 569 (peptidase M04.012 in accordance with the MEROPS peptidase database, http://merops.sanger.ac.uk) is a 583-aa zinc metalloprotease of the M4 family. The M4 family is composed of secreted eubacterial endopeptidases that contain an HEXXH motif (where X is any amino acid), which has been shown in crystallographic studies to form part of the metal-binding site (Gomis-Ruth, 2003). The B. cereus 569 NprB contains a typical 26-aa signal peptide (http://www.cbs.dtu.dk/services/SignalP/) and a Zn2+-binding site with the sequence HEFGH. The most similar orthologue of B. cereus 569 NprB is that of B. cereus ATCC 14579 (identities = 574/583 [98%], positives = 578/583 [99%]) (Ivanova et al., 2003). The B. anthracis orthologue BAS2543 (567 aa) is less similar (identities = 32%, positives = 50%) and has a slightly different 29-aa signal peptide and Zn2+-binding site, HEFTH (GenBank and SWISS protein Database). Although the B. cereus NprB resembles this and other close relatives over its entire length, it is interesting to note that an extended sequence at the Zn+2-binding site, HEFGHAV, exactly matches that of an otherwise more distantly related protease, the anthrax toxin lethal factor (Bragg & Robertson, 1989).
In the course of this work, we also noted that overexpression of NprB is not tolerated in either B. cereus or B. anthracis when PlcR and PapR are highly expressed from a plasmid. Thus, the average efficiency of B. cereus 569 and B. cereus 536 transformation with pUTE29-plcR-papR, pFP12 or pUTE29-nprB was similar for all indicated plasmids (100 ± 20 for B. cereus 569 and 600 ± 100 cfu/μg for B. cereus 536) whereas in the case of p102 and p182 the efficiency was only 6 ± 2 cfu/μg for both host strains. Both p102 (pUTE29-nprB-plcR-papR) and p182 (pUTE29-nprB-plcR fusion to papR) plasmids encode nprB in the same gene cluster with plcR and papR. The nprB gene is overexpressed both in p102 and p182 transformants due to high copy number of nprB along with plcR and papR. In the case of B. anthracis Ames 33, the average efficiency of transformation with pUTE29-plcR-papR, pFP12 and pUTE29-nprB was 900 ± 200 cfu/μg. The average efficiency of transformation was only 150 ± 30 cfu/μg with the p102 plasmid and no transformants were obtained with p182. Thus, the introduction of a transcriptionally active nrpB gene was not tolerated when its expression was controlled by a fused PlcR-PapR protein (p182, constitutive synthesis). In the case of the p102 plasmid the synthesis of NprB was apparently diminished due to PapR regulation and therefore it was better tolerated in B. anthracis.
The calculated molecular masses of NprB are 64.4 and 61.6 kDa for the protein with and without the signal peptide, respectively. However, analysis of the 2D-gel of the B. cereus 569 secretome (Fig. 5), along with data from Gohar et al. on the B. cereus 14579 secretome (Gohar et al., 2005), demonstrated that NprB migrates as if it were approximately 37 kDa. To determine the true size of the mature form of the NprB we produced the protease in the sporulation negative B. anthracis BH450 strain which has reduced proteolytic activity. B. anthracis BH450 transformed with pB148 or its parent plasmid pDG148-Stu were spotted on casein agar plates containing 50 μg mL-1 kanamycin. Large zones of casein hydrolysis were observed only for the pB148 transformant (Fig. 6A). This production of NprB occurred even without induction of the Pspac promoter. Addition of IPTG even up to 2 mM did not greatly enhance expression of the nprB gene from the Pspac promoter (Fig. 6B, upper and lower panels). To better understand the behavior of this plasmid we eliminated the lacI repressor gene from pB148, producing pB148Lac-. Expression remained high (data not shown). These data suggest that expression of nprB in these plasmids may depend on either the Pspac promoter operating even in the presence of the LacI repressor, or else on the bleomycin resistance gene promoter that is located immediately upstream of the Pspac promoter. Regardless of the mechanism, the resulting plasmid, pB148Lac-, provided expression levels that were sufficient for isolation and purification of NprB from B. anthracis BH450. The purified protein was transferred to a PVDF membrane and subjected to N-terminal sequence determination. The sequence of the first 11 residues at the N-terminus was determined to be VTSPRLSPASE, a sequence corresponding to that encoded beginning at nucleotide 703 of Genbank accession number AY195601. This shows that NprB is synthesized as a preproenzyme with a 234-amino acid-long prepro sequence (Fig. 6C). The mature enzyme is therefore composed of 349 amino acids and has a calculated mass of 38.2 kDa, corresponding well to that determined by SDS-polyacrylamide gel electrophoresis. The enzyme is resistant to 10 mM phenylmethylsulphonyl fluoride and can be completely inhibited by the metal chelator EDTA (10 mM) (data not shown).
Figure 6.
Characterization of B. cereus 569 NprB. (A) 2.5, 5 and 7.5 μl of overnight cultures of B. anthracis BH450 transformed with either pDG148-Stu (upper row) or pB148 (lower row) were spotted on casein agar plates containing 50 μg mL-1 kanamycin. Plates were incubated overnight at 37°C. (B) B. anthracis BH450 (pB148) was cultured in FA broth in the presence of kanamycin (50 μg mL-1) either in the absence or presence of 2 mM IPTG. Supernatants collected at T2 (2 h after the end of exponential growth) were concentrated and applied to SDS-polyacrylamide (10 to 20%) gel using the PhastGel System (Amersham Pharmacia). The gel was stained with Coomassie blue. M designates molecular mass markers. The arrowhead indicates the 38.2-kDa neutral protease B. Results are presented in the upper panel. Protease activities of the corresponding supernatants were determined by spotting them on casein agar (lower panel). The zones of casein proteolysis were observed after 2 h incubation at 37°C. (C) Graphic presentation of the three functional segments of the B. cereus 569 NprB preproenzyme. The protein includes the signal peptide (SP), propeptide region (Pro) and mature protease.
NprB/PapR cleavage
Because PapR is cleaved outside the cells, we wished to characterize its secretion and the initial processing by the signal peptidase. For this purpose the amino acid sequence of the theoretical 48-aa PapR polypeptide was submitted to the SignalP V1.1 Prediction Server, Center for Biological Sequence Analysis, Department of Biotechnology, Technical University of Denmark (http://www.cbs.dtu.dk/services/SignalP/). The SignalP-Neutral Networks prediction for Gram-positive bacteria yielded nearly equal probability scores for 21- and 26-aa signal peptides. However, the SignalP-Hidden Markov Models prediction that was also developed for Gram-positive bacteria demonstrated a much higher score for the 21-aa signal peptide. Therefore, we synthesized a 27-aa C-terminal peptide as a target for cleavage by the purified, recombinant NprB protease. In general, the NprB protease cleaved the synthetic PapR peptide very slowly. We found that only 30-40% of the peptide was digested after 6 h and approximately 70-80% after 24 h of incubation at 37°C. The efficiency of the synthetic PapR cleavage was estimated as a percent ratio of HPLC peak areas for cleaved/uncleaved PapR (data not shown). It was not possible to discriminate between primary and secondary cleavage sites since all cleavage peptides could be detected after 6 h of digestion. Analysis of the cleavage products by reverse phase HPLC and mass spectrometry showed that NprB processed the synthetic 27-aa PapR propeptide to produce a set of C-terminal peptides 11, 8, 7, and 5 amino acids in length (Table 3). The following cleavage sites of the synthetic pro-PapR were obtained: E16-↓-V17, L19-↓-A20, A20-↓-S21 and D22-↓-M23.
TABLE 3.
Summary of peptides identified during MS of NprB/PapR reaction mixture
| Observed mass | Proposed structure |
|
|---|---|---|
| Calculated mass | Sequence | |
| 3079.35 ± 1.12 | 3079.38 | DTALEKSKVISHDNQEVQLASDMPFEF |
| 2426.58 ± 2.42 | 2427.60 | DTALEKSKVISHDNQEVQLASD |
| 2224.46 ± 1.97 | 2225.43 | DTALEKSKVISHDNQEVQLA |
| 2154.12 ± 1.45 | 2154.36 | DTALEKSKVISHDNQEVQL |
| 1813.85 ± 1.25 | 1813.93 | DTALEKSKVISHDNQE |
| 1282.63 ± 1.65 | 1283.46 | VQLASDMPFEF |
| 942.79 ± 1.38 | 943.04 | ASDMPFEF |
| 871.44 ± 1.14 | 871.96 | SDMPFEF |
| 671.07 ± 1.49 | 669.79 | MPFEF |
Extracellular peptides increase PlcR-mediated PC-PLC activity
Study of B. cereus group member sequences identified four individual PlcR clusters with which a corresponding C-terminal PapR pentapeptide is associated (Slamti & Lereclus, 2005). The B. cereus 569 strain that is the subject of this work belongs to PlcR group II. Because NprB digestion of PapR produced several different C-terminal peptides, we synthesized several and compared their ability to activate PlcR (Table 2). As a reporter of activity, we used plcB (PC-PLC) expression, which is strongly PlcR-dependent (Pomerantsev et al., 2003). Strong activation of B. cereus 535 (PapR-) PC-PLC was found with the 7-, 8-, and 27-aa C-teminal PapR peptides, whereas the 5-aa peptide had lower activity (Fig. 7). In contrast, B. cereus 536 (NprB-) was activated only by the 5- and 7-aa peptides, indicating that peptides larger than 7 residues must be cleaved by NprB to produce the smaller active peptides. These data are consistent with a recent report that the 7-aa peptide is most effective activator of PlcR (Bouillaut et al., 2008).
Figure 7.
Comparison of the activity of the B. cereus 569 PlcR in the absence of either PapR (B. cereus 535) or NprB (B. cereus 536) in association with cognate penta-, hepta-, octa- and 27aa C-terminal PapR peptides (Table 2). Synthetic peptides (5 μM) were added to a culture of the B. cereus strains at stationary phase (OD600 1.5 ± .02). PC-PLC assays were performed 1 h after peptide addition.
To demonstrate the universal ability of the PlcR/PapR quorum sensing system to work in the entire B. cereus group we attempted to replace the disrupted B. anthracis system with the corresponding system from B. cereus 569. For this purpose, we employed the previously described strain SdT2, which produces full-size active B. cereus 569 PlcR intracellularly (Pomerantsev et al., 2004). As reported before, the intracellular PlcR is not sufficient to activate enzymes hydrolyzing lecithin outside the cells. The same four C-terminal PapR peptides used above were added to cultures of B. anthracis SdT2 to activate plcB expression. Dilution studies showed that the maximum activity was found for the B. cereus 569 heptapeptide SDMPFEF. The longer or shorter peptides were less active, especially at the lower concentrations (Fig. 8A, top).
Figure 8.
Heterologous B. cereus 569 PlcR/PapR system activates B. anthracis plcB expression while B. cereus 569 PlcR with a C-terminal truncation of six amino acids has lost the ability to activate the B. anthracis plcB in response to PapR addition. (A, upper panel) Phosphatidylcholine hydrolyzing activity of the recombinant B. anthracis SdT2 strain (producing intact PlcR of B. cereus 569). (A, lower panel) Phosphatidylcholine hydrolyzing activity of the recombinant B. anthracis SdT251 strain (producing truncated PlcR of B. cereus 569). Both B. anthracis SdT2 and SdT251 strains were grown at 37°C in LB broth with the indicated B. cereus 569 PapR C-terminal peptides added to final concentrations of 1, 10 or 100 μM. The synthetic peptides were added to a culture of the B. anthracis strains at stationary phase (OD600 1.3 ± .02). PC-PLC assays were performed 1 h after peptide addition. (B) PlcR proteins were analyzed by Western blot of whole-cell proteins from B. anthracis SdT2 and SdT251 using antiserum 1451 as described previously (Pomerantsev et al., 2004). The strains were grown for 10 h in LB broth at 37°C. M, molecular mass markers.
However, even the heptapeptide could not activate plcB expression in B. anthracis SdT251 strain containing PlcR with a C-terminal truncation of six amino acids (Fig. 8A, bottom). Although Western blot analysis of SdT251 cell lysates showed production of the truncated B. cereus 569 PlcR in amounts similar to that of full-length B. cereus 569 PlcR protein in B. anthracis SdT2 (Fig. 8B, both samples contain the expected ≈ 33-34-kDa bands for full-length and truncated monomers as well as slowly migrating aggregates formed by over-produced proteins stuck to themselves or to other materials), supplementation of cultures with the peptides induced PC-PLC activity in B. anthracis SdT2 but not in B. anthracis SdT251, demonstrating the importance of not only PapR but also the PlcR C-terminal region. The presence of this C-terminal region could be important either in PlcR structural stability or in recruiting other members of the transcriptional complex.
Discussion
The functional dependence of the PlcR regulon on the export and retrieval of peptides derived from the PapR precursor peptide was first recognized in studies on B. thuringiensis 407 Cry- and B. cereus by Slamti and Lereclus (Slamti & Lereclus, 2002) and was confirmed in our studies for B. anthracis (Pomerantsev et al., 2003; Pomerantsev et al., 2004). The PapR peptide contains a predicted 21-amino acid signal peptide typical of SecA-dependant secreted proteins (Okstad et al., 1999). Thus, it was expected that secretion and processing of the 48-amino acid PapR by signal peptidase cleavage after A21 in the sequence SLA-DT would generate an extracellular 27-amino acid propeptide. The demonstration that synthetic peptides corresponding to the C-terminus of this 27-amino acid propeptide could activate gene expression (Slamti & Lereclus, 2002), together with the preference of the oligopeptide permease for shorter peptides ranging in size from 5 to 20 amino acids (Pottathil & Lazazzera, 2003), implies that additional proteolytic processing must take place extracellularly. The requirement that both the peptide substrate and the processing protease accumulate to sufficient concentrations outside the cell for effective processing and subsequent pentapeptide uptake to occur are characteristics of a system intended to sense bacterial density, i.e., a quorum-sensing system (Schauder & Bassler, 2001).
In this study, we have shown that B. cereus 569 NprB is a key component of this density-sensing gene regulation system. In the absence of NprB, it appears that peptides able to activate PlcR are not generated from PapR for import by the oligopeptide permease (Opp) complex (Gominet et al., 2001). This is clearly indicated in both Fig. 2C and Fig. 7: the B. cereus 536 nprB mutant did not express PC-PLC activity when treated with a 27-aa PapR propeptide, whereas the B. cereus 535 papR mutant did respond to the same peptide.
Like other bacillus metalloproteases, NprB is secreted by the SecA system and its 26-amino acid signal peptide is removed by a signal peptidase. Furthermore, NprB may become active only after removal of an N-terminal pro-region, as is common for secreted metalloproteases. This activation could be either autocatalytic or dependent on other secreted proteases (Park et al., 2004). Thus, it may be that additional secreted proteases are required to act cooperatively or in a cascade that includes NprB and leads to processing of the PapR propeptide. A similar involvement of multiple proteases (subtilisin, Epr, and Vpr) has been demonstrated in peptide processing in the B. subtilis Phr peptide system. However, in the Phr peptide system each protease could produce the mature peptide (Lanigan-Gerdes et al., 2007). Our work indicates that NprB plays a dominant role in the regulation of B. cereus 569 genes by PlcR/PapR, but does not rule out a contribution by other proteases.
However, the presence of multiple copies of nprB in combination with plcR and papR in multicopy plasmids p102 and p182 could lead to a PlcR-dependent NprB overexpression that could inhibit the growth of or even kill bacteria. The growth inhibition and death could be a consequence of enhanced expression of the peptide gene that is co-expressed with clo and is reported to have antibacterial activity (Brillard & Lereclus, 2007). The fact that overexpression of the nprB-plcR-papR gene cluster in either B. cereus 569 or B. anthracis SdT appears to be toxic to the host cells may explain why some B. cereus and all B. anthracis strains lost the nprB gene from the nprB-plcR-PapR gene cluster. A type M4 neutral protease such as BAS2543 may have played the role of NprB in the putative B. cereus ancestor of B. anthracis because this was the only protease found in exponential phase culture supernatants of B. anthracis SdT12 (Fig. 5), and, similar to nprB, this protease gene contains a PlcR-binding site in the promoter region. Accordingly, the C-terminal sequence of PapR of B. anthracis, ASDVPFEY, would have been expected to be a good substrate for this hypothetical protease. Given these considerations, one can suggest that each of the four PlcR-PapR sequence groups may have depended on a particular protease selected for the ability to correctly cleave the corresponding PapR.
The general scheme of PlcR/PapR regulation is presented in Fig. 9. Transcription of the plcR is self-regulated and takes place simultaneously with transcription of the nprB gene. The papR gene is also transcribed under control of the PlcR protein within the same gene cluster as plcR. The papR transcription is enhanced by an additional PlcR-binding site immediately before papR. Low levels of constitutive transcription of all three genes must occur, perhaps maintained by small amounts of PlcR even in the absence of the preferred C-terminal PapR peptides (Fig. 9, I; i.e., a PlcR-dependent but PapR peptide-independent basal transcription of PlcR-dependent genes). Alternatively, a low basal level of expression of the nprB, plcR and papR genes in the absence of any PlcR might occur (i.e., a PlcR-independent basal level of transcription from “leaky” or alternative promoters.) Regardless of the mechanism involved, the resulting limited synthesis of all the three proteins would prime the system to respond when bacterial densities increased (Fig. 9, II). While the model presented in Fig. 9 is supported by extensive studies, not every aspect of it has been proven. However, the central role of PlcR is evident. For example, disruption of plcR gene completely abolished NprB production in B. cereus 14579 (Gohar et al., 2002).
Figure 9.
Model of PlcR/PapR regulon activation in B. cereus 569 strain. (I) Schematic diagram of intracellular synthesis of PlcR (indicated by horseshoes), NprB and PapR preproenzymes (both indicated by line figures with dashed N-terminal sequences). The plcR, papR and nprB transcription is initiated by RNA polymerases (indicated by hatched figures) activated by only a PlcR dimer bound to the recognition sites. (II) Extracellular NprB processing of pro-PapR into a set of C-terminal peptides with the consequent transport of the peptides into the cell via oligopeptide permease complex Opp ABCDE. (III) Intracellular assembly of the transcriptional complex including PlcR protein with corresponding C-terminal peptide, PlcR-binding site and RNA polymerase. RNA polymerase is activated when the C-terminal peptide is bound to PlcR and the corresponding PlcR-binding site that is located in the vicinity of the RNA polymerase sigma subunit recognition site. Without the corresponding C-terminal peptide the initiating transcriptional complex is not assembled and RNA polymerase is not activated as illustrated in region I. See text for details.
As mentioned above, shorter peptides ranging in size from 5 to 20 amino acids are preferred for transport inside the cell via the Opp complex. However, it is not known what are the optimum and maximum sizes of peptides transported by the Opp complex of the B. cereus group. The role of this complex in PlcR/PapR activation was well established previously by Gominet for B. thuringiensis (Gominet et al., 2001). In this study we demonstrate that the NprB protease is also involved in this activation. On the basis of this fact we suggest that both PapR and NprB interact extracellularly to produce a set of C-terminal peptides as shown in Fig. 9, II. The OppA component of the OppABCDE complex accepts the peptides and delivers them to the membrane-bound BCDE core subunits that transfer the peptides inside the cell. It is currently unknown whether any single part of the system shown in Fig. 9, II is the dominant element determining the extent of PapR peptide activation of PlcR. Critical steps could be the rate of PapR cleavage, Opp complex selectivity for different peptides, rate of peptide uptake, or the resulting concentration of the peptides inside the cell.
Both the size of the peptides and their sequence are important. The requirement for a particular sequence was known from the identification of the four complementary PlcR-PapR groups. Previous suggestions that pentapeptides are optimal must now be reexamined in view of the finding that a heptapeptide was most active in the system studied here (Fig. 8). The same conclusion was made by Bouillaut et al. in a recent paper (Bouillaut et al., 2008). The difference between the heptapeptide and pentapeptide was explained by a slightly lower affinity of the pentapeptide for its cognate PlcR molecule. It has been also demonstrated that lysine at position 278 of the group II PlcR is very important for specific PapR-mediated activation. This K278 is well positioned to form a salt-bridge with the heptapeptide aspartic acid (Bouillaut et al., 2008). Nevertheless, even PlcR with C-terminal lysine (seven C-terminal amino acids were eliminated) could not activate transcription from the plcA promoter in response to PapR addition (Bouillaut et al., 2008). These data along with our finding that PlcR with a C-terminal truncation of six amino acids could not activate transcription of plcB in response to PapR addition (Fig. 8) demonstrate the importance of not only PapR but also the PlcR C-terminal region.
On the basis of these observations we suggest that to achieve maximal transcriptional activation in the B. cereus group each gene requires a particular combination of PlcR type with a corresponding PapR heptapeptide rather than with peptides of various sizes and sequences. The different affinities of PlcR-binding sites to active PlcR:PapR heptapeptide complexes could explain the differences in gene expression kinetics and magnitude. This would not be surprising given the complexity of the transcriptional complex, in which the DNA elements recognized by the PlcR:PapR heptapeptide and various sigma subunits will differ in sequence and spacing. Schematically, the interaction between PlcR, PapR peptide, PlcR-binding site and RNA-polymerase is presented in Fig. 9, III. The detailed description of these interactions demands further study of specific protein-protein and DNA-protein contacts.
Acknowledgments
We thank M. Garfield and J. Lukszo from the NIAID core facility for peptide synthesis and protein sequencing as well as Dr. Nga Nguyen from the FDA core facility for assistance with mass spectrometry analysis. We also acknowledge T. Read for the disclosure of the B. cereus 569 chromosomal sequences and K. Beliakov for assistance with the figure formatting. This research was supported by the Intramural Research Program of the NIH, National Institute of Allergy and Infectious Diseases.
Supplementary Material
References
- Agaisse H, Gominet M, Okstad OA, Kolsto AB, Lereclus D. PlcR is a pleiotropic regulator of extracellular virulence factor gene expression in Bacillus thuringiensis. Mol Microbiol. 1999;32:1043–1053. doi: 10.1046/j.1365-2958.1999.01419.x. [DOI] [PubMed] [Google Scholar]
- Aronson AI, Angelo N, Holt SC. Regulation of extracellular protease production in Bacillus cereus T: characterization of mutants producing altered amounts of protease. J Bacteriol. 1971;106:1016–1025. doi: 10.1128/jb.106.3.1016-1025.1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouillaut L, Perchat S, Arold S, Zorrilla S, Slamti L, Henry C, Gohar M, Declerck N, Lereclus D. Molecular basis for group-specific activation of the virulence regulator PlcR by PapR heptapeptides. Nucleic Acids Res. 2008;36:3791–3801. doi: 10.1093/nar/gkn149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bragg TS, Robertson DL. Nucleotide sequence and analysis of the lethal factor gene (lef) from Bacillus anthracis. Gene. 1989;81:45–54. doi: 10.1016/0378-1119(89)90335-1. [DOI] [PubMed] [Google Scholar]
- Brillard J, Lereclus D. Characterization of a small PlcR-regulated gene co-expressed with cereolysin O. BMC Microbiol. 2007;7:52. doi: 10.1186/1471-2180-7-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Declerck N, Bouillaut L, Chaix D, Rugani N, Slamti L, Hoh F, Lereclus D, Arold ST. Structure of PlcR: Insights into virulence regulation and evolution of quorum sensing in Gram-positive bacteria. Proc Natl Acad Sci U S A. 2007;104:18490–18495. doi: 10.1073/pnas.0704501104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drobniewski FA. Bacillus cereus and related species. Clin Microbiol Rev. 1993;6:324–338. doi: 10.1128/cmr.6.4.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gohar M, Gilois N, Graveline R, Garreau C, Sanchis V, Lereclus D. A comparative study of Bacillus cereus, Bacillus thuringiensis and Bacillus anthracis extracellular proteomes. Proteomics. 2005;5:3696–3711. doi: 10.1002/pmic.200401225. [DOI] [PubMed] [Google Scholar]
- Gohar M, Okstad OA, Gilois N, Sanchis V, Kolsto AB, Lereclus D. Two-dimensional electrophoresis analysis of the extracellular proteome of Bacillus cereus reveals the importance of the PlcR regulon. Proteomics. 2002;2:784–791. doi: 10.1002/1615-9861(200206)2:6<784::AID-PROT784>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- Gominet M, Slamti L, Gilois N, Rose M, Lereclus D. Oligopeptide permease is required for expression of the Bacillus thuringiensis plcR regulon and for virulence. Mol Microbiol. 2001;40:963–975. doi: 10.1046/j.1365-2958.2001.02440.x. [DOI] [PubMed] [Google Scholar]
- Gomis-Ruth F. Structural aspects of the metzincin clan of metalloendopeptidases. Mol Biotechnol. 2003;24:157–202. doi: 10.1385/MB:24:2:157. [DOI] [PubMed] [Google Scholar]
- Helgason E, Okstad OA, Caugant DA, Johansen HA, Fouet A, Mock M, Hegna I, Kolsto AB. Bacillus anthracis, Bacillus cereus, and Bacillus thuringiensis -One species on the basis of genetic evidence. Appl Environ Microbiol. 2000;66:2627–2630. doi: 10.1128/aem.66.6.2627-2630.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanova N, Sorokin A, Anderson I, Galleron N, Candelon B, Kapatral V, Bhattacharyya A, Reznik G, Mikhailova N, Lapidus A, et al. Genome sequence of Bacillus cereus and comparative analysis with Bacillus anthracis. Nature. 2003;423:87–91. doi: 10.1038/nature01582. [DOI] [PubMed] [Google Scholar]
- Ivins BE, Ezzell JW, Jr, Jemski J, Hedlund KW, Ristroph JD, Leppla SH. Immunization studies with attenuated strains of Bacillus anthracis. Infect Immun. 1986;52:454–458. doi: 10.1128/iai.52.2.454-458.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph P, Fantino JR, Herbaud ML, Denizot F. Rapid orientated cloning in a shuttle vector allowing modulated gene expression in Bacillus subtilis. FEMS Microbiol Lett. 2001;205:91–97. doi: 10.1111/j.1574-6968.2001.tb10930.x. [DOI] [PubMed] [Google Scholar]
- Lanigan-Gerdes S, Dooley AN, Faull KF, Lazazzera BA. Identification of subtilisin, Epr and Vpr as enzymes that produce CSF, an extracellular signalling peptide of Bacillus subtilis. Mol Microbiol. 2007;65:1321–1333. doi: 10.1111/j.1365-2958.2007.05869.x. [DOI] [PubMed] [Google Scholar]
- Lereclus D, Agaisse H, Gominet M, Salamitou S, Sanchis V. Identification of a Bacillus thuringiensis gene that positively regulates transcription of the phosphatidylinositol-specific phospholipase C gene at the onset of the stationary phase. J Bacteriol. 1996;178:2749–2756. doi: 10.1128/jb.178.10.2749-2756.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lereclus D, Agaisse H, Grandvalet C, Salamitou S, Gominet M. Regulation of toxin and virulence gene transcription in Bacillus thuringiensis. Int J Med Microbiol. 2000;290:295–299. doi: 10.1016/S1438-4221(00)80024-7. [DOI] [PubMed] [Google Scholar]
- Okstad OA, Gominet M, Purnelle B, Rose M, Lereclus D, Kolsto AB. Sequence analysis of three Bacillus cereus loci carrying PIcR-regulated genes encoding degradative enzymes and enterotoxin. Microbiology. 1999;145(Pt 11):3129–3138. doi: 10.1099/00221287-145-11-3129. [DOI] [PubMed] [Google Scholar]
- Park CH, Lee SJ, Lee SG, Lee WS, Byun SM. Hetero- and autoprocessing of the extracellular metalloprotease (Mpr) in Bacillus subtilis. J Bacteriol. 2004;186:6457–6464. doi: 10.1128/JB.186.19.6457-6464.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S, Leppla SH. Optimized production and purification of Bacillus anthracis lethal factor. Protein Expr Purif. 2000;18:293–302. doi: 10.1006/prep.2000.1208. [DOI] [PubMed] [Google Scholar]
- Perego M. A peptide export-import control circuit modulating bacterial development regulates protein phosphatases of the phosphorelay. Proc Natl Acad Sci U S A. 1997;94:8612–8617. doi: 10.1073/pnas.94.16.8612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomerantsev AP, Kalnin KV, Osorio M, Leppla SH. Phosphatidylcholine-specific phospholipase C and sphingomyelinase activities in bacteria of the Bacillus cereus group. Infect Immun. 2003;71:6591–6606. doi: 10.1128/IAI.71.11.6591-6606.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomerantsev AP, Pomerantseva OM, Leppla H. A spontaneous translational fusion of Bacillus cereus PlcR and PapR activates transcription of PlcR-dependent genes in Bacillus anthracis via binding with a specific palindromic sequence. Infect Immun. 2004;72:5814–5823. doi: 10.1128/IAI.72.10.5814-5823.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomerantsev AP, Sitaraman R, Galloway CR, Kivovich V, Leppla SH. Genome engineering in Bacillus anthracis using Cre recombinase. Infect Immun. 2006;74:682–693. doi: 10.1128/IAI.74.1.682-693.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pottathil M, Lazazzera BA. The extracellular Phr peptide-Rap phosphatase signaling circuit of Bacillus subtilis. Front Biosci. 2003;8:D32–D45. doi: 10.2741/913. [DOI] [PubMed] [Google Scholar]
- Pritzlaff CA, Chang JC, Kuo SP, Tamura GS, Rubens CE, Nizet V. Genetic basis for the beta-haemolytic/cytolytic activity of group B Streptococcus. Mol Microbiol. 2001;39:236–247. doi: 10.1046/j.1365-2958.2001.02211.x. [DOI] [PubMed] [Google Scholar]
- Radnedge L, Agron PG, Hill KK, Jackson PJ, Ticknor LO, Keim P, Andersen GL. Genome differences that distinguish Bacillus anthracis from Bacillus cereus and Bacillus thuringiensis. Appl Environ Microbiol. 2003;69:2755–2764. doi: 10.1128/AEM.69.5.2755-2764.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy A, Battisti L, Thorne CB. Identification of self-transmissible plasmids in four Bacillus thuringiensis subspecies. J Bacteriol. 1987;169:5263–5270. doi: 10.1128/jb.169.11.5263-5270.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J, Russell DW. Molecular Cloning A Laboratory Manual. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, New York: 2001. [Google Scholar]
- Schauder S, Bassler BL. The languages of bacteria. Genes Dev. 2001;15:1468–1480. doi: 10.1101/gad.899601. [DOI] [PubMed] [Google Scholar]
- Singh Y, Chaudhary VK, Leppla SH. A deleted variant of Bacillus anthracis protective antigen is non-toxic and blocks anthrax toxin action in vivo. J Biol Chem. 1989;264:19103–19107. [PubMed] [Google Scholar]
- Slamti L, Lereclus D. Specificity and polymorphism of the PlcR-PapR quorum-sensing system in the Bacillus cereus group. J Bacteriol. 2005;187:1182–1187. doi: 10.1128/JB.187.3.1182-1187.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slamti L, Lereclus D. A cell-cell signaling peptide activates the PlcR virulence regulon in bacteria of the Bacillus cereus group. EMBO J. 2002;21:4550–4559. doi: 10.1093/emboj/cdf450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephenson S, Mueller C, Jiang M, Perego M. Molecular analysis of Phr peptide processing in Bacillus subtilis. J Bacteriol. 2003;185:4861–4871. doi: 10.1128/JB.185.16.4861-4871.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.