

Abstract
Axonal degeneration is a prominent feature of many neurological disorders that are associated with mitochondrial dysfunction, including Parkinson's disease, motor neuron disease, and inherited peripheral neuropathies. Studies of the Wlds mutant mouse, which undergoes delayed Wallerian degeneration in response to axonal injury, suggest that axonal degeneration is an active process. Wlds mice also have slower axonal degeneration and disease progression in numerous models of neurodegenerative disease. The Wlds mutation results in the production of a chimeric protein that contains the full-length coding sequence of nicotinamide mononucleotide adenylyltransferase 1 (Nmnat1), which alone is sufficient for axonal protection in vitro. To test the effects of increased Nmnat expression on axonal degeneration induced by mitochondrial dysfunction, we examined dorsal root ganglion (DRG) neurons treated with rotenone. Rotenone induced profound axonal degeneration in DRG neurons; however, this degeneration was delayed by expression of Nmnat. Nmnat-mediated protection resulted in decreased axonal accumulation and sensitivity to reactive oxygen species (ROS) but did not affect the change in the rate of rotenone-induced loss in neuronal ATP. Nmnat also prevented axonal degeneration caused by exposure to exogenous oxidants and reduced the level of axonal ROS after treatment with vincristine, further supporting the idea that Nmnat promotes axonal protection by mitigating the effects of ROS.
Keywords: Nmnat, Wlds, axonal degeneration, mitochondria, ROS, ATP
Introduction
Mitochondrial dysfunction caused by mutations or toxic agents has been implicated in many neurodegenerative diseases, including those associated with axonal degeneration (Lin and Beal, 2006). It results in a host of cellular abnormalities, including decreased ATP synthesis, disrupted mitochondrial fusion and transport, and the increased generation of reactive oxygen species (ROS) (Lin and Beal, 2006; Baloh et al., 2007). The association of mitochondrial dysfunction with neurodegenerative diseases is perhaps strongest in Parkinson's disease (PD), which can be associated with mutations in mitochondria-associated proteins (e.g., DJ-1 and PINK-1) (Martin, 2006) and ingestion of 1-methyl 4-phenyl 1,2,3,6-tetrahydropyridine (MPTP), an inhibitor of mitochondrial complex I (Langston et al., 1983; Betarbet et al., 2000). It is generally accepted that environmental factors play an important role in the pathogenesis of PD (Sherer et al., 2002a); in particular, there is a strong association between PD and exposure to pesticides, including many that inhibit mitochondrial complex I (Semchuk et al., 1992; Gorell et al., 1998, 2004; Priyadarshi et al., 2000, 2001; Ascherio et al., 2006; Brown et al., 2006). Further studies have shown that the pesticide rotenone leads to dopaminergic neuron loss and PD-like symptoms in animals (Betarbet et al., 2000).
Neurodegenerative disease is commonly associated with neuronal cell death, but it is becoming increasingly apparent that axonopathy is also a major component of many neurodegenerative diseases, including amyotrophic lateral sclerosis, PD, and Alzheimer's disease (Coleman, 2005). Unfortunately, the molecular mechanisms underlying axonal degeneration remain poorly characterized. One model of axonal degeneration derives from studies of anoxic injury to axons, which leads to mitochondrial failure, decreased levels of ATP, reduced Na+/K+ ATPase activity, and increased axonal Na+ and Ca2+ (Stys et al., 1992). These changes are followed by calpain activation and eventual axonal protein degradation (Coleman, 2005). Studies of the Wlds mutant mouse, which exhibits slow axonal Wallerian degeneration in response to nerve injury, have led to the conclusion that axonal degeneration, like apoptosis, is an active process (Lunn et al., 1989; Raff et al., 2002). Axonal degeneration is delayed by the wlds mutation in a wide range of disease models, such as the pmn mice, a model of motor neuron degeneration; vincristine treatment, a model of chemotherapy-induced neuropathy; and MPTP, a model of PD (Wang et al., 2001b; Ferri et al., 2003; Hasbani and O'Malley, 2006), suggesting that a common “degenerative” pathway is activated by these insults that can be inhibited by this mutation.
The Wlds mutant mouse harbors a triplication of a gene fusion that produces a chimeric protein composed of the N-terminal 70 aa of ubiquitination factor 4b, a unique 18 residue linker region, and the full-length nicotinamide mononucleotide adenylyltransferase 1 (Nmnat1) protein (Conforti et al., 2000). Using dorsal root ganglion (DRG) neuronal cultures, we previously demonstrated that the Nmnat1 portion of the Wlds protein is the moiety responsible for axonal protection (Araki et al., 2004). Many studies in both rodents and Drosophila have confirmed these results (Hoopfer et al., 2006; Macdonald et al., 2006; Zhai et al., 2006), whereas other investigators have suggested the Ube4b fragment may modulate the Nmnat protective effect (Wang et al., 2005; Conforti et al., 2007). Notably, we found that Nmnat3, which is localized to mitochondria, also provides axonal protection from mechanical injury (Sasaki et al., 2006). In the present study, we show that increased Nmnat protein expression effectively delayed rotenone-induced axonal degeneration in DRG neuronal cultures. Nmnat3 expression decreased ROS accumulation in neurons treated with rotenone but had little effect on the rate of ATP loss in these cells. We also demonstrate that Nmnat3 can prevent axonal damage resulting from exogenous application of oxidants, suggesting that Nmnat expression prevents axonal degeneration by decreasing the accumulation or damage caused by ROS generated during mitochondrial and/or oxidative injury. We show that vincristine, a known cause of axonal degeneration that is preventable by Nmnat (Araki et al., 2004), induces ROS and that this increase can be prevented by Nmnat. Finally, we demonstrate that antioxidants are sufficient for axonal protection from rotenone-induced axonal degeneration, further supporting the idea that Nmnat-mediated protection occurs via mitigating ROS-mediated damage.
Materials and Methods
Reagents.
All reagents are from Sigma-Aldrich (St. Louis, MO) unless otherwise noted.
Dissociated DRG cultures.
DRGs were dissected from embryonic day 14.5 (E14.5)–E15.5 Sprague Dawley rat embryos. DRGs were dissociated with collagenase followed by trypsin and subsequent trituration through a fire-polished glass pipette. Cells were seeded onto Matrigel (BD Biosciences, San Jose, CA)-coated 24-well dishes and grown in DMEM containing 10% FBS, penicillin and streptomycin, and 3 μm aphidicolin or, alternatively, in Neurobasal with B27 (Invitrogen, Carlsbad, CA) and 25 ng/ml nerve growth factor followed 24 h later by the addition of 1 μm 5-fluoro-2′-deoxyuridine and 1 μm uridine (Harlan Bioproducts, Indianapolis, IN). After 4 d in culture, all of the neuronal cultures were maintained in DMEM containing 10% FBS, penicillin and streptomycin, 1 μm 5-fluoro-2′-deoxyuridine, 1 μm uridine, and NGF.
Construction of lentiviral expression plasmids.
The GFP plasmid (FUGW) was a gift from D. Baltimore (California Institute of Technology, Pasadena, CA). Nmnat1 (FCIV-Nmnat1) and Nmnat3 (FCIV-Nmnat3) were described previously (Sasaki et al., 2006). Hexahistidine-tagged-Nmnat1 (FUW-Nmnat1) and Nmnat3 (FUW-Nmnat3) (Araki et al., 2004; Sasaki et al., 2006) were cloned into the FUW plasmid without GFP (gift from D. Baltimore) to produce lentiviral transfer vectors without GFP expression for studies using ROS-sensitive fluorescent dyes.
Lentiviral infection of DRG neurons.
Lentiviruses expressing transgenes were generated as previously described (Araki et al., 2004). For infection of DRG neurons, lentivirus (105 to 106 infectious units) was added to an individual well of a 24-well plate containing ∼5 × 104 neurons. Transgene expression from the lentivirus was allowed to proceed for 4–10 d before using the infected neurons for experiments. Viral infection and transgene expression was monitored, where applicable, by monitoring the GFP reporter via fluorescent microscopy.
Neuronal cell body death.
Neurons were treated with rotenone or vehicle control and monitored for cell body damage by ethidium homodimer exclusion from Biotium (Hayward, CA). Ethidium homodimer was added to the cultures at the indicated time at a final concentration of 100 nm. For each condition, four fields with an average of 35 cells per well in each of four wells were examined using phase-contrast and red fluorescence merged images. Data were collected from two independent experiments.
Morphometric analysis of axonal degeneration.
Dissociated DRG neurons were treated with the indicated chemical agents and imaged using phase-contrast microscopy with a 40× lens at the indicated time points. A grid was created over each image using NIH ImageJ software using the grid plugin (line area = 100,000). The cell counting plugin was used to score each neurite. Degenerating and healthy axons were counted in at least three high-power fields per image (∼20 neurites) for each well (the observer was blinded to the condition). Axonal segments were considered degenerated if they showed evidence of swellings and/or blebbing (n ≥ 6 wells per condition from duplicate experiments).
Analysis of ATP levels.
ATP assays were performed on neuronal cultures seeded at the same density lysed in 1% Triton X-100, 0.1 m Tris, pH 7.8, and 0.5 mm EDTA. Cell lysates were used in a luciferase-based ATP assay (Promega, Madison, WI), where ATP is expressed as a percentage of control based on arbitrary units of luminescence and then converted to micromolar ATP based on a standard curve (n ≥ 6 wells per condition; results are from two independent experiments) (Chang et al., 2003). ATP was measured under all conditions at the end of the treatment period.
Quantitation of ROS levels.
ROS was measured using CM-H2DCFDA dye at 1.85 μm (Invitrogen). The dye was added to neurons in serum-free DMEM without phenol red for 1 h, and the cells were washed twice with PBS. Fields containing exclusively axons were identified using phase-contrast microscopy. These fields were imaged using epifluorescence with a FITC filter and equal exposure times on an Eclipse TE300 microscope with a 40× Nikon (Tokyo, Japan) Pan-Fluor lens. The mean intensity of the field minus the minimum intensity (to remove the background fluorescence of the field) was recorded; three fields per well were imaged from three wells per condition. Results are from two independent experiments. All conditions were measured at the end of the treatment period.
Visualization of mitochondrial potential.
The methodology we used has been previously described (Ikegami and Koike, 2003). Briefly, DRG cultures were treated with either vehicle control or rotenone for 6 h. Cultures were then changed to phenol red-free DMEM that contained the mitochondrial potential-sensitive dye Mitotracker Red CMXRos (final concentration, 25 nm; Invitrogen) and then costained with calcein AM (final concentration, 500 nm; Invitrogen), a small fluorescein-based molecule that is cleaved by nonspecific esterases and trapped inside the cell, to visualize the axons. Cultures were treated with 100 μm FCCP for 1 h before imaging and during exposure to the Mitotracker Red and calcein AM dyes to depolarize the mitochondrial membrane. Cultures were incubated at 37° for 30 min and then washed twice with phenol red-free DMEM and visualized by fluorescence microscopy using the FITC filter for calcein AM and the rhodamine filter for Mitotracker Red.
Quantitation of antioxidant enzyme expression.
RNA was collected from dissociated rat DRGs after 14 DIV. Five wells from a 24-well plate were pooled, and three pools for each condition were collected. RNA was prepared using Trizol (Invitrogen). First-strand cDNA templates were prepared using 1 μg of RNA template from each pool using standard methods. Quantitative reverse transcription (qRT)-PCR was performed by monitoring in real time the increase in fluorescence of the SYBR-green dye on a TaqMan 7700 Sequence Detection System (Applied Biosystems, Foster City, CA). The expression levels were normalized to 18S ribosomal RNA to account for variations between the levels of total cDNA template across different samples. Each qRT-PCR experiment was performed in triplicate using three pools of cDNA template. The primers used are as follows: glutathione peroxidase 1 (GPX1), (Fernandez-Gomez et al., 2006) forward, 5′-GCAGATACACCAGGCGCTTT-3′; reverse, 5′-GGCTTCTATATCGGGTTCGA-3′; catalase (CATA), forward, 5′-GCACACTTTGACAGAGAGCGG-3′; reverse, 5′-CTTTGCCTTGGAGTATCTGGTAATATC-3′; superoxide dismutase 1 (SOD1), forward, 5′-TCAGGAGAGCATTCCATCATTG-3′; reverse, 5′-CAGCATTTCCAGTCTTTGTACTTTCTT-3′; SOD2, forward, 5′-CACAGCATTTTCTGGACAAACC-3′; reverse, 5′-CCTTAAACTTCTCAAAAGACCCAAAG-3′; 18S, forward, 5′-CGCCGCTAGAGGTGAAATTCT-3′; reverse, 5′-CGGCTACCACATCCAAGGAA-3′.
Statistics.
ANOVA was used followed by individual comparisons with Student's two-tailed, unpaired t tests. Comparisons were considered significant with a p > 0.05 after Bonferroni's correction for multiple comparisons. Values are reported as mean ± SEM unless otherwise noted.
Results
Rotenone treatment of DRG neurons results in axonal degeneration
The expression of Wlds, Nmnat1, or Nmnat3 in neurons protects against axonal degeneration resulting from damage caused by mechanical, genetic, or chemical injury (Gillingwater and Ribchester, 2001; Wang et al., 2001a,b; Ferri et al., 2003; Samsam et al., 2003; Araki et al., 2004; Gillingwater et al., 2004; Mi et al., 2005; Sasaki et al., 2006; Zhai et al., 2006). The association between mitochondrial dysfunction and axonal degeneration in multiple neurological disorders encouraged us to investigate whether Nmnat expression could protect axons from damage caused by mitochondrial dysfunction. We used DRG sensory neurons, which we previously used (Araki et al., 2004; Sasaki et al., 2006) in our studies of Wlds- and Nmnat-mediated axonal protection after mechanical injury, to study axonal degeneration caused by mitochondrial inhibitors. We tested the effects of rotenone, a lipophilic complex I inhibitor that can be used in nondopaminergic neurons (Betarbet et al., 2000; Sherer et al., 2003), on DRG neurons in concentrations ranging from 0.1 to 10 μm. We assessed axonal degeneration using phase-contrast microscopy after rotenone addition and found that 1–5 μm rotenone caused significant axonal degeneration by 24 h that continued to progress until, by 72 h, most axons had largely degenerated (Fig. 1 A,B and data not shown).
Figure 1.
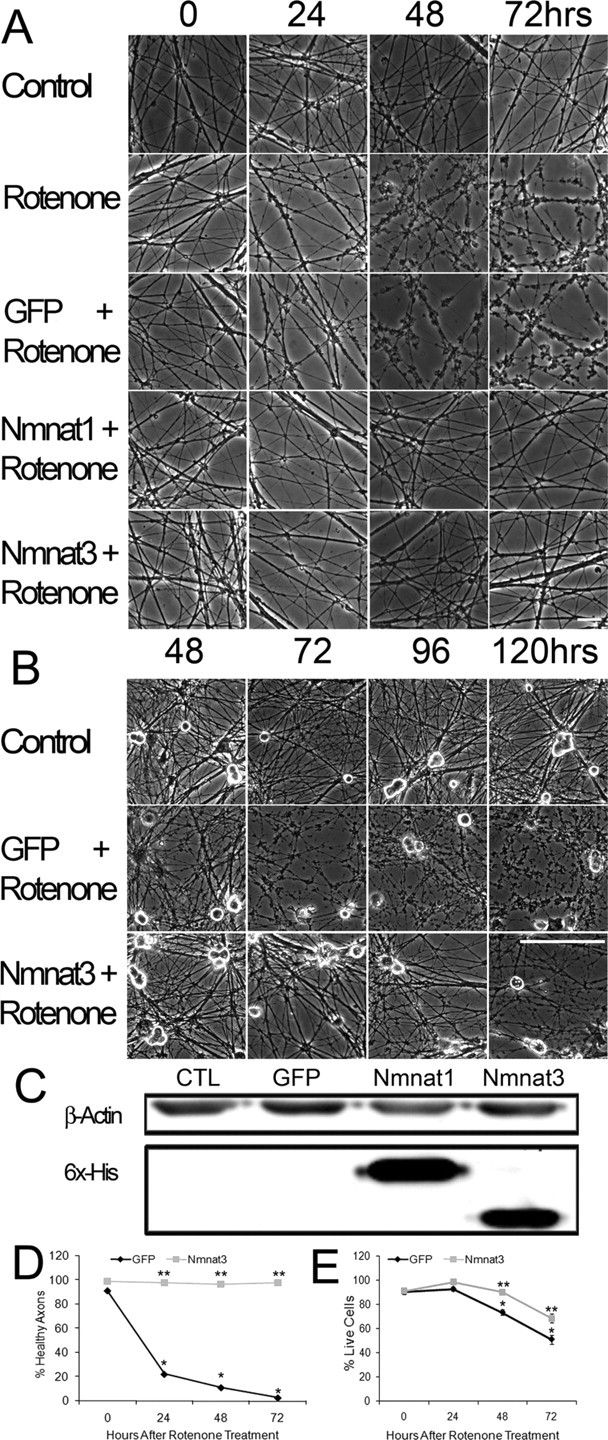
Nmnat expression protects against rotenone-mediated axonal degeneration in DRG neurons. A, B, DRG neurons were treated with rotenone (2.5 μm) for the indicated time, and phase-contrast microscopy was used to assess axonal degeneration. Degeneration is subtle after 24 h, but note the extensive axonal damage caused by rotenone by 48 h. Nmnat1- and Nmnat3-expressing neurons do not show axonal degenerative changes until after 96 h. Scale bars: A, 20 μm; B, 100 μm. C, Western-blot analysis of lysates of DRG neurons infected with lentivirus expressing GFP alone, Nmnat1, or Nmnat3 using a monoclonal antibody to the hexahistidine tag. D, Quantification of rotenone-induced axonal degeneration at 0, 24, 48, and 72 h in DRG neurons expressing GFP or Nmnat3 (4 fields per well, 6 wells per condition from duplicate experiments; see Materials and Methods). E, Quantification of surviving DRG neurons after treatment with rotenone for 24, 48, or 72 h using ethidium homodimer exclusion (6 wells per condition from duplicate experiments). *p < 0.01 compared with GFP-expressing neurons at 0 h; **p < 0.01 compared with rotenone-treated GFP-expressing neurons at corresponding time point. Error bars represent ±SEM.
To determine whether axonal degeneration preceded cell death in neurons exposed to rotenone, we performed a time course experiment in which we monitored cell body damage using an ethidium homodimer exclusion assay in conjunction with microscopic analysis of axonal degeneration. After 72 h, when 100% of axonal segments showed signs of degeneration, 51% survival of cell bodies were still intact (Fig. 1 E). These results, which indicate that axons are more susceptible to rotenone toxicity than neuronal cell bodies, are consistent with previous reports (Betarbet et al., 2000; Testa et al., 2005). Additionally, because soma membrane permeability clearly lagged behind axonal degeneration, these results also demonstrate that cell death was not a prerequisite for axonal degeneration.
Nmnat proteins protect axons and cell bodies from rotenone toxicity
To determine whether Nmnat activity could protect axons and/or cell bodies against rotenone-induced mitochondrial inhibition, we infected DRG neurons with lentivirus expressing either GFP alone or hexahistidine-tagged Nmnat1 or Nmnat3 along with GFP. After infection, when >95% of neurons express GFP (as monitored by fluorescence microscopy), Nmnat transgene expression was clearly demonstrable by Western analysis (Fig. 1 C). Similarly infected neuronal cultures were treated with 2.5 μm rotenone, and axonal degeneration and cell death were monitored using phase-contrast microscopy and ethidium homodimer exclusion, respectively. Whereas neurons expressing GFP alone manifested extensive axonal degeneration within 72 h, axons of Nmnat-expressing neurons showed no evidence of degradation until 96 h after rotenone addition (Fig. 1 A,B,D). Because the rotenone site of action and Nmnat3 localization are both in the mitochondria, Nmnat3 was used for the remainder of the experiments. The axonal protection afforded by Nmnat proteins in neurons treated with mitochondrial inhibitors, in addition to previous work demonstrating protection against mechanical injury or vincristine exposure, suggests that Nmnat is acting at a point in the degeneration process that is common to multiple types of damage.
To confirm that rotenone inhibition of complex I was specifically responsible for the axonal degeneration in these experiments, we treated DRG cultures with two additional complex I inhibitors, pyridaben (2.5 μm) and fenpyroximate (2.5 μm) (Sherer et al., 2007). We found that both of these complex I inhibitors also caused severe axonal degeneration by 24 h. Furthermore, we found that Nmnat3 expression could clearly prevent this axonal degeneration, just as it had inhibited axonal degeneration caused by rotenone (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Nmnat expression also had a modest protective effect on cell death, although it was less impressive than the protection provided to axons. For example, whereas 51% of neurons expressing GFP alone excluded ethidium homodimers after 72 h of rotenone treatment, 68% of Nmnat3-expressing neurons were viable at 72 h (Fig. 1 E). It should be noted that at 72 h, there is almost complete protection of axons, even though 32% of the cells no longer exclude ethidium homodimers (a late marker of cell death). This dichotomy is similar to results obtained using axotomy or growth factor deprivation, and emphasizes that the mechanism of axonal protection is different from (and perhaps independent of) those involved in the prevention of cell death.
Nmnat3 prevents oxidant-induced axonal degeneration
The accumulation of ROS as a result of mitochondrial inhibition has been implicated in the pathogenesis of several neurodegenerative diseases (Lin and Beal, 2006). Excessive ROS production has multiple detrimental effects, such as DNA damage; oxidation of proteins, which leads to their subsequent inhibition; and oxidation of lipids, which creates toxic products, including 4-hydroxynonenal (Geller et al., 2001; Sherer et al., 2002b; Duarte et al., 2005; Keeney et al., 2006; Ramachandiran et al., 2007). Because it is well known that rotenone induces an increase in ROS, we hypothesized that rotenone damage is initiated by ROS and that Nmnat might protect against ROS damage caused by exposure to exogenous oxidants. To test this idea, we treated DRG neurons infected with lentivirus expressing either GFP alone or Nmnat3 with 100–300 μm hydrogen peroxide (H2O2). In cultures infected with GFP alone, there was significant dose-dependent axonal degeneration after 24 h (Fig. 2 A). In contrast, axonal degeneration from H2O2 exposure was inhibited in neurons expressing Nmnat3 (Fig. 2 A). To confirm the ability of Nmnat to prevent oxidant-induced damage, we also examined the effects of the organic oxidant tert-butyl hydroperoxide (TBHP). In GFP-expressing (control) neurons, TBHP caused extensive axonal degeneration within 48 h (supplemental Fig. 2, available at www.jneurosci.org as supplemental material), whereas neurons expressing Nmnat3 showed no evidence of TBHP-mediated axonal degeneration during this time period (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). These data demonstrate that Nmnat can prevent axonal degeneration triggered by ROS and suggest that Nmnat-mediated protection is accomplished by decreasing the toxicity of ROS produced either endogenously or exogenously.
Figure 2.

Nmnat activity delays axonal degeneration induced by treatment with the oxidant H2O2. A, Quantification of axonal degeneration induced by H2O2 treatment. DRG neurons were treated with the indicated dose of H2O2, and axonal degeneration was monitored after 24 h using phase-contrast microscopy (*p < 0.01 compared with GFP infected cultures; 3 fields per well, 6 wells per condition from duplicate experiments; error bars represent ±SEM). B, Representative images of DRG axons treated with 0–300 μm H2O2 after 24 h and visualized by phase-contrast microscopy. C, Expression of genes encoding antioxidant enzymes. Quantitative RT-PCR analysis was performed using mRNA templates prepared from DRG neurons infected with GFP or Nmnat3 for 10 DIV. The mRNA levels of the antioxidant genes examined were not significantly altered by Nmnat3 (n = 3; each sample represents 5 pooled wells; p > 0.1 in all comparisons; data are from a representative experiment).
Protection of oxidant-mediated axonal damage by Nmnat suggested that it might act via increasing neuronal expression of antioxidant proteins, including superoxide dismutase, glutathione peroxidase, and/or catalase (Thiruchelvam et al., 2005; Fernandez-Gomez et al., 2006; Jung et al., 2007). We used qRT-PCR to compare their mRNA levels in Nmnat3-expressing versus control DRG neurons. However, we found no significant differences in the RNA levels of these genes (Fig. 2 C), suggesting that Nmnat-mediated protection does not involve transcriptional regulation of these enzymes.
Nmnat3 expression decreases axonal ROS levels
The axonal protective activity of Nmnat3 against ROS supplied by exogenous oxidant or generated intracellularly via inhibition of mitochondrial complex I led us to hypothesize that Nmnat expression may reduce rotenone-induced ROS accumulation and/or toxicity. To test this idea, we measured relative ROS levels in axons using a ROS-sensitive fluorescent dye, CM-H2DCFDA, and found that ROS levels were significantly increased in axons 6 h after rotenone treatment (Fig. 3 A,B). The level of ROS continued to rise until 96 h after rotenone treatment, when the severely degenerated state of most axons in the cultures precluded a quantitative analysis of axonal ROS levels (Fig. 3 A). Interestingly, the increase in ROS, which is detectable after 6 h of rotenone treatment, substantially preceded any observable axonal degeneration (Fig. 3 A,B).
Figure 3.

Nmnat expression in DRG neurons decreases rotenone-mediated ROS accumulation. A, Representative images of DRG neurons treated with rotenone or vehicle for 0, 6, 48, or 96 h. ROS levels were monitored using the ROS-sensitive dye CM-H2DCFDA and fluorescence microscopy (left), and axonal degeneration was monitored using phase-contrast microscopy (right). Scale bar, 20 μm. B, Quantification of ROS levels in DRG neurons treated as above. Rotenone-treated DRG neurons expressing Nmnat3 showed decreased CM-H2DCFDA dye fluorescence (i.e., ROS) compared with control neurons. ROS levels do increase in Nmnat3-expressing neurons when axonal degeneration becomes visible (96 h; # p < 0.05 compared with time 0 of each condition; *p < 0.05 compared with control culture at corresponding time point; n = 18 fields from duplicate experiments; data are mean ± SEM). C, Visualization of mitochondrial membrane potential. Neuronal cultures were treated with DMSO (control), FCCP for 1 h, or rotenone for 6 h, and then Mitotracker Red (mitochondrial membrane potential-dependent dye) and calcein AM (intracellular dye to stain axons) were added and visualized by fluorescence microscopy. FCCP decreases the staining of Mitotracker Red, consistent with a drop in mitochondrial membrane potential, whereas the staining in rotenone-treated neurons showed minimal changes compared with control neurons. Shown are representative images from three fields per well, three wells per condition (duplicated experiments). Scale bar, 20 μm.
To determine the effects of Nmnat expression on rotenone-induced ROS accumulation, neurons were infected with Nmnat3 lentivirus that lacks the GFP reporter (to avoid interference with the CM-H2DCFDA fluorescence). The infected neurons were treated with rotenone (or vehicle), and ROS levels were measured 6, 48, or 96 h later. We found that neurons expressing Nmnat3 had statistically significant lower axonal ROS levels after rotenone treatment compared with controls (Fig. 3 A,B) and that this decrease in ROS was correlated with the slower axonal degeneration observed in these neurons.
Mitochondrial dysfunction is often accompanied by a decrease in mitochondrial membrane potential (Nicholls, 2004), and others have suggested that dissipation of mitochondrial membrane potential precedes axonal degeneration and that the degeneration can be blocked if the mitochondrial membrane potential is preserved (Ikegami and Koike, 2003; Nicholls, 2004; Yang et al., 2007). To determine whether rotenone caused a loss of mitochondrial membrane potential, we stained cultures with Mitotracker Red, a mitochondrial potential-dependent dye. However, consistent with other studies with rotenone (Johnson-Cadwell et al., 2007; Marella et al., 2007), we did not observe a collapse of mitochondrial membrane potential after rotenone treatment (6 h) at a time when rotenone-induced increases in ROS are already observed (Fig. 3 C). These observations demonstrate that Nmnat expression decreases ROS accumulation, suggesting that inhibition of ROS accumulation and/or toxicity is a principal mechanism by which Nmnat proteins protect against rotenone-induced axonal degeneration.
Nmnat3 inhibits ROS accumulation in vincristine-treated neurons
In addition to their axonal protective effects after mechanical injury or mitochondrial inhibition, Wlds and Nmnat proteins protect against vincristine-mediated axonal degeneration (Wang et al., 2001b; Araki et al., 2004). Recent studies have shown that vincristine toxicity in cancer cells may be mediated by ROS and that treatment with antioxidants can inhibit vincristine-induced cell death (Groninger et al., 2002; Tsai et al., 2007). We therefore tested whether vincristine treatment resulted in increased axonal ROS and whether Nmnat axonal protection of vincristine-treated neurons was associated with decreased ROS accumulation. We used CM-H2DCFDA fluorescence to monitor ROS levels in DRG neurons treated with 0.04 μm vincristine for 0, 6, 24, and 48 h. Vincristine caused a modest, but significant, increase in ROS after 6 h (Fig. 4 A,B) that continued to increase as axonal degeneration became apparent at 24 and 48 h after vincristine addition. In contrast, Nmnat3-expressing neurons treated with vincristine showed decreased accumulation of ROS at 24 and 48 h (Fig. 4 A,B). Cultures were maintained for up to 96 h to demonstrate that Nmnat3 protected against vincristine-mediated axonal degeneration (Fig. 4 C). Although the primary effect of vincristine is microtubule disruption, these results suggest that it also stimulates ROS production and that ROS generation may play a role in vincristine axonal toxicity. Together, these data suggest that Nmnat-mediated axonal protection against a variety of insults results from its ability to inhibit ROS-mediated damage or signaling. This would imply, as suggested by others, that ROS-stimulated processes could be a point of convergence for multiple pathways that culminate in axonal degeneration (Arundine et al., 2004; Alexandre et al., 2006; Holtz et al., 2006).
Figure 4.

Nmnat expression in DRG neurons inhibits vincristine-mediated increases in ROS levels. A, DRG neurons were treated with vincristine (0.04 μm) or vehicle, and ROS levels were measured at the indicated time points using the ROS-sensitive dye CM-H2DCFDA and visualization with fluorescence (left) and phase-contrast microscopy (right). Scale bar, 20 μm. B, Quantification of ROS levels in DRG neurons demonstrated that neurons expressing Nmnat3 had decreased CM-H2DCFDA fluorescence (i.e., ROS; # p < 0.05 compared with control at time 0; *p < 0.05 compared with vehicle treated control cultures; n = 18 fields from duplicate experiments; mean ± SEM). C, DRG neurons (control vs Nmnat3 expressing) were treated with vincristine for 96 h to demonstrate the axonal protection afforded by Nmnat3. Representative images are shown.
Nmnat3-mediated axonal protection occurs despite decreases in neuronal ATP levels
Cells treated with inhibitors of mitochondrial electron transport have decreased levels of ATP, along with an increased accumulation of ROS. Several studies have indicated that increased ROS levels, and not the loss of ATP, are the proximal causes for neuronal death induced by mitochondrial inhibition (Sherer et al., 2003; Watabe and Nakaki, 2006). However, others have shown that Nmnat1-mediated axonal protection involves the maintenance of axonal ATP levels (Wang et al., 2005). Although dampening the increases in ROS accumulation is clearly an important feature of Nmnat-mediated protection, we also investigated the effects of Nmnat expression on ATP levels in DRG neurons. ATP levels of DRG neurons treated with rotenone for various lengths of time were measured using an ATP-dependent luciferase-based assay. We found that rotenone caused 55.3 and 83.4% decreases in ATP levels in DRG neurons treated for 6 and 96 h, respectively (Fig. 5 A).
Figure 5.

Nmnat3 does not slow rotenone-induced decreases in ATP levels. A, DRG neurons expressing Nmnat3 or GFP were treated with rotenone (2.5 μm) for the indicated times, and ATP levels were determined using a luciferase-based assay. Note that Nmnat3 expression had minimal effects on the rate of ATP loss (*p < 0.05 compared with control at the indicated time; n = 6 wells per condition read in duplicate from duplicate experiments). B, Left, DRG neurons were treated with 1 mm TTX for 6 h with or without 2.5 μm rotenone, and ATP levels were determined. TTX increased ATP in both conditions (*p < 0.05 compared with non-TTX control; **p < 0.05 compared with non-TTX and nonrotenone control; n = 6 wells per condition read in duplicate from 2 independent experiments; error bars represent ±SEM). Right, Axonal degeneration was observed in the absence or presence of TTX at 24 h. C, Left, DRG neurons were treated with 2-DG (20 mm) for 24 h, and ATP levels were measured (*p < 0.05; n ≥ 6; error bars represent ±SEM). Right, DRG neurons were treated with 2-DG (20 mm) for 48 h. Representative phase-contrast images show no evidence of axonal degeneration.
To test whether Nmnat3 promoted axonal protection via prevention of these rotenone-induced decreases in ATP, we treated Nmnat3-expressing DRG neurons with rotenone and monitored ATP levels. Nmnat3-expressing neurons had a small increase in basal ATP levels (control, 100 ± 1.3% ATP; Nmnat3, 118 ± 2.7% ATP). Rotenone treatment caused a rapid fall in ATP levels in both control and Nmnat3-expressing neurons, but ATP levels were always slightly higher in Nmnat3-expressing neurons (Fig. 5 A). We also calculated the rate of ATP loss for both control and Nmnat3-expressing neurons. During the first 6 h, ATP loss was rapid in both GFP- and Nmnat3-expressing neurons (GFP, 9.2 ± 0.39% ATP/h; Nmnat3, 9.6 ± 0.39% ATP/h). The rate of ATP loss slowed at later time points but was very similar in GFP- and Nmnat3-expressing neurons (0.3 ± 0.07% ATP/h and 0.17 ± 0.19% ATP/h, respectively). The initial decrease in ATP is likely a result of the inhibition of oxidative phosphorylation by rotenone, whereas the later and slower losses of ATP are presumably caused by axonal and cellular degeneration and death. Thus, even though axons of Nmnat3-expressing neurons were intact, their ATP levels decreased to a degree similar to those of degenerating axons of control neurons (Fig. 1 B, 2A, 5A).
Although the differences in ATP levels between Nmnat3-expressing and control neurons did not correlate well with the extent of axonal degeneration, ATP levels were slightly higher when Nmnat3 was overexpressed. To further investigate the possibility that ATP levels were key to axonal degeneration, we treated cultures with tetrodotoxin (TTX; a voltage-gated sodium channel blocker). TTX treatment should serve to increase neuronal ATP levels, because it inhibits the influx of sodium and negates the need to expend ATP for maintaining axonal membrane potential via the Na+/K+ ATPase (Stys, 2004). In accord, DRG cultures treated for 6 h with TTX had a 20% increase in ATP (Fig. 5 B). In cultures treated with rotenone and TTX simultaneously, the presence of TTX dramatically decreased the rotenone-stimulated drop in ATP (Fig. 5 B). Despite this effect on ATP levels, TTX had no effect on rotenone-induced axonal degeneration (Fig. 5 B and data not shown). These data indicate that increasing the ATP levels via inhibition of voltage-stimulated sodium influxes does not inhibit axonal degeneration.
To further explore the relationship between ATP levels and axonal degeneration, we treated DRG neurons with 2-deoxyglucose (2-DG), which inhibits glycolysis by blocking glucose uptake and phosphorylation but does not interfere with the mitochondrial electron transport chain or increase ROS levels (Sherer et al., 2003). Treatment with 2-DG resulted in a 37.0 ± 5.3% decrease in ATP after 24 h, but there was no evidence of axonal degeneration or increased ROS levels after 48 h (Fig. 5 C and data not shown). Even long-term treatment of DRG neurons with 2-DG (18 d) did not cause axonal degeneration, although the low ATP level observed at 24 h remained throughout the treatment period (data not shown).
Antioxidants inhibit rotenone-mediated axonal degeneration without altering ATP levels.
The suppression of ROS accumulation by Nmnat proteins is reminiscent of that observed in cells treated with antioxidants, whose utility in preventing axonal degeneration secondary to mitochondrial inhibition has been suggested by others (Sherer et al., 2003, 2007; Testa et al., 2005). To determine whether antioxidants can protect against rotenone-induced axonal degeneration, we administered vitamin E (α-tocopherol) in doses from 125 to 1000 μm (corresponding to 0.052–0.47 IU/ml) to DRG neurons treated with rotenone and assessed axonal degeneration. We found that whereas axonal degeneration in control cultures (rotenone alone) began within 24–48 h, axons from neurons treated with vitamin E remained intact for up to 120 h (Fig. 6 A,B). To determine whether vitamin E also affected ATP levels, perhaps by directly blocking the actions of rotenone, we measured ATP levels at various times after rotenone treatment. As previously observed in Nmnat3-expressing neurons, vitamin E did not significantly suppress the initial loss of ATP (Fig. 6 C); however, as axons began to break down (in the untreated neurons), a difference in ATP levels was observed.
Figure 6.
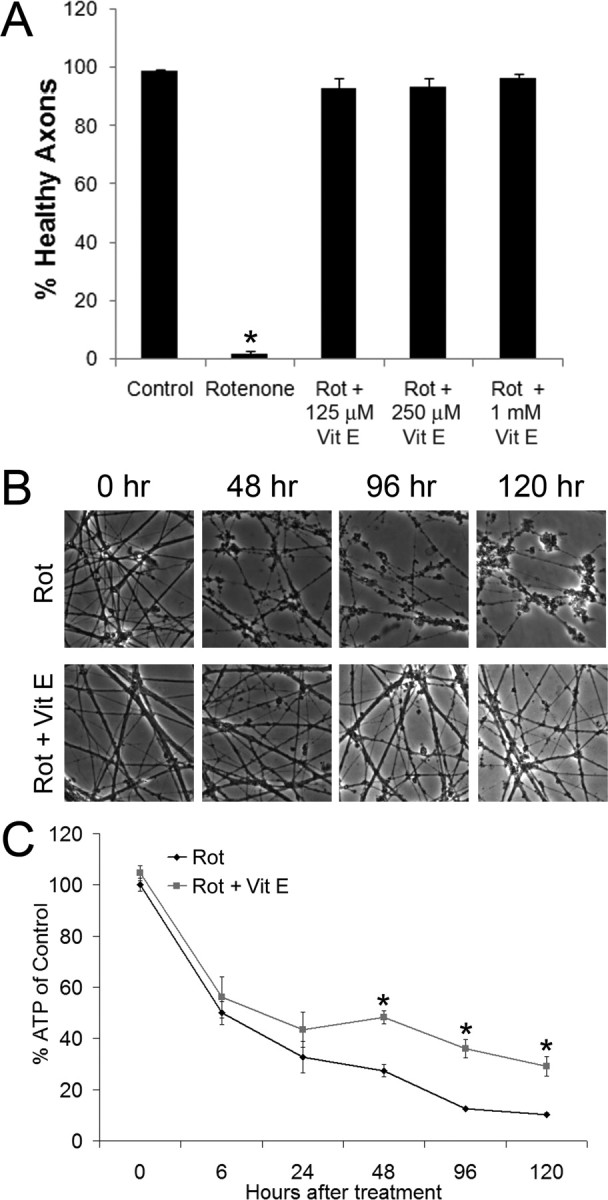
Vitamin E prevents axonal degeneration but does not affect initial ATP losses. A, DRG neurons were treated with rotenone (2.5 μm) and the indicated doses of vitamin E. Axonal degeneration was quantified after 6 d of treatment using images obtained by phase-contrast microscopy (*p < 0.05 compared with control; n = 6 wells per condition from duplicate experiments; error bars represent ±SEM). B, DRG neurons were treated with rotenone (2.5 μm) in the presence or absence of vitamin E (1 mm), and axonal degeneration was assessed by phase-contrast microscopy. Whereas axons in cultures treated with rotenone only were visibly damaged by 48 h, neurons treated with rotenone and vitamin E were intact at 120 h. C, DRG cultures were treated with rotenone (2.5 μm) in the presence or absence of vitamin E (1 mm), and ATP levels were measured at the indicated time points. Vitamin E failed to prevent the initial loss of ATP (*p < 0.05; n ≥ 6 wells from duplicate experiments; error bars represent ±SEM).
Together, these results indicate that a decrease in ATP levels is insufficient to cause axonal degeneration. Furthermore, the degree of ATP loss, unlike ROS levels, did not correlate with axonal degeneration temporally or with Nmnat3 expression, suggesting that inhibition of ROS-initiated axonal degeneration rather than maintenance of ATP levels is the primary driver of Nmnat3-mediated protection against rotenone-induced axonal degeneration.
Discussion
Elucidating the mechanism and role of axonal degeneration in neurodegeneration is an important step in developing strategies for preventing and treating neurodegenerative diseases. We recently demonstrated that Nmnat1 and Nmnat3 expression can prevent axonal degeneration against a variety of insults, including microtubule disruption and mechanical injury (Sasaki et al., 2006). This protection requires the enzymatic activity of Nmnat, which converts nicotinamide mononucleotide to nicotinamide adenine dinucleotide (NAD+) (Araki et al., 2004; Berger et al., 2005), although recent studies in Drosophila neurodegeneration have suggested that additional Nmnat activities may also be important for synaptic protection (Zhai et al., 2006). The importance of mitochondrial dysfunction in neurological disorders involving axonal degeneration, and the ability of the Wlds mutation to protect against MPTP-induced parkinsonism (Hasbani and O'Malley, 2006), suggested that Nmnat proteins might prevent degeneration caused by mitochondrial inhibition. Here, we demonstrated that Nmnat expression robustly inhibits rotenone-induced degeneration of DRG axons and provides modest protection to the neuronal soma. In exploring the mechanism of this protection, we found that Nmnat expression did not affect the rate of decrease in ATP levels caused by rotenone-mediated mitochondrial inhibition. Furthermore, we found that 50–60% losses of ATP were neither necessary nor sufficient for axonal degeneration. However, Nmnat proteins inhibited axonal degeneration caused by external oxidants and decreased the accumulation of axonal ROS during rotenone and vincristine treatment, suggesting that they protect axons by reducing ROS accumulation or toxicity.
Interestingly, studies with the acute 6-OHDA and MPTP rodent PD models showed that the Wlds mutation provided robust protection against TH+ fiber loss but little or no protection of dopaminergic neuronal cell bodies (Sajadi et al., 2004; Hasbani and O'Malley, 2006). These in vivo results are consistent with our findings and suggest that Nmnat-mediated protection is focused primarily on axons. The effects of Wlds have not been tested in the chronic rotenone model of PD, which more closely mimics the degenerative progression observed in PD patients, because this model only works in rats (Hornykiewicz, 1966; Betarbet et al., 2000; Richter et al., 2007); however, our results suggest that increasing Nmnat expression in this model may rescue axonal function and thereby prevent dopaminergic neuron death.
ROS has been implicated in the pathogenesis of neurodegenerative diseases, in particular PD, as well as in the deficits that occur in normal aging (Lin and Beal, 2006). For example, complex I inhibition and oxidative damage to mitochondrial proteins have been observed in brains of PD patients, changes that can be replicated by treatment of isolated mitochondria with rotenone (Keeney et al., 2006). Indeed, antioxidants provide protection for dopaminergic neurons in the MPTP, 6-OHDA, rotenone, and genetic models of PD in vivo and in vitro (Schulz et al., 1995; Matthews et al., 1999; Bahat-Stroomza et al., 2005; Testa et al., 2005; Wang et al., 2006). In addition, RNAi-mediated inhibition of Drosophila PINK1, a kinase mutated in familial PD, resulted in losses of dopaminergic neurons and eye defects. These deficits could be rescued by expression of superoxide dismutase or treatment with the antioxidant vitamin E, suggesting that ROS was responsible for the damage (Wang et al., 2006). Moreover, organotypic midbrain cultures treated with rotenone display loss of TH+ fibers, increased protein oxidation, and to a much lesser extent cell body shrinkage; deficits that are consistent with an axonopathy in which axonal degeneration precedes cell body loss. Similar to our findings, these deficits appear to involve ROS, because they could be prevented by coadministration with vitamin E (Testa et al., 2005). Together, these results suggest that halting ROS-induced damage can prevent the damage caused by mitochondrial dysfunction.
Inhibition of the electron transport chain can cause apoptosis in human dopaminergic cells (Watabe and Nakaki, 2006). Further study showed that ATP loss could not account for the induction of apoptosis in this model; instead, ROS production was necessary and sufficient to promote apoptosis of these cells. Axonal degeneration was not directly addressed in that study; however, the results are consistent with the idea that moderate mitochondrial inhibition causes damage primarily via excess ROS production rather than through abnormalities associated with energetic deficits (i.e., decreased ATP) (Bao et al., 2005). On the other hand, some studies have suggested that preventing axonal ATP loss is a crucial aspect of axonal protection by Nmnat (Ikegami and Koike, 2003; Wang et al., 2005). However, our results indicate that the rate of ATP loss is comparable in control and Nmnat-expressing neurons as well as vitamin E-treated neurons, suggesting that this is not the mechanism by which Nmnat protects against axonal degeneration. Furthermore, neurons treated with 2-DG for prolonged periods of time have up to 40% lower levels of ATP but show no evidence of axonal degeneration, providing additional support for the idea that decreases in ATP levels are unlikely to be the only driver of axonal degeneration. Finally, we showed that treatment with tetrodotoxin could mitigate rotenone-induced decreases in ATP yet could not prevent axonal degeneration. It appears that moderate ATP losses are insufficient for axonal degeneration; however, lower ATP levels could affect axonal stability by increasing neuronal susceptibility to ROS-mediated damage.
Others have shown that the Wlds protein maintains the mitochondrial membrane potential in vinblastine-treated SCG neurons, and they suggested that this may play a role in Wlds-mediated axonal protection (Ikegami and Koike, 2003). Our results indicate that rotenone causes an increase in ROS without a collapse of the mitochondrial membrane potential. It is possible that rotenone does cause small changes in the mitochondrial membrane potential, but the physiological significance of small changes in mitochondrial membrane potential is unclear (Nicholls, 2004; Nicholls et al., 2007).
We cannot exclude the possibility that ROS is simply a trigger to initiate axonal degeneration, much like axonal severing, and that Nmnat protects axons by blocking downstream targets of this signaling cascade. It is also possible that ROS accumulation is secondary to the initiation of this degeneration pathway; however, this is less likely, because axonal degeneration induced by exogenous oxidants can also be blocked by Nmnat expression.
Furthermore, Nmnat expression can protect axons from diverse insults, suggesting that the axonal damage in many of these paradigms may ultimately involve ROS, or ROS-mediated signaling. Although mitochondria are considered to be the major source of ROS, other sources of ROS production include membrane-bound NADPH-oxidase, a source of oxidative products induced in neurons by signaling intermediates, and xanthine oxidase, which produces ROS in neurons in response to hypoxia and glucose deprivation (Ibi et al., 2006; Abramov et al., 2007). Other insults not directly linked to mitochondrial dysfunction, including taxol, vincristine, and mechanical injury, also induce ROS and neurodegeneration (Hall, 1987; Groninger et al., 2002; Arundine et al., 2004; Alexandre et al., 2006; Holtz et al., 2006; Tsai et al., 2007).
The mechanism by which Nmnat proteins prevent the accumulation and/or toxicity of ROS is unclear. It does not appear that transcriptional changes for previously identified antioxidant genes are involved (Gillingwater et al., 2006). Interestingly, the production and usage of NAD+ and its metabolites can play important roles in antioxidant responses (Mack et al., 2001). One possibility is that Nmnat proteins increase NAD+ availability without increasing NADH breakdown via oxidation, thus providing more NADH for conversion to NADPH, a cofactor for many ROS-scavenging enzymes (Mazzio and Soliman, 2003). Alternatively, Nmnat proteins may have additional activities, such as antioxidant activity, much like peroxidases or dismutases. An undiscovered function of Nmnat, in addition to its role in NAD+ biosynthesis, could explain why Nmnat enzymatic activity is not required to block vacuole formation and neurodegeneration in the Drosophila retina (Zhai et al., 2006). However, this is inconsistent with observations indicating that NAD+ and NAD+ precursors as well as other enzymes involved in NAD+ biosynthesis can also provide axonal protection after axotomy (Araki et al., 2004; Wang et al., 2005; Sasaki et al., 2006). Identifying how increased Nmnat expression limits ROS damage may yield clues as to how it promotes axonal protection in general, as well as opening up new avenues for understanding how ROS causes axonal degeneration.
Footnotes
This work was supported by National Institutes of Health (NIH) Neuroscience Blueprint Core Grant NS057105 to Washington University, the Hope Center for Neurological Disorders, the Muscular Dystrophy Association (J.M.), and NIH Grants AG13730 and NS39358 (J.M.). We thank Eugene Johnson, Biplab Dasgupta, Albert Misko, Jason Gustin, Sanjay Jain, Robert Baloh, Robert Schmidt, Yo Sasaki, Bhupinder Vohra, and Elizabeth Ryu for fruitful discussions and reading of this manuscript and Amber Neilson for assistance with virus production.
J.M. and Washington University have a financial interest in Sirtris Pharmaceuticals. Sirtris Pharmaceuticals did not support this work.
References
- Abramov AY, Scorziello A, Duchen MR. Three distinct mechanisms generate oxygen free radicals in neurons and contribute to cell death during anoxia and reoxygenation. J Neurosci. 2007;27:1129–1138. doi: 10.1523/JNEUROSCI.4468-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandre J, Batteux F, Nicco C, Chéreau C, Laurent A, Guillevin L, Weill B, Goldwasser F. Accumulation of hydrogen peroxide is an early and crucial step for paclitaxel-induced cancer cell death both in vitro and in vivo . Int J Cancer. 2006;119:41–48. doi: 10.1002/ijc.21685. [DOI] [PubMed] [Google Scholar]
- Araki T, Sasaki Y, Milbrandt J. Increased nuclear NAD biosynthesis and SIRT1 activation prevent axonal degeneration. Science. 2004;305:1010–1013. doi: 10.1126/science.1098014. [DOI] [PubMed] [Google Scholar]
- Arundine M, Aarts M, Lau A, Tymianski M. Vulnerability of central neurons to secondary insults after in vitro mechanical stretch. J Neurosci. 2004;24:8106–8123. doi: 10.1523/JNEUROSCI.1362-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ascherio A, Chen H, Weisskopf MG, O'Reilly E, McCullough ML, Calle EE, Schwarzschild MA, Thun MJ. Pesticide exposure and risk for Parkinson's disease. Ann Neurol. 2006;60:197–203. doi: 10.1002/ana.20904. [DOI] [PubMed] [Google Scholar]
- Bahat-Stroomza M, Gilgun-Sherki Y, Offen D, Panet H, Saada A, Krool-Galron N, Barzilai A, Atlas D, Melamed E. A novel thiol antioxidant that crosses the blood brain barrier protects dopaminergic neurons in experimental models of Parkinson's disease. Eur J Neurosci. 2005;21:637–646. doi: 10.1111/j.1460-9568.2005.03889.x. [DOI] [PubMed] [Google Scholar]
- Baloh RH, Schmidt RE, Pestronk A, Milbrandt J. Altered axonal mitochondrial transport in the pathogenesis of Charcot-Marie-Tooth disease from mitofusin 2 mutations. J Neurosci. 2007;27:422–430. doi: 10.1523/JNEUROSCI.4798-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao L, Avshalumov MV, Rice ME. Partial mitochondrial inhibition causes striatal dopamine release suppression and medium spiny neuron depolarization via H2O2 elevation, not ATP depletion. J Neurosci. 2005;25:10029–10040. doi: 10.1523/JNEUROSCI.2652-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger F, Lau C, Dahlmann M, Ziegler M. Subcellular compartmentation and differential catalytic properties of the three human nicotinamide mononucleotide adenylyltransferase isoforms. J Biol Chem. 2005;280:36334–36341. doi: 10.1074/jbc.M508660200. [DOI] [PubMed] [Google Scholar]
- Betarbet R, Sherer TB, MacKenzie G, Garcia-Osuna M, Panov AV, Greenamyre JT. Chronic systemic pesticide exposure reproduces features of Parkinson's disease. Nat Neurosci. 2000;3:1301–1306. doi: 10.1038/81834. [DOI] [PubMed] [Google Scholar]
- Brown TP, Rumsby PC, Capleton AC, Rushton L, Levy LS. Pesticides and Parkinson's disease—is there a link? Environ Health Perspect. 2006;114:156–164. doi: 10.1289/ehp.8095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang LK, Schmidt RE, Johnson EM., Jr Alternating metabolic pathways in NGF-deprived sympathetic neurons affect caspase-independent death. J Cell Biol. 2003;162:245–256. doi: 10.1083/jcb.200302109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman M. Axon degeneration mechanisms: commonality amid diversity. Nat Rev Neurosci. 2005;6:889–898. doi: 10.1038/nrn1788. [DOI] [PubMed] [Google Scholar]
- Conforti L, Tarlton A, Mack TG, Mi W, Buckmaster EA, Wagner D, Perry VH, Coleman MP. A Ufd2/D4Cole1e chimeric protein and overexpression of Rbp7 in the slow Wallerian degeneration (WldS) mouse. Proc Natl Acad Sci USA. 2000;97:11377–11382. doi: 10.1073/pnas.97.21.11377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conforti L, Fang G, Beirowski B, Wang MS, Sorci L, Asress S, Adalbert R, Silva A, Bridge K, Huang XP, Magni G, Glass JD, Coleman MP. NAD(+) and axon degeneration revisited: Nmnat1 cannot substitute for Wld(S) to delay Wallerian degeneration. Cell Death Differ. 2007;14:116–127. doi: 10.1038/sj.cdd.4401944. [DOI] [PubMed] [Google Scholar]
- Duarte AI, Santos MS, Oliveira CR, Rego AC. Insulin neuroprotection against oxidative stress in cortical neurons—involvement of uric acid and glutathione antioxidant defenses. Free Radic Biol Med. 2005;39:876–889. doi: 10.1016/j.freeradbiomed.2005.05.002. [DOI] [PubMed] [Google Scholar]
- Fernandez-Gomez FJ, Pastor MD, Garcia-Martinez EM, Melero-Fernandez de Mera R, Gou-Fabregas M, Gomez-Lazaro M, Calvo S, Soler RM, Galindo MF, Jordan J. Pyruvate protects cerebellar granular cells from 6-hydroxydopamine-induced cytotoxicity by activating the Akt signaling pathway and increasing glutathione peroxidase expression. Neurobiol Dis. 2006;24:296–307. doi: 10.1016/j.nbd.2006.07.005. [DOI] [PubMed] [Google Scholar]
- Ferri A, Sanes JR, Coleman MP, Cunningham JM, Kato AC. Inhibiting axon degeneration and synapse loss attenuates apoptosis and disease progression in a mouse model of motoneuron disease. Curr Biol. 2003;13:669–673. doi: 10.1016/s0960-9822(03)00206-9. [DOI] [PubMed] [Google Scholar]
- Geller HM, Cheng K-Y, Goldsmith NK, Romero AA, Zhang A-L, Morris EJ, Grandison L. Oxidative stress mediates neuronal DNA damage and apoptosis in response to cytosine arabinoside. J Neurochem. 2001;78:265–275. doi: 10.1046/j.1471-4159.2001.00395.x. [DOI] [PubMed] [Google Scholar]
- Gillingwater TH, Ribchester RR. Compartmental neurodegeneration and synaptic plasticity in the Wld(s) mutant mouse. J Physiol (Lond) 2001;534:627–639. doi: 10.1111/j.1469-7793.2001.00627.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillingwater TH, Haley JE, Ribchester RR, Horsburgh K. Neuroprotection after transient global cerebral ischemia in Wld(s) mutant mice. J Cereb Blood Flow Metab. 2004;24:62–66. doi: 10.1097/01.WCB.0000095798.98378.34. [DOI] [PubMed] [Google Scholar]
- Gillingwater TH, Wishart TM, Chen PE, Haley JE, Robertson K, MacDonald SH, Middleton S, Wawrowski K, Shipston MJ, Melmed S, Wyllie DJ, Skehel PA, Coleman MP, Ribchester RR. The neuroprotective WldS gene regulates expression of PTTG1 and erythroid differentiation regulator 1-like gene in mice and human cells. Hum Mol Genet. 2006;15:625–635. doi: 10.1093/hmg/ddi478. [DOI] [PubMed] [Google Scholar]
- Gorell JM, Johnson CC, Rybicki BA, Peterson EL, Richardson RJ. The risk of Parkinson's disease with exposure to pesticides, farming, well water, and rural living. Neurology. 1998;50:1346–1350. doi: 10.1212/wnl.50.5.1346. [DOI] [PubMed] [Google Scholar]
- Gorell JM, Peterson EL, Rybicki BA, Johnson CC. Multiple risk factors for Parkinson's disease. J Neurol Sci. 2004;217:169–174. doi: 10.1016/j.jns.2003.09.014. [DOI] [PubMed] [Google Scholar]
- Groninger E, Meeuwsen-De Boer GJ, De Graaf SS, Kamps WA, De Bont ES. Vincristine induced apoptosis in acute lymphoblastic leukaemia cells: a mitochondrial controlled pathway regulated by reactive oxygen species? Int J Oncol. 2002;21:1339–1345. doi: 10.3892/ijo.21.6.1339. [DOI] [PubMed] [Google Scholar]
- Hall ED. Intensive anti-oxidant pretreatment retards motor nerve degeneration. Brain Res. 1987;413:175–178. doi: 10.1016/0006-8993(87)90166-1. [DOI] [PubMed] [Google Scholar]
- Hasbani DM, O'Malley KL. Wld(S) mice are protected against the parkinsonian mimetic MPTP. Exp Neurol. 2006;202:93–99. doi: 10.1016/j.expneurol.2006.05.017. [DOI] [PubMed] [Google Scholar]
- Holtz WA, Turetzky JM, Jong YJ, O'Malley KL. Oxidative stress-triggered unfolded protein response is upstream of intrinsic cell death evoked by parkinsonian mimetics. J Neurochem. 2006;99:54–69. doi: 10.1111/j.1471-4159.2006.04025.x. [DOI] [PubMed] [Google Scholar]
- Hoopfer ED, McLaughlin T, Watts RJ, Schuldiner O, O'Leary DD, Luo L. Wld(s) protection distinguishes axon degeneration following injury from naturally occurring developmental pruning. Neuron. 2006;50:883–895. doi: 10.1016/j.neuron.2006.05.013. [DOI] [PubMed] [Google Scholar]
- Hornykiewicz O. Dopamine (3-hydroxytyramine) and brain function. Pharmacol Rev. 1966;18:925–964. [PubMed] [Google Scholar]
- Ibi M, Katsuyama M, Fan C, Iwata K, Nishinaka T, Yokoyama T, Yabe-Nishimura C. NOX1/NADPH oxidase negatively regulates nerve growth factor-induced neurite outgrowth. Free Radic Biol Med. 2006;40:1785–1795. doi: 10.1016/j.freeradbiomed.2006.01.009. [DOI] [PubMed] [Google Scholar]
- Ikegami K, Koike T. Non-apoptotic neurite degeneration in apoptotic neuronal death: pivotal role of mitochondrial function in neurites. Neuroscience. 2003;122:617–626. doi: 10.1016/j.neuroscience.2003.08.057. [DOI] [PubMed] [Google Scholar]
- Johnson-Cadwell LI, Jekabsons MB, Wang A, Polster BM, Nicholls DG. ‘Mild Uncoupling’ does not decrease mitochondrial superoxide levels in cultured cerebellar granule neurons but decreases spare respiratory capacity and increases toxicity to glutamate and oxidative stress. J Neurochem. 2007;101:1619–1631. doi: 10.1111/j.1471-4159.2007.04516.x. [DOI] [PubMed] [Google Scholar]
- Jung TW, Lee JY, Shim WS, Kang ES, Kim SK, Ahn CW, Lee HC, Cha BS. Rosiglitazone protects human neuroblastoma SH-SY5Y cells against MPP+ induced cytotoxicity via inhibition of mitochondrial dysfunction and ROS production. J Neurol Sci. 2007;253:53–60. doi: 10.1016/j.jns.2006.11.020. [DOI] [PubMed] [Google Scholar]
- Keeney PM, Xie J, Capaldi RA, Bennett JP., Jr Parkinson's disease brain mitochondrial complex I has oxidatively damaged subunits and is functionally impaired and misassembled. J Neurosci. 2006;26:5256–5264. doi: 10.1523/JNEUROSCI.0984-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langston JW, Ballard P, Tetrud JW, Irwin I. Chronic parkinsonism in humans due to a product of meperidine-analog synthesis. Science. 1983;219:979–980. doi: 10.1126/science.6823561. [DOI] [PubMed] [Google Scholar]
- Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443:787–795. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- Lunn ER, Perry VH, Brown MC, Rosen H, Gordon S. Absence of Wallerian degeneration does not hinder regeneration in peripheral nerve. Eur J Neurosci. 1989;1:27–33. doi: 10.1111/j.1460-9568.1989.tb00771.x. [DOI] [PubMed] [Google Scholar]
- Macdonald JM, Beach MG, Porpiglia E, Sheehan AE, Watts RJ, Freeman MR. The Drosophila cell corpse engulfment receptor draper mediates glial clearance of severed axons. Neuron. 2006;50:869–881. doi: 10.1016/j.neuron.2006.04.028. [DOI] [PubMed] [Google Scholar]
- Mack TG, Reiner M, Beirowski B, Mi W, Emanuelli M, Wagner D, Thomson D, Gillingwater T, Court F, Conforti L, Fernando FS, Tarlton A, Andressen C, Addicks K, Magni G, Ribchester RR, Perry VH, Coleman MP. Wallerian degeneration of injured axons and synapses is delayed by a Ube4b/Nmnat chimeric gene. Nat Neurosci. 2001;4:1199–1206. doi: 10.1038/nn770. [DOI] [PubMed] [Google Scholar]
- Marella M, Seo BB, Matsuno-Yagi A, Yagi T. Mechanism of cell death caused by complex I defects in a rat dopaminergic cell line. J Biol Chem. 2007;282:24146–24156. doi: 10.1074/jbc.M701819200. [DOI] [PubMed] [Google Scholar]
- Martin LJ. Mitochondriopathy in Parkinson disease and amyotrophic lateral sclerosis. J Neuropathol Exp Neurol. 2006;65:1103–1110. doi: 10.1097/01.jnen.0000248541.05552.c4. [DOI] [PubMed] [Google Scholar]
- Matthews RT, Klivenyi P, Mueller G, Yang L, Wermer M, Thomas CE, Beal MF. Novel free radical spin traps protect against malonate and MPTP neurotoxicity. Exp Neurol. 1999;157:120–126. doi: 10.1006/exnr.1999.7045. [DOI] [PubMed] [Google Scholar]
- Mazzio EA, Soliman KF. Cytoprotection of pyruvic acid and reduced β-nicotinamide adenine dinucleotide against hydrogen peroxide toxicity in neuroblastoma cells. Neurochem Res. 2003;28:733–741. doi: 10.1023/a:1022813817743. [DOI] [PubMed] [Google Scholar]
- Mi W, Beirowski B, Gillingwater TH, Adalbert R, Wagner D, Grumme D, Osaka H, Conforti L, Arnhold S, Addicks K, Wada K, Ribchester RR, Coleman MP. The slow Wallerian degeneration gene, WldS, inhibits axonal spheroid pathology in gracile axonal dystrophy mice. Brain. 2005;128:405–416. doi: 10.1093/brain/awh368. [DOI] [PubMed] [Google Scholar]
- Nicholls DG. Mitochondrial dysfunction and glutamate excitotoxicity studied in primary neuronal cultures. Curr Mol Med. 2004;4:149–177. doi: 10.2174/1566524043479239. [DOI] [PubMed] [Google Scholar]
- Nicholls DG, Johnson-Cadwell L, Vesce S, Jekabsons M, Yadava N. Bioenergetics of mitochondria in cultured neurons and their role in glutamate excitotoxicity. J Neurosci Res. 2007;85:3206–3212. doi: 10.1002/jnr.21290. [DOI] [PubMed] [Google Scholar]
- Priyadarshi A, Khuder SA, Schaub EA, Shrivastava S. A meta-analysis of Parkinson's disease and exposure to pesticides. Neurotoxicology. 2000;21:435–440. [PubMed] [Google Scholar]
- Priyadarshi A, Khuder SA, Schaub EA, Priyadarshi SS. Environmental risk factors and Parkinson's disease: a metaanalysis. Environ Res. 2001;86:122–127. doi: 10.1006/enrs.2001.4264. [DOI] [PubMed] [Google Scholar]
- Raff MC, Whitmore AV, Finn JT. Axonal self-destruction and neurodegeneration. Science. 2002;296:868–871. doi: 10.1126/science.1068613. [DOI] [PubMed] [Google Scholar]
- Ramachandiran S, Hansen JM, Jones DP, Richardson JR, Miller GW. Divergent mechanisms of paraquat, MPP+, and rotenone toxicity: oxidation of thioredoxin and caspase-3 activation. Toxicol Sci. 2007;95:163–171. doi: 10.1093/toxsci/kfl125. [DOI] [PubMed] [Google Scholar]
- Richter F, Hamann M, Richter A. Chronic rotenone treatment induces behavioral effects but no pathological signs of parkinsonism in mice. J Neurosci Res. 2007;85:681–691. doi: 10.1002/jnr.21159. [DOI] [PubMed] [Google Scholar]
- Sajadi A, Schneider BL, Aebischer P. Wlds-mediated protection of dopaminergic fibers in an animal model of Parkinson disease. Curr Biol. 2004;14:326–330. doi: 10.1016/j.cub.2004.01.053. [DOI] [PubMed] [Google Scholar]
- Samsam M, Mi W, Wessig C, Zielasek J, Toyka KV, Coleman MP, Martini R. The Wlds mutation delays robust loss of motor and sensory axons in a genetic model for myelin-related axonopathy. J Neurosci. 2003;23:2833–2839. doi: 10.1523/JNEUROSCI.23-07-02833.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki Y, Araki T, Milbrandt J. Stimulation of nicotinamide adenine dinucleotide biosynthetic pathways delays axonal degeneration after axotomy. J Neurosci. 2006;26:8484–8491. doi: 10.1523/JNEUROSCI.2320-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz JB, Henshaw DR, Matthews RT, Beal MF. Coenzyme Q10 and nicotinamide and a free radical spin trap protect against MPTP neurotoxicity. Exp Neurol. 1995;132:279–283. doi: 10.1016/0014-4886(95)90033-0. [DOI] [PubMed] [Google Scholar]
- Semchuk KM, Love EJ, Lee RG. Parkinson's disease and exposure to agricultural work and pesticide chemicals. Neurology. 1992;42:1328–1335. doi: 10.1212/wnl.42.7.1328. [DOI] [PubMed] [Google Scholar]
- Sherer TB, Betarbet R, Greenamyre JT. Environment, mitochondria, and Parkinson's disease. Neuroscientist. 2002a;8:192–197. doi: 10.1177/1073858402008003004. [DOI] [PubMed] [Google Scholar]
- Sherer TB, Betarbet R, Stout AK, Lund S, Baptista M, Panov AV, Cookson MR, Greenamyre JT. An in vitro model of Parkinson's disease: linking mitochondrial impairment to altered α-synuclein metabolism and oxidative damage. J Neurosci. 2002b;22:7006–7015. doi: 10.1523/JNEUROSCI.22-16-07006.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherer TB, Betarbet R, Testa CM, Seo BB, Richardson JR, Kim JH, Miller GW, Yagi T, Matsuno-Yagi A, Greenamyre JT. Mechanism of toxicity in rotenone models of Parkinson's disease. J Neurosci. 2003;23:10756–10764. doi: 10.1523/JNEUROSCI.23-34-10756.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherer TB, Richardson JR, Testa CM, Seo BB, Panov AV, Yagi T, Matsuno-Yagi A, Miller GW, Greenamyre JT. Mechanism of toxicity of pesticides acting at complex I: relevance to environmental etiologies of Parkinson's disease. J Neurochem. 2007;100:1469–1479. doi: 10.1111/j.1471-4159.2006.04333.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stys PK. White matter injury mechanisms. Curr Mol Med. 2004;4:113–130. doi: 10.2174/1566524043479220. [DOI] [PubMed] [Google Scholar]
- Stys PK, Waxman SG, Ransom BR. Ionic mechanisms of anoxic injury in mammalian CNS white matter: role of Na+ channels and Na+–Ca2+ exchanger. J Neurosci. 1992;12:430–439. doi: 10.1523/JNEUROSCI.12-02-00430.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Testa CM, Sherer TB, Greenamyre JT. Rotenone induces oxidative stress and dopaminergic neuron damage in organotypic substantia nigra cultures. Brain Res Mol Brain Res. 2005;134:109–118. doi: 10.1016/j.molbrainres.2004.11.007. [DOI] [PubMed] [Google Scholar]
- Thiruchelvam M, Prokopenko O, Cory-Slechta DA, Richfield EK, Buckley B, Mirochnitchenko O. Overexpression of superoxide dismutase or glutathione peroxidase protects against the paraquat + maneb-induced Parkinson disease phenotype. J Biol Chem. 2005;280:22530–22539. doi: 10.1074/jbc.M500417200. [DOI] [PubMed] [Google Scholar]
- Tsai SY, Sun NK, Lu HP, Cheng ML, Chao CC. Involvement of reactive oxygen species in multidrug resistance of a vincristine-selected lymphoblastoma. Cancer Sci. 2007;98:1206–1214. doi: 10.1111/j.1349-7006.2007.00513.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Qian L, Xiong H, Liu J, Neckameyer WS, Oldham S, Xia K, Wang J, Bodmer R, Zhang Z. Antioxidants protect PINK1-dependent dopaminergic neurons in Drosophila . Proc Natl Acad Sci USA. 2006;103:13520–13525. doi: 10.1073/pnas.0604661103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Zhai Q, Chen Y, Lin E, Gu W, McBurney MW, He Z. A local mechanism mediates NAD-dependent protection of axon degeneration. J Cell Biol. 2005;170:349–355. doi: 10.1083/jcb.200504028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang MS, Wu Y, Culver DG, Glass JD. The gene for slow Wallerian degeneration (Wld(s)) is also protective against vincristine neuropathy. Neurobiol Dis. 2001a;8:155–161. doi: 10.1006/nbdi.2000.0334. [DOI] [PubMed] [Google Scholar]
- Wang MS, Fang G, Culver DG, Davis AA, Rich MM, Glass JD. The WldS protein protects against axonal degeneration: a model of gene therapy for peripheral neuropathy. Ann Neurol. 2001b;50:773–779. doi: 10.1002/ana.10039. [DOI] [PubMed] [Google Scholar]
- Watabe M, Nakaki T. ATP depletion does not account for apoptosis induced by inhibition of mitochondrial electron transport chain in human dopaminergic cells. Neuropharmacology. 2006;52:536–541. doi: 10.1016/j.neuropharm.2006.07.037. [DOI] [PubMed] [Google Scholar]
- Yang Y, Fukui K, Koike T, Zheng X. Induction of autophagy in neurite degeneration of mouse superior cervical ganglion neurons. Eur J Neurosci. 2007;26:2979–2988. doi: 10.1111/j.1460-9568.2007.05914.x. [DOI] [PubMed] [Google Scholar]
- Zhai RG, Cao Y, Hiesinger PR, Zhou Y, Mehta SQ, Schulze KL, Verstreken P, Bellen HJ. Drosophila NMNAT maintains neural integrity independent of its NAD synthesis activity. PLoS Biol. 2006;4:e416. doi: 10.1371/journal.pbio.0040416. [DOI] [PMC free article] [PubMed] [Google Scholar]