

Abstract
Human immunodeficiency virus (HIV)-1 infection of the CNS produces changes in dendritic morphology that correlate with cognitive decline in patients with HIV-1 associated dementia (HAD). Here, we investigated the effects of HIV-1 transactivator of transcription (Tat), a protein released by virus-infected cells, on synapses between hippocampal neurons using an imaging-based assay that quantified clusters of the scaffolding protein postsynaptic density 95 fused to green fluorescent protein (PSD95–GFP). Tat (24 h) decreased the number of PSD95–GFP puncta by 50 ± 7%. The decrease was concentration-dependent (EC50 = 6 ± 2 ng/ml) and preceded cell death. Tat acted via the low-density lipoprotein receptor-related protein (LRP) because the specific LRP blocker, receptor associated protein (RAP), prevented the Tat-induced decrease in the number of PSD95–GFP puncta. Ca2+ influx through the NMDA receptor was necessary for Tat-induced synapse loss. Expression of an ubiquitin ligase inhibitor protected synapses, implicating the ubiquitin–proteasome pathway. In contrast to synapse loss, Tat induced cell death (48 h) required activation of nitric oxide synthase. The ubiquitin ligase-inhibitor nutlin-3 prevented synapse loss but not cell death induced by Tat. Thus, the pathways diverged, consistent with the hypothesis that synapse loss is a mechanism to reduce excess excitatory input rather than a symptom of the neuron's demise. Furthermore, application of RAP to cultures treated with Tat for 16 h reversed synapse loss. These results suggest that the impaired network function and decreased neuronal survival produced by Tat involve distinct mechanisms and that pharmacologic targets, such as LRP, might prove useful in restoring function in HAD patients.
Keywords: Tat, LRP, PSD95, proteasome, NeuroAIDS, neurotoxicity
Introduction
Human immunodeficiency virus (HIV)-associated dementia (HAD) is one of the most important complications associated with AIDS because this neuropsychiatric disorder eventually impairs the patient's ability to perform even the most simple functions of daily living (Price et al., 1988; Kaul and Lipton, 2006; Hult et al., 2008; Minagar et al., 2008). Because HIV-1 does not infect neurons, HIV-1 neurotoxicity is indirect resulting from the release of various factors (Genis et al., 1992; Speth et al., 2001), including viral products such as HIV transactivator of transcription (Tat) (Nath et al., 1999). Tat is actively secreted by infected astrocytes, microglia and macrophages (King et al., 2006). HIV Tat mRNA is elevated in the brains of patients with HAD (Hudson et al., 2000) and when introduced into the CNS, Tat produces neuropathologies similar to those seen in HAD (Kim et al., 2003; Maragos et al., 2003).
Tat induces neuronal death by binding to the low-density lipoprotein receptor-related protein (LRP) (Liu et al., 2000) with subsequent activation of the NMDA receptor (Haughey et al., 2001; Song et al., 2003; Self et al., 2004). The resulting Ca2+ rise (Bonavia et al., 2001; Perez et al., 2001; Haughey and Mattson, 2002) activates neuronal nitric oxide synthase (nNOS) leading to cell death (Kruman et al., 1998). Indeed, the formation of a macromolecular complex involving LRP, postsynaptic density protein 95 (PSD95), NMDA receptor and nNOS was shown to be an early step in Tat-induced apoptosis (Eugenin et al., 2007).
Dendritic pruning, loss of spines and degradation of synaptic proteins precede cell death in many neurodegenerative disorders, including HAD (Masliah and Terry, 1993; Swann et al., 2000; Fiala et al., 2002; Sa et al., 2004). These morphological and biochemical changes correlate with neurological and cognitive decline better than cell death (Sa et al., 2004; Buttini et al., 2005). Tat causes a proteasome-mediated degradation of microtubule-associated protein 2 (MAP2) and the collapse of cytoskeletal filaments (Aprea et al., 2006). This loss of dendritic structure could account for the impaired synaptic plasticity observed in neural networks exposed to Tat (Li et al., 2004). How Tat-induced changes in dendritic morphology relate to Tat-induced death is unclear. Do LRP and the NMDA receptor participate in dendritic damage as they do in Tat-induced death? Activation of the ubiquitin–proteasome pathway does not generally lead to cell death; could synaptic loss be independent from the agonal event?
Here, we investigated the effect of HIV-1 Tat on the number of synaptic connections between rat hippocampal neurons in culture using an imaging-based assay that detected clusters of PSD95 fused to green fluorescent protein (PSD95–GFP). We found that Tat-induced cell death and synapse loss were initiated by LRP-dependent activation of the NMDA receptor. However, the downstream pathway leading to synapse loss was distinct from that leading to cell death. Furthermore, Tat-induced synapse loss was reversed by the LRP-specific chaperone, receptor-associated protein (RAP). These findings suggest that Tat-induced synapse loss might be a cellular mechanism to cope with excitotoxic stress and that RAP can modulate changes in synaptic stability.
Materials and Methods
Materials.
Materials were obtained from the following sources: the PSD95–GFP expression vector (pGW1-CMV-PSD95-EGFP) was kindly provided by Donald B. Arnold (University of Southern California, Los Angeles, CA) (Arnold and Clapham, 1999); the alternative reading frame polypeptide (ARF) expression vector (pcDNA3-myc-ARF) was kindly provided by Yanping Zhang (University of North Carolina, Chapel Hill, NC) (Zhang et al., 1998); the expression vector for DsRed2 (pDsRed2-N1) from Clontech; Dulbecco's modified Eagle medium (DMEM), 1,2-bis-(o-aminophenoxy)-ethane-N,N,N′,N′-tetraacetic acid-acetoxymethyl ester (BAPTA-AM), fetal bovine serum and horse serum from Invitrogen; Rat RAP, from Fitzgerald Industries International; dizocilpine (MK801), and all other reagents from Sigma.
HIV-1 Tat protein was obtained through the National Institutes of Health (NIH) AIDS Research and Reference Reagent Program, Division of AIDS, National Institute of Allergy and Infectious Diseases, NIH from Dr. John Brady. Recombinant HIV-1 Tat (86 aa) was produced in the Escherichia coli expression system and was determined to be at least 95% pure by SDS-PAGE. For control for experiments, Tat was heat-inactivated by incubation at 85°C for 30 min. Unless otherwise indicated the concentration of Tat in all experiments was 50 ng/ml (3.6 nm). HIV-1 BRU Tat (amino acids 32–62) (Tat32–62) was obtained from the Centre for AIDS Reagents supported by the European Union Programme European Vaccine Against AIDS/Medical Research Council and the United Kingdom Medical Research Council.
PSD95–GFP lacking the PEST sequence (PSD95ΔPEST-GFP) was produced by site-directed mutagenesis using the QuikChange Multi Kit (Stratagene). A mutagenic primer with the sequence: 5′ATAGTGACAACCAAGAAATACAGCCCGGCCCACCTCCCCAAC-3′ was designed to span nucleotides 16–93 of the rat PSD95 coding region but contained no complementarity to nucleotides 37–72 (coding for amino acids 13RYQDEDTPPLEH24), effectively deleting of this sequence upon mutagenic amplification. The construct was verified by DNA sequencing.
Cell culture.
Rat hippocampal neurons were grown in primary culture as described previously (Shen and Thayer, 1998b) with minor modifications. Fetuses were removed on embryonic day 17 from maternal rats, anesthetized with CO2, and killed by decapitation. Hippocampi were dissected and placed in Ca2+ and Mg2+-free HEPES-buffered Hanks salt solution (HHSS), pH 7.45. HHSS was composed of the following (in mm): HEPES 20, NaCl 137, CaCl2 1.3, MgSO4 0.4, MgCl2 0.5, KCl 5.0, KH2PO4 0.4, Na2HPO4 0.6, NaHCO3 3.0, and glucose 5.6. Cells were dissociated by trituration through a 5 ml pipette and a flame-narrowed Pasteur pipette, pelleted and resuspended in DMEM without glutamine, supplemented with 10% fetal bovine serum and penicillin/streptomycin (100 U/ml and 100 μg/ml, respectively). Dissociated cells were then plated at a density of 10,000–20,000 cells/dish onto a 25 mm-round cover glass (#1) glued to cover a 19 mm diameter opening drilled through the bottom of a 35 mm Petri dish. The coverglass was precoated with matrigel (200 μl, 0.2 mg/ml). Neurons were grown in a humidified atmosphere of 10% CO2 and 90% air, pH 7.4, at 37°C, and fed at days 1 and 6 by exchange of 75% of the media with DMEM, supplemented with 10% horse serum and penicillin/streptomycin. Cells used in these experiments were cultured without mitotic inhibitors for a minimum of 12 d.
Transfection.
Rat hippocampal neurons were transfected between 10 and 13 d in vitro using a modification of a protocol described previously (Waataja et al., 2008). Briefly, hippocampal cultures were incubated for at least 20 min in DMEM supplemented with 1 mm kynurenic acid, 10 mm MgCl2, and 5 mm HEPES, to reduce neurotoxicity. A DNA/calcium phosphate precipitate containing 1 μg plasmid DNA per well was prepared, allowed to form for 30 min at room temperature and added to the culture. After a 90 min incubation, cells were washed once with DMEM supplemented with MgCl2 and HEPES and then returned to conditioned media, saved at the beginning of the procedure.
Confocal imaging.
Transfected neurons were transferred to the stage of a confocal microscope (Olympus; Fluoview 300) and viewed through a 60× oil-immersion objective (NA. 1.40). For experiments in which the same neurons were imaged before and after a 24-h interval, the locations of individual cells were recorded using micrometers attached to the stage of the microscope. Multiple optical sections spanning 8 μm in the z-dimension were collected (1 μm steps), and these optical sections were combined through the z-axis into a compressed z stack. GFP was excited at 488 nm with an argon ion laser and emission collected at 530 nm (10 nm bandpass). The excitation (HeNe laser) and emission wavelengths for DsRed2 were 543 nm and >605 nm, respectively.
Image processing.
To count and label PSD95–GFP puncta, an automated algorithm was created using MetaMorph 6.2 image processing software described previously (Waataja et al., 2008). Briefly, maximum z-projection images were created from the DsRed2 and GFP image stacks. Next, a threshold set 1 SD above the image mean was applied to the DsRed2 image. This created a 1-bit image that was used as a mask via a logical AND function with the GFP maximum z-projection. A top-hat filter (80 pixels) was applied to the masked PSD95–GFP image. A threshold set 1.5 SD above the mean intensity inside the mask was then applied to the contrast enhanced image. Structures between 8 and 80 pixels (∼0.37–3.12 μm in diameter) were counted as PSDs. The structures were then dilated and superimposed on the DsRed2 maximum z-projection for visualization. PSD counts were presented as mean ± SEM where n is the number of cells, each from a separate cover glass over multiple cultures. We used Student's t test for single or ANOVA with Bonferoni post test for multiple statistical comparisons.
Toxicity.
Cell death was quantified using propidium iodide (PI) fluorescence as previously described (Gilbert et al., 2007). Cell culture was performed as described above except that 10,000 cells/well were plated in 96-well plates and grown for 12–14 d in vitro. The experiment was started by replacing 100 μl (∼2/3 volume) of the cell culture medium with fresh DMEM containing 10% horse serum, penicillin/streptomycin, 70 μm PI and either neurotoxin (1 mm glutamate or Tat at various concentrations) or vehicle (control). The plate was placed in a FluoStar Galaxy multiwell fluorescent plate scanner (BMG Technologies) and maintained at 37°C. PI fluorescence intensity measurements (excitation 544 ± 15 nm, emission 620 ± 15 nm) were taken at time 0, 24 and 48 h. Between measurements, cells were returned to the incubator and kept at 37°C in 10% CO2. Drugs, when present, were applied 15 min before application of the neurotoxin and included in the media exchange. Each treatment was performed in triplicate; thus, a set of 3 wells from a single plating of cells was defined as an individual experiment (n = 1).
Immunocytochemistry.
Rat hippocampal neurons were labeled using a protocol described previously (Kim et al., 2008). Briefly, PSD95–GFP-transfected hippocampal neurons were fixed with cooled methanol for 10 min at −20°C, blocked with 10% BSA and incubated for 16 h at 4°C with the following primary antibodies: mouse anti-NR2A (1:200; Millipore), mouse anti-NR2B (1:200; Transduction Laboratories) and rabbit anti-DsRed (1:100; Clontech). Cells were then incubated in tetramethylrhodamine isothiocyanate-labeled anti-rabbit antiserum (DAKO) and Alexa Fluor 647-anti-mouse IgG (Invitrogen) for 1 h at room temperature. After washing in phosphate buffered saline, coverslips were inverted on slides over a drop of fluoromount-G (Southern Biotechnology Associates). GFP (excitation = 488 nm; emission = 530 nm (10 nm bandpass), tetramethylrhodamine isothiocyanate (excitation = 543 nm; emission = >605 nm) and Alexa647 (excitation = 633 nm; emission = > 645 nm) labeled neurons were imaged using confocal microscopy.
Results
HIV-1 Tat-induced PSD loss
Changes in the number of synapses between rat hippocampal neurons in culture were monitored by imaging cells expressing PSD95–GFP and DsRed2 as previously described (Waataja et al., 2008). In Figure 1 A, we show representative images of neurons 48 h after transfection with expression plasmids for PSD95–GFP and DsRed2. PSD95–GFP expressed as discrete puncta that contrasted well from diffuse green fluorescence found throughout the cell. DsRed2 expression filled the soma and dendrites and was used to track morphological changes, as a mask for image processing and to determine cell viability based on cytoplasmic retention of the fluorescent protein. Image processing identified and counted puncta by locating intensity peaks of the appropriate size (mean diameter = 0.52 μm) in contact with the DsRed2 mask. We demonstrated previously that PSD95–GFP puncta colocalize with neurotransmitter release sites, NMDA-induced Ca2+ increases and NMDA receptor immunoreactivity (Waataja et al., 2008).
Figure 1.
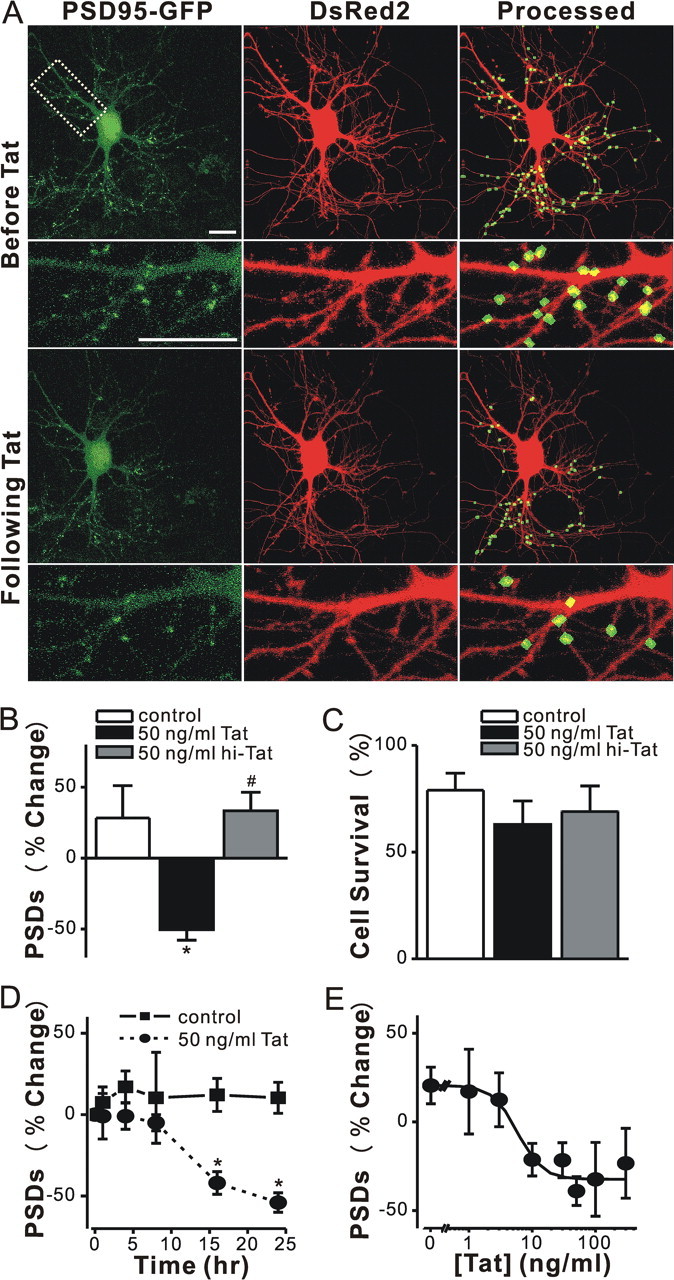
HIV-1 Tat induced PSD loss in a time and concentration dependent manner. A, Confocal fluorescent images display maximum z-projections of neurons expressing PSD95–GFP and DsRed2 before and 24 h after treatment with 50 ng/ml Tat. Processing of PSD95–GFP images identified PSDs as fluorescent puncta meeting intensity and size criteria and in contact with a mask derived from the DsRed2 image. Labeled PSDs were dilated and overlaid on the DsRed2 image for visualization purposes (processed). The insets are enlarged images of the boxed region. Scale bar, 10 μm. B, C, Bar graphs summarize the effects of Tat on changes in PSD95–GFP puncta (B, PSDs) and cell viability (C, cell survival) after 24 h treatment under control conditions (control, open bars) or following treatment with 50 ng/ml Tat (solid bars) or 50 ng/ml heat inactivated Tat (hi-Tat, gray bars). Data are mean ± SEM; *p < 0.01 relative to control; # p < 0.01 relative to Tat (ANOVA with Bonferroni post test). D, Graph shows time-dependent changes in the number of PSD95–GFP puncta for untreated cells (control, squares) and cells treated with 50 ng/ml Tat (circles). Data are expressed as mean ± SEM; *p < 0.01 relative to PSDs counted before the addition of Tat (0 h). ANOVA with Bonferroni post test. E, Graph shows concentration-dependent changes in the number PSD95–GFP puncta for cells treated with the indicated concentration of Tat. The mean ± SEM of the net change in PSD95–GFP puncta 24 h after treatment with Tat are plotted for the concentrations indicated (n ≥ 4 for each data point). The curve was fit by a logistic equation of the form percentage PSD change = [(A1 − A2)/(1 + (X/EC50)p)]+A2 where X = Tat concentration, A1 = 21 ± 7% PSD change without Tat, A2 = −32 ± 5% PSD change at a maximally effective Tat concentration and p = slope factor. EC50 was calculated using a nonlinear, least-squares curve fitting program. EC50 and p were 6 ± 2 ng/ml and 2 ± 1, respectively.
Treatment with 50 ng/ml HIV-1 Tat for 24 h decreased the number of fluorescent puncta by 50 ± 7% (n = 7) (Fig. 1 A,B). This finding is consistent with Tat-induced dendritic pruning and spine loss described previously (Maragos et al., 2003). Tat-induced PSD loss was significantly different from untreated cultures or cultures treated with heat-inactivated Tat (Fig. 1 B). In contrast, treatment with Tat for 24 h did not significantly affect cell survival relative to control or cells treated with heat-inactivated Tat (Fig. 1 C). Previous studies have noted that Tat-induced cell death develops over several days (Bonavia et al., 2001). Under control conditions, 21 ± 9% (n = 24) of the neurons died, consistent with the previous observation that as CNS cultures mature in vitro increased synaptic activity causes spontaneous cell death (Peterson et al., 1989; Shen and Thayer, 1998a). These results indicate that Tat-induced loss of synaptic connections precedes overt cell death.
Exposure to Tat caused a significant loss of PSD95–GFP puncta by 16 h (−42 ± 7%, p < 0.05, n = 12) and the magnitude of the loss increased to −54 ± 6% (n = 7) by 24 h (Fig. 1 D). Tat-induced loss of PSDs was concentration dependent (EC50 = 6 ± 2 ng/ml) (Fig. 1 E). We used the intensity of green fluorescence in the puncta and whole cell to assess changes in PSD95–GFP protein levels following 24 h treatment with Tat. The algorithm for counting puncta was used to define postsynaptic regions and the DsRed mask to define the whole-cell region. We then determined the area and intensity for these two regions before and after treatment with Tat. The whole-cell area and integrated fluorescence intensity did not significantly change following treatment with Tat, consistent with stable overall expression of PSD95–GFP. The total area encompassed by postsynaptic sites decreased, as expected from the decrease in the number of puncta (Δ area= −30.4 ± 4.0%, n = 11). However, the size of individual puncta did not change significantly after Tat treatment (Δ area = 6.4 ± 5.6%, n = 11). The integrated intensity of individual puncta remaining after treatment with Tat was not changed significantly (Δ intensity = −5.8 ± 4.8%, n = 11). In untreated cells, neither region changed in area or intensity over the course of a 24 h experiment.
Tat-induced synapse loss is both NMDA receptor- and LRP-dependent
Eugenin et al. (2007) have shown that Tat produces cell death via a pathway initiated by Tat binding to LRP with subsequent activation of NMDA receptors and nNOS-mediated apoptosis (Fig. 2 A). Alternatively, Tat was shown to induce morphological changes in dendrites by recruiting the proteasome to microtubules (Aprea et al., 2006) although, the role of LRP and the NMDA receptor were not investigated. Because activation of NMDA receptors induced the loss of PSDs via the ubiquitin–proteasome pathway (Waataja et al., 2008), we examined the possibility that Tat-induced PSD loss might also be initiated by this pathway. Pretreatment with MK801 (10 μm) for 30 min abolished Tat-induced synapse loss (n = 6) (Fig. 2 B), indicating that Tat-induced loss of PSD95–GFP is dependent on the NMDA receptor. Furthermore, chelating intracellular Ca2+ with BAPTA-AM (100 μm; 30 min pretreat) completely blocked Tat-induced synapse loss (n = 6) (Fig. 2 B). Previous studies have shown that the neurotoxic epitope of Tat resides between amino acids 31–61 (Nath et al., 1996). Tat32–62 (3.6 nm) induced synapse loss comparable with full-length Tat (Fig. 2 C), consistent with the idea that synapse loss and toxicity are initiated by the same events. To determine whether binding of Tat to LRP was necessary for Tat-induced synapse loss, we treated the cells with recombinant LRP receptor-associated protein (RAP), which binds to LRP (Bu, 2001) and antagonizes the neuronal uptake of exogenous Tat. Pretreatment with RAP (50 nm) for 15 min completely blocked Tat-induced synapse loss (n = 5) (Fig. 2 C). Tat-induced synapse loss requires LRP and NMDA receptor mediated Ca2+ influx, yet changes in PSDs (Waataja et al., 2008) and dendritic morphology (Aprea et al., 2006) have been linked to the ubiquitin–proteasome pathway. We next examined the possibility that this pathway might bifurcate with one path leading to synapse loss and the other to cell death.
Figure 2.
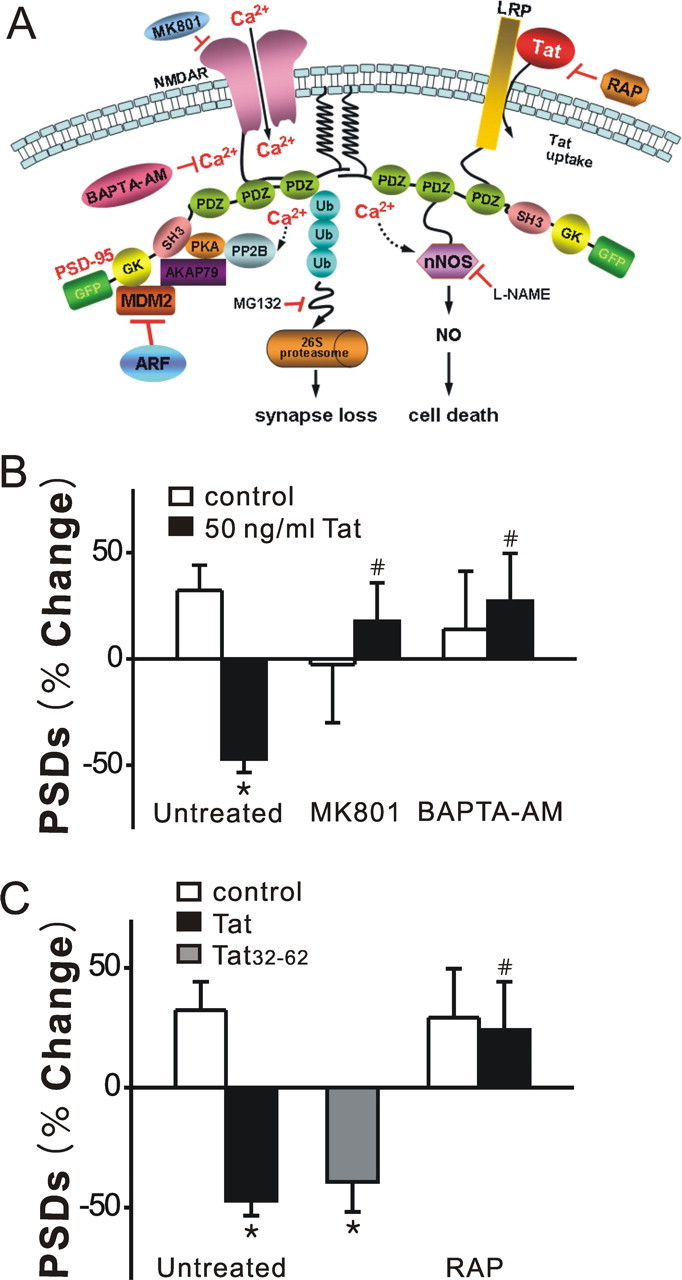
Tat-induced synapse loss is both NMDA receptor- and LRP-dependent. A, Proposed mechanism of Tat-induced synapse loss and death. B, Inhibition of NMDA receptor-mediated increases in [Ca2+]i prevent Tat-induced synapse loss. Bar graph summarizes the effects of inhibitors on changes in PSD95–GFP puncta (PSDs) after 24 h treatment under control (open bars) or Tat-treated (solid bars) conditions. Cultures were treated with 10 μm MK801 or 100 μm BAPTA-AM for 30 min before addition of Tat. C, The neurotoxic epitotope of Tat induced synapse loss via an LRP-dependent mechanism. HIV-1 BRU Tat amino acids 32–62 (Tat32–62) induced synapse loss. Tat-induced synapse loss was prevented by the LRP inhibitor, RAP (50 nm) applied 15 min before Tat treatment. Data are expressed as mean ± SEM; *p < 0.01 relative to control; # p < 0.01 relative to Tat alone (Untreated) (ANOVA with Bonferroni post test).
Tat induced loss of PSD95–GFP puncta is mediated by the ubiquitin–proteasome pathway not the nNOS apoptotic pathway
Activation of NMDA receptors has been shown to induce PSD95 degradation via the ubiquitin–proteasome pathway (Colledge et al., 2003; Waataja et al., 2008). Tat causes a proteasome-mediated degradation of MAP2 and the collapse of cytoskeletal filaments (Aprea et al., 2006). We determined whether Tat might induce the loss of PSD95–GFP puncta by activation of the ubiquitin–proteasome pathway. Twenty-four hour exposure to micromolar concentrations of the proteasome inhibitor MG132 (Z-Leu-Leu-Leu-aldehyde) was toxic to hippocampal cultures (data not shown). Thus, we used an alternative genetic approach to inhibit the proteasome pathway. ARF binds to and inhibits MDM2 (murine double minute 2) (Zhang et al., 1998) which is an ubiquitin E3 ligase known to ubiquitinate PSD95 targeting it for proteasomal degradation (Colledge et al., 2003). Cultured hippocampal neurons were transfected with expression vectors for ARF (pcDNA3-myc-ARF) (Zhang et al., 1998), PSD95–GFP and DsRed2 as demonstrated previously (Waataja et al., 2008). Expression of ARF protected hippocampal neurons from Tat-induced synapse loss (n = 9, p < 0.01) (Fig. 3 A,B). The MDM2 inhibitor nutlin-3 binds to MDM2 and stabilizes its downstream targets (Vassilev et al., 2004; Uo et al., 2007). Nutlin-3 (1 μm) also reduced the loss of PSDs (n = 4, p < 0.01) (Fig. 3 B). These data suggest that Tat activates an E3 ligase leading to the loss of synaptic connections.
Figure 3.
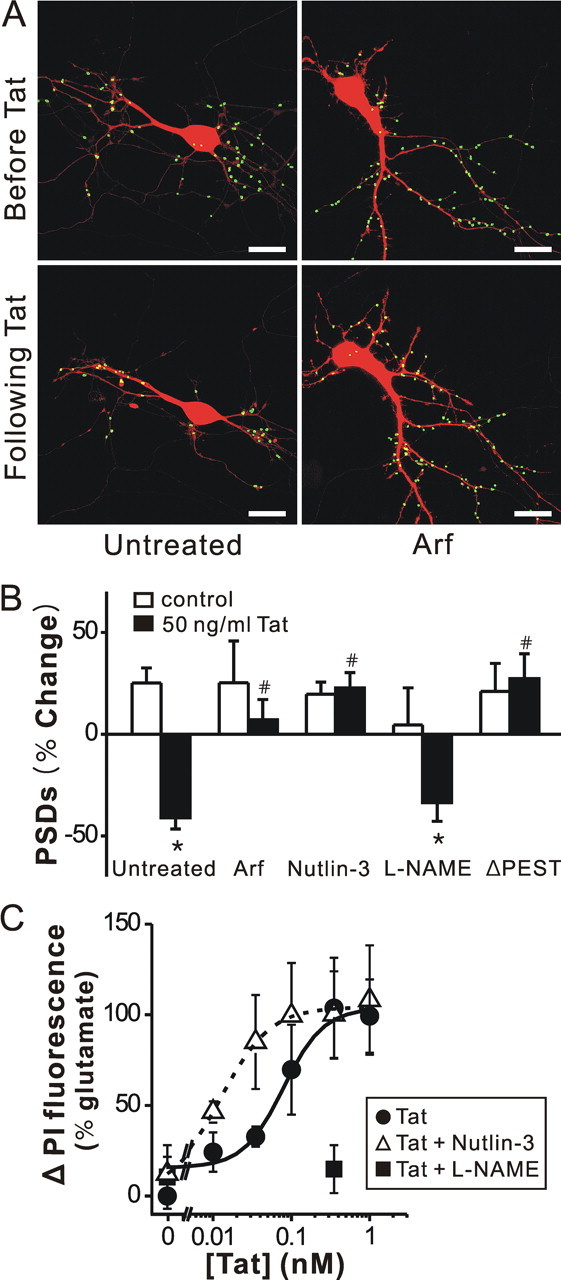
Tat induced PSD loss via the ubiquitin–proteasome pathway, and Tat induced cell death by activating NOS. A, Processed images acquired before and after 24 h treatment with 50 ng/ml Tat in cells coexpressing ARF display labeled PSDs superimposed on DsRed2 fluorescence. Scale bar, 10 μm. B, Bar graph summarizes the effects of inhibition of the ubiquitin–proteasome pathway and inhibition of nNOS on changes in PSD–GFP puncta (PSDs) after 24 h treatment under control (open bars) or 50 ng/ml Tat-treated (solid bars) conditions. Cells were treated with ARF (expression plasmid cotransfected with DsRed2 and PSD95–GFP), 1 μm nutlin-3 or 100 μm l-NAME as indicated. In cells expressing PSD95ΔPEST-GFP (ΔPEST), Tat did not affect the number of synaptic sites. Data are expressed as mean ± SEM. *p < 0.01 relative to control; # p < 0.01 relative to Tat alone (untreated) (ANOVA with Bonferroni post test). C, Inhibition of an ubiquitin ligase increased and inhibition of NOS decreased, Tat-induced cell death. Cell death was measured using the PI fluorescence assay detailed in Materials and Methods. Cell death was measured after 48 h treatment with the indicated concentrations of Tat in the absence (control, circles) or presence of 1 μm nutlin-3 (open triangles) or 100 μm l-NAME (squares). PI fluorescence was normalized to that measured from cells treated for 48 h with 1 mm glutamate (100%). PI fluorescence from untreated wells was subtracted from each curve (0%). Concentration response curves were generated by fitting a logistic equation to the data using a nonlinear, least-squares curve fitting program (Origin 6.0) and EC50 values calculated. A logistic equation of the form Δ PI Fluorescence = [(A2 − A1)/(1 + (X/EC50)p)]+A1 where X = Tat concentration, A1 = percentage change in PI fluorescence without Tat, A2 = percentage change in PI fluorescence at maximal Tat concentration and p = slope factor. A set of triplicate wells from a single plating of cells was defined as a single experiment (n = 1). All data are presented as mean ± SEM.
Tat could induce ubiquitination of PSD95 directly (Colledge et al., 2003) or it could ubiquitinate cytoskeletal proteins leading to a secondary loss of PSD95–GFP puncta (Aprea et al., 2006). PEST sequences regulate protein stability and ubiquitination (Rechsteiner and Rogers, 1996) and a PEST sequence at the N terminus of PSD95 is required for ubiquitination (Colledge et al., 2003). We generated a PSD95–GFP deletion mutant lacking this PEST sequence (amino acids 13–23 in rat PSD-95). Cells expressing PSD95ΔPEST-GFP displayed punctate fluorescence comparable with those expressing wild type PSD95–GFP. However, exposure to Tat did not significantly reduce the number of PSD95ΔPEST-GFP puncta (Fig. 3 B), consistent with the idea that Tat induced ubiquitination of PSD95 is required for loss of synaptic sites.
Activation of the ubiquitin–proteasome pathway is not generally thought to lead to neuronal cell death. However, activation of nNOS is clearly linked to Tat-induced neuronal death (Eugenin et al., 2007). Application of NG-nitro-l-arginine methyl ester hydrochloride (l-NAME) 15 min before and during 24 h exposure to Tat did not significantly affect PSD loss (34 ± 9%; n = 7) relative to Tat treatment alone (Fig. 3 B).
If Tat-induced synapse loss is mediated by a different path than Tat-induced neuronal death, then perhaps synapse loss is a coping mechanism rather than part of the agonal event. We tested this hypothesis by performing a series of cell survival assays on hippocampal cultures (Fig. 3 C). Cell death induced by various concentrations of Tat was quantified by uptake of propidium iodide measured after 48 h exposure. Consistent with the results of Liu et al. (2000), l-NAME (100 μm) completely blocked Tat-induced neurotoxicity. In contrast, treatment with nutlin-3, at a concentration that when applied alone had no effect on cell survival (1 μm), shifted significantly the Tat concentration response curve to the left (EC50 = 13 ± 0.1 pm) relative to control (EC50 = 83 ± 29 pm) (p < 0.05). This concentration of nutlin-3 completely blocked Tat-induced PSD loss (Fig. 3 B). These findings suggest that the synapse loss induced by Tat may actually improve survival, possibly by reducing NMDA receptor-mediated Ca2+ influx.
Tat-induced PSD loss is reversible
If PSD loss proceeds via a path that is independent from that leading to cell death, then synapse loss might be reversible. We next examined whether hippocampal neurons could recover from downregulation of PSD95–GFP puncta following exposure to Tat. Recovery from synapse loss induced by epileptic activity was limited to relatively short exposures to the aberrant synaptic activity (Kim et al. 2008). Thus, we chose to study recovery from Tat after the shortest exposure that produced reliable synapse loss. Hippocampal neurons exposed to Tat for 16 h lost a significant fraction of their PSDs (34 ± 9%; n = 8; p < 0.001) (Fig. 4). After exposure to Tat for 16 h, the protein was removed (washout) as described in the scheme shown in Figure 4 A. Cells were imaged again 8 and 32 h following application of the inhibitors (24 and 48 h elapsed time in Fig. 4 C). Tat induced a gradual decrease in the number of PSDs that was not reversed by withdrawal of Tat after 16 h of treatment. Removing Tat from the bathing media would not necessarily remove Tat adhering to cells or internalized Tat. Thus, we tested the inhibitors that were effective when applied to the cells before treatment with Tat. Application of MK801 or BAPTA-AM after treatment with Tat for 16 h failed to reverse the decline in the number of synapses measured at 24 h (Fig. 4 D). Both of these agents prevented synapse loss when applied before Tat, suggesting that a window during which these agents are effective had closed. Thus, we were surprised by the results of experiments in which RAP was added after 16 h treatment with Tat (Fig. 4 C). RAP not only prevented the further decline in PSD number but actually enabled recovery of PSD95–GFP puncta in the maintained presence of Tat. Neurons displayed a significant recovery in the number of PSDs (p < 0.001, n = 5) 8 h after the addition of RAP. This recovery persisted for 32 h following RAP treatment. These results demonstrate that synapses can recover after exposure to Tat and that LRP might be an important target for inducing the recovery of synaptic function.
Figure 4.
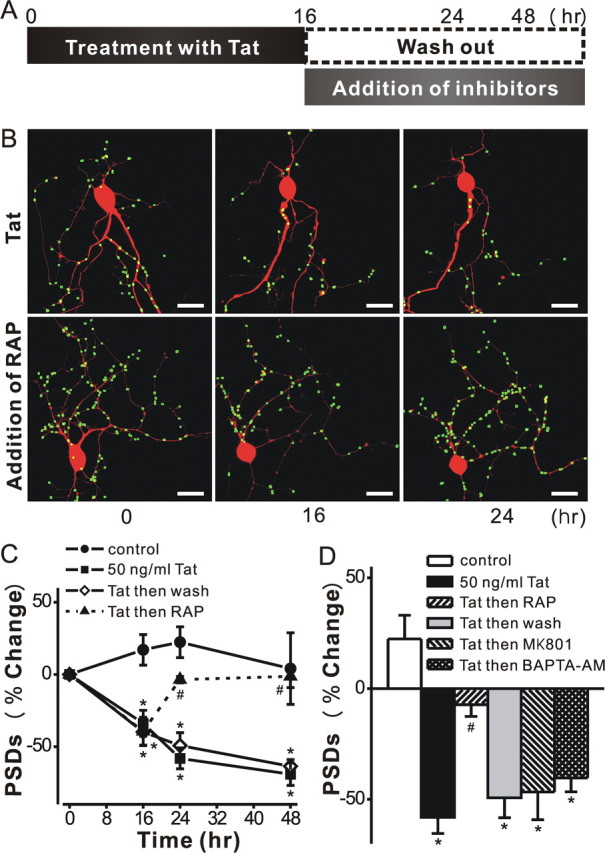
Tat-induced PSD loss is reversible. A, Schematic shows time line for experiments. B, Representative images display labeled PSDs on neurons expressing PSD95–GFP and DsRed2 before (0 h) and during (16 and 24 h) treatment with 50 ng/ml Tat. The LRP inhibitor RAP (50 nm) was added after 16 h (bottom frames). Note that PSDs lost after 16 h exposure to Tat recovered following treatment with RAP. C, Graph shows significant PSD loss after 16 h exposure to Tat which was sustained for 48 h in the continued presence of Tat (squares) or if Tat was removed after 16 h (open diamonds). PSDs recovered to control (circles) levels following addition of RAP at 16 h (triangles). Data are expressed as mean ± SEM *p < 0.01 relative to control; # p < 0.001 relative to Tat alone for 16 h (ANOVA with Bonferroni post test). D, Bar graph summarizes the net changes in the number of PSD95–GFP puncta (PSDs) 24 h after no treatment (control, open bar) or the addition of Tat (solid bars). The indicated treatments were applied after 16 h exposure to Tat and changes were measure 8 h later (24 h elapsed time). Data are expressed as mean ± SEM. *p < 0.01 relative to control; # p < 0.05 relative to Tat alone for 24 h (ANOVA with Bonferroni post test).
Functional PSDs are composed of many proteins, including NMDA receptors, which participate in synaptic transmission. To determine if the new PSD95–GFP puncta formed during RAP-induced recovery represent synaptic sites, we induced synapse loss and recovery and then fixed neurons expressing PSD95–GFP and labeled them with antibodies to the NMDA receptor. Individual neurons were imaged before treatment, after treatment with Tat for 16 h and again after treatment with RAP for 8 h (24 h elapsed time) (Fig. 5 A). The cells were fixed and labeled with antibodies to the NR2A and 2B subunits of the NMDA receptor. In naive cultures immunocytochemistry revealed that 70 ± 2% (n = 7) of PSD95–GFP puncta colocalize with NR2A or 2B (Waataja et al., 2008). In cultures treated with RAP in the continuous presence of Tat, 74.5 ± 2.5% (n = 4) of PSD95–GFP puncta colocalize with NR2A or 2B immunoreactivity. Because the same percentage of puncta colabel with NR2 immunoreactivity, we conclude that the new puncta are comparable with those in naive cells. It is difficult to track the loss and recovery of a single puncta through a 24 h experiment and through the fixing process, because the cell's morphology changes over the course of the experiment. However, as shown in the inset to Figure 5, it is clear that in a region of dendrite that underwent loss and clear repopulation with new PSD95–GFP puncta, the new puncta contain NR2-immunoreativity.
Figure 5.
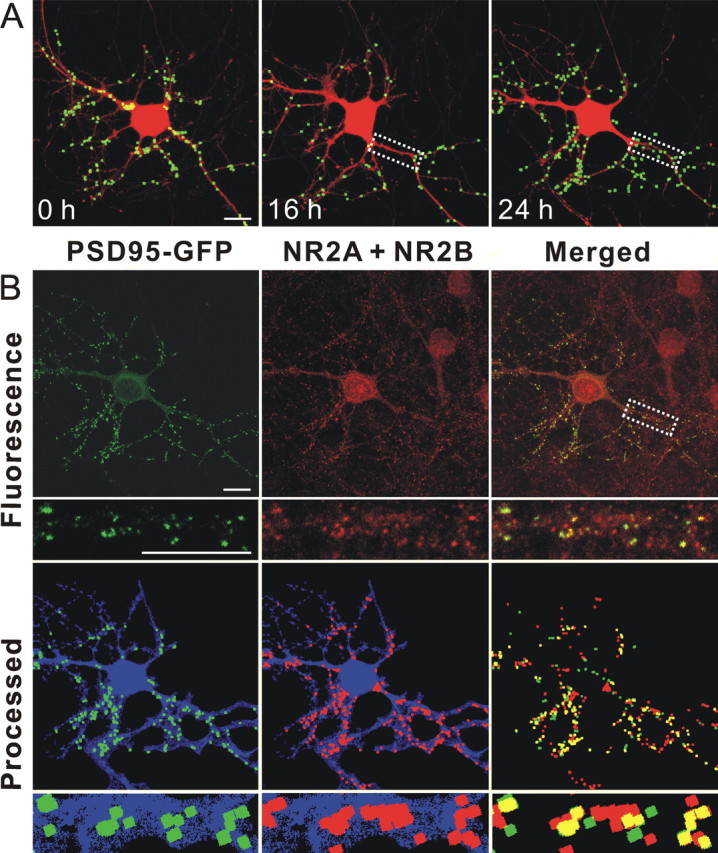
PSDs recovered after RAP-induced reversal of Tat-induced synapse loss contain NMDA receptor immunoreactivity. A, Representative images display labeled PSDs on a neuron expressing PSD95–GFP and DsRed2 before (0 h) and during (16 and 24 h) treatment with 50 ng/ml Tat. The LRP inhibitor RAP (50 nm) was added after 16 h. Eight hours after administering RAP, the number of synapses recovered (24 h frame). B, After collecting the live cell image at 24 h (as shown in A 24 h frame) the cells were fixed and labeled with antibodies to DsRed and the NR2A and 2B subunits of the NMDA receptor as described in Materials and Methods. Confocal micrographs display PSD95–GFP fluorescence (green) and NR2A and NR2B (red) immunoreactivity. Merged images display overlapping puncta (yellow). Processed images display puncta within the DsRed2 mask (blue). The same image-processing algorithm described in Materials and Methods was used to identify both NR2 immunoreactive puncta and PSD95–GFP puncta. The insets are enlarged images of the boxed region. Note that NR2A and NR2B immunoreactivity includes nontransfected cells in the field. Scale bar, 10 μm.
Discussion
HIV-1 infection of the CNS results in dendritic pruning and a reduction in spine density (Sa et al., 2004), changes that precede neuronal death and are thought to result in cognitive decline (Zink et al., 2002; Buttini et al., 2005). Here, we used an imaging-based assay to study the effects HIV-1 Tat, a protein released by virus-infected cells, on synaptic connections between rat hippocampal neurons in vitro. Tat induced a dramatic loss of synapses. The initial steps leading to synapse loss, binding to LRP, activation of NMDA receptors and a rise in [Ca2+]i, were similar to those previously reported for Tat-induced cell death (Liu et al., 2000; Self et al., 2004; Eugenin et al., 2007). However, while subsequent activation of nNOS was required for cell death, synapse loss was mediated by the ubiquitin–proteasome pathway, consistent with a previous study that showed that Tat altered spine morphology via the proteasomal pathway (Aprea et al., 2006). This bifurcation in the pathway is consistent with the hypothesis that synapse loss is a coping mechanism rather than a symptom of the neuron's demise. Indeed, Tat-induced synapse loss was reversible, suggesting that rapid, dynamic changes in the number of synapses might balance excitatory input with survival. Perhaps the synapse loss that presents early in HAD can be reversed to improve neurocognitive outcomes in NeuroAIDS patients.
We used an in vitro assay to track changes in the number of individual synapses within a neural network over time (Kim et al., 2008; Waataja et al., 2008). [Ca2+]i imaging and immunocytochemistry had previously confirmed that PSD95–GFP puncta represent functional postsynaptic sites and their close apposition to functional neurotransmitter release sites indicated that they corresponded to functional synapses (Waataja et al., 2008). This imaging approach has several strengths. Longitudinal imaging enabled detection of synapse loss and recovery from the same cell and automated processing was objective and quantitative, enabling hypothesis testing.
HIV-1 Tat is a regulatory protein that is released by acutely infected cells and can be taken up rapidly by other cells (Frankel and Pabo, 1988). Tat enters neurons via LRP expressed on the cell surface (Liu et al., 2000). LRP is an endocytic receptor that binds multiple lipoprotein ligands (May et al., 2007). It is also a signal transduction receptor that participates in neurodevelopment and synaptic plasticity (Herz and Chen, 2006). LRP is associated with neurodegenerative disease. Polymorphisms in the genes for LRP, and its ligands apolipoprotein E and α-macroglobulin are linked to Alzheimer's disease (Schellenberg et al., 2000) and HAD (Corder et al., 1998). A role for LRP in Tat-induced synapse loss was established by inhibition of synapse loss by RAP. RAP is a molecular chaperone that prevents LRP from interacting with ligands and facilitates LRP folding and trafficking within the secretory pathway (Bu, 2001). Recombinant RAP has been used extensively as an antagonist in the study of LRP function. There is precedent for LRP ligands such as activated α2-macroglobulin activating NMDA receptors to increase [Ca2+]i (Bacskai et al., 2000). That Tat activates NMDA receptors is well established (Mattson et al., 2005) although, several possible mechanisms of activation have been proposed. Activation of kinases that phosphorylate the NMDA receptor (Haughey et al., 2001) or protein-protein interactions (Eugenin et al., 2007) are most compatible with the LRP-mediated effects described here. However, direct activation of the receptor (Prendergast et al., 2002), modulation of Zn interactions with NMDA receptors (Song et al., 2003) and recruitment and activation of the 20S proteasome (Aprea et al., 2006) have all been described. NMDA receptor mediated increases in [Ca2+]i initiate both synaptic plasticity and cell death (Albensi, 2007). Thus, the initial stages of Tat-induced synapse loss closely parallel those reported for initiating cell death pathways.
The path leading to synapse loss diverged from that leading to cell death; the mechanism of synapse loss shared some features in common to those that produce synaptic plasticity. Downstream from the Tat-induced [Ca2+]i increase, the activation of an ubiquitin ligase was required for synapse loss. Expression of ARF inhibits the E3 ubiquitin ligase MDM2, also known as Hdm2 (Zhang et al., 1998), and was shown here to prevent synapse loss. That the Tat-induced synapse loss was mediated by the ubiquitin–proteasome pathway is in good agreement with previous reports of Tat-induced changes in dendritic morphology. In neurons transfected with a Tat expression vector, Tat recruited proteasomes to microtubules with subsequent MAP2 degradation (Aprea et al., 2006). The path leading to activation of the ubiquitin–proteasome pathway appears most similar to that described for long-term depression of synaptic transmission (Colledge et al., 2003) and excitotoxicity (Waataja et al., 2008). Interestingly, Tat itself is a substrate for MDM2 although, ubiquitination does not target Tat to the proteasome but instead, plays a regulatory role (Bres et al., 2003). Because PSD95ΔPEST-GFP puncta were resistant to Tat-induced loss, it appears that PSD95 is the relevant substrate for synapse loss induced by ubiquitin ligase activity. The ubiquitin–proteasome pathway mediates many of the dynamic changes in spine structure and function that underlie synaptic plasticity (Ehlers, 2003; Bingol and Schuman, 2006).
Significant neurological improvement accompanies initiation of highly active antiretroviral therapy in patients with HAD (Bellizzi et al., 2006), consistent with the idea that cognitive impairment is due at least in part to reversible actions of the virus. RAP enabled a dramatic recovery of PSD95–GFP puncta in the continued presence of Tat. In contrast, withdrawal of Tat, addition of the Ca2+ chelator BAPTA or blocking NMDA receptors with MK801, treatments that prevented PSD loss when applied before Tat, failed to reverse the decline in the number of synapses. Thus, there appears to be a window during which these agents may protect from Tat-induced synapse loss. Alternatively, BAPTA and MK801 might fail to induce recovery of synapses because NMDA receptor-mediated [Ca2+]i increases are required for synapse formation. The reversal of synapse loss by RAP is intriguing. RAP is known to prevent the entry of Tat but, presumably Tat is already in the cell when RAP was applied at 16 h. The observation that removing Tat did not reverse or even halt the continued loss of synapses suggests that RAP does more than prevent further entry of Tat into the cell. RAP binds to LRP with high affinity (Bu, 2001) potentially disrupting protein-protein interactions that are required for synapse loss. Another intriguing question is whether synapses recover because RAP induces their formation or does RAP enable the cell to re-equilibrate to its original number of synapses (Desai, 2003) by removing the signal driving synapse loss. A better understanding of the processes that initiate the formation of synapses following neurotoxic insult could guide the development of drugs that improve cognitive function in HAD.
Tat-induced synapse loss differed temporally and mechanistically from neuronal death. That these pathways are distinct was supported by the failure of NOS inhibitors to prevent synapse loss while completely preventing Tat-induced cell death. This observation is consistent with previous studies that have demonstrated that activation of nNOS mediates Tat-induced neuronal apoptosis (Eugenin et al., 2007). In contrast, Tat-induced synapse loss was prevented by treatment with the ubiquitin ligase inhibitor nutlin-3 at concentrations that were without effect on cell survival. Thus, synapse loss is not necessarily a step on the path toward death; instead it may be a coping mechanism that enables the cell to adapt to excessive excitatory input (Hasbani et al., 2001; Waataja et al., 2008). This conclusion is supported by experiments in which the development of ischemic tolerance resulted from ubiquitin proteasome-mediated synaptic remodeling that rendered neurons refractory to NMDA receptor-mediated toxicity (Meller et al., 2008). Many of the hallmark signs and symptoms of neurodegenerative disease might actually be coping mechanisms (Finkbeiner et al., 2006).
The initial stages of Tat-induced synapse loss closely parallel those reported for triggering cell death pathways and have been explored as potential targets for neuroprotective drugs. Modulation of LRP (Eugenin et al., 2007), block of NMDA receptors (Anderson et al., 2004; Gendelman, 2007), and chelation of [Ca2+]i (Tymianski et al., 1994) produced encouraging results in vitro but did not generally translate into high neuroprotective efficacy in the clinic. Because neuronal loss has consistently failed to correlate with neurological impairment (Adle-Biassette et al., 1999), protecting synapses from HIV-1 neurotoxicity, possibly at targets distinct from those that improve neuronal survival (Bellizzi et al., 2006), might be an effective strategy to improve cognitive function in HAD. In preliminary studies, we found that lithium, a drug shown to improve neurocognitive function in HAD patients (Letendre et al., 2006), increases the number of synapses between hippocampal neurons (H. J. Kim and S. A. Thayer unpublished observations). A potential drawback to protecting synapses is that if synapse loss is a coping mechanism, then preventing downregulation of synapses may actually increase neuronal death. This cautions that optimal neuroprotective agents would act upstream of the bifurcation in the neurotoxicity pathway, before the synapse loss and cell death pathways diverge, or act at dual targets to improve survival and retain connectivity.
Footnotes
This work was supported by National Institute on Drug Abuse–National Institutes of Health (NIH) Grants DA07304, DA024428, and DA021743. We thank Drs. Donald B. Arnold and Yanping Zhang for providing expression plasmids for PSD95–GFP and p14ARF, respectively. HIV-1 Tat was provided by the NIH AIDS Research and Reference Reagent Program, and Tat32–62 was provided by the Centre for AIDS Reagents supported by the European Union Programme European Vaccine Against AIDS/Medical Research Council (MRC) (contract QLKZ-CT-1999-00609) and the United Kingdom MRC.
References
- Adle-Biassette H, Chrétien F, Wingertsmann L, Héry C, Ereau T, Scaravilli F, Tardieu M, Gray F. Neuronal apoptosis does not correlate with dementia in HIV infection but is related to microglial activation and axonal damage. Neuropathol Appl Neurobiol. 1999;25:123–133. doi: 10.1046/j.1365-2990.1999.00167.x. [DOI] [PubMed] [Google Scholar]
- Albensi BC. The NMDA receptor/ion channel complex: a drug target for modulating synaptic plasticity and excitotoxicity. Curr Pharm Des. 2007;13:3185–3194. doi: 10.2174/138161207782341321. [DOI] [PubMed] [Google Scholar]
- Anderson ER, Gendelman HE, Xiong H. Memantine protects hippocampal neuronal function in murine human immunodeficiency virus type 1 encephalitis. J Neurosci. 2004;24:7194–7198. doi: 10.1523/JNEUROSCI.1933-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aprea S, Del Valle L, Mameli G, Sawaya BE, Khalili K, Peruzzi F. Tubulin-mediated binding of human immunodeficiency virus-1 Tat to the cytoskeleton causes proteasomal-dependent degradation of microtubule-associated protein 2 and neuronal damage. J Neurosci. 2006;26:4054–4062. doi: 10.1523/JNEUROSCI.0603-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold DB, Clapham DE. Molecular determinants for subcellular localization of PSD-95 with an interacting K+ channel. Neuron. 1999;23:149–157. doi: 10.1016/s0896-6273(00)80761-8. [DOI] [PubMed] [Google Scholar]
- Bacskai BJ, Xia MQ, Strickland DK, Rebeck GW, Hyman BT. The endocytic receptor protein LRP also mediates neuronal calcium signaling via N-methyl-D-aspartate receptors. Proc Natl Acad Sci U S A. 2000;97:11551–11556. doi: 10.1073/pnas.200238297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellizzi MJ, Lu SM, Gelbard HA. Protecting the synapse: evidence for a rational strategy to treat HIV-1 associated neurologic disease. J Neuroimmune Pharmacol. 2006;1:20–31. doi: 10.1007/s11481-005-9006-y. [DOI] [PubMed] [Google Scholar]
- Bingol B, Schuman EM. Activity-dependent dynamics and sequestration of proteasomes in dendritic spines. Nature. 2006;441:1144–1148. doi: 10.1038/nature04769. [DOI] [PubMed] [Google Scholar]
- Bonavia R, Bajetto A, Barbero S, Albini A, Noonan DM, Schettini G. HIV-1 Tat causes apoptotic death and calcium homeostasis alterations in rat neurons. Biochem Biophys Res Commun. 2001;288:301–308. doi: 10.1006/bbrc.2001.5743. [DOI] [PubMed] [Google Scholar]
- Brès V, Kiernan RE, Linares LK, Chable-Bessia C, Plechakova O, Tréand C, Emiliani S, Peloponese JM, Jeang KT, Coux O, Scheffner M, Benkirane M. A non-proteolytic role for ubiquitin in Tat-mediated transactivation of the HIV-1 promoter. Nat Cell Biol. 2003;5:754–761. doi: 10.1038/ncb1023. [DOI] [PubMed] [Google Scholar]
- Bu G. The roles of receptor-associated protein (RAP) as a molecular chaperone for members of the LDL receptor family. Intl Rev Cytology. 2001;209:79–116. doi: 10.1016/s0074-7696(01)09011-8. [DOI] [PubMed] [Google Scholar]
- Buttini M, Masliah E, Barbour R, Grajeda H, Motter R, Johnson-Wood K, Khan K, Seubert P, Freedman S, Schenk D, Games D. Beta-amyloid immunotherapy prevents synaptic degeneration in a mouse model of Alzheimer's disease. J Neurosci. 2005;25:9096–9101. doi: 10.1523/JNEUROSCI.1697-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colledge M, Snyder EM, Crozier RA, Soderling JA, Jin Y, Langeberg LK, Lu H, Bear MF, Scott JD. Ubiquitination regulates PSD-95 degradation and AMPA receptor surface expression. Neuron. 2003;40:595–607. doi: 10.1016/s0896-6273(03)00687-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corder EH, Robertson K, Lannfelt L, Bogdanovic N, Eggertsen G, Wilkins J, Hall C. HIV-infected subjects with the E4 allele for APOE have excess dementia and peripheral neuropathy. Nat Med. 1998;4:1182–1184. doi: 10.1038/2677. [DOI] [PubMed] [Google Scholar]
- Desai NS. Homeostatic plasticity in the CNS: synaptic and intrinsic forms. J Physiol Paris. 2003;97:391–402. doi: 10.1016/j.jphysparis.2004.01.005. [DOI] [PubMed] [Google Scholar]
- Ehlers MD. Activity level controls postsynaptic composition and signaling via the ubiquitin-proteasome system. Nat Neurosci. 2003;6:231–242. doi: 10.1038/nn1013. [DOI] [PubMed] [Google Scholar]
- Eugenin EA, King JE, Nath A, Calderon TM, Zukin RS, Bennett MV, Berman JW. HIV-Tat induces formation of an LRP-PSD-95- NMDAR-nNOS complex that promotes apoptosis in neurons and astrocytes. Proc Natl Acad Sci U S A. 2007;104:3438–3443. doi: 10.1073/pnas.0611699104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiala JC, Spacek J, Harris KM. Dendritic spine pathology: cause or consequence of neurological disorders? Brain Res Rev. 2002;39:29–54. doi: 10.1016/s0165-0173(02)00158-3. [DOI] [PubMed] [Google Scholar]
- Finkbeiner S, Cuervo AM, Morimoto RI, Muchowski PJ. Disease-modifying pathways in neurodegeneration. J Neurosci. 2006;26:10349–10357. doi: 10.1523/JNEUROSCI.3829-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankel AD, Pabo CO. Cellular uptake of the tat protein from human immunodeficiency virus. Cell. 1988;55:1189–1193. doi: 10.1016/0092-8674(88)90263-2. [DOI] [PubMed] [Google Scholar]
- Gendelman HE. Biomarkers, laboratory, and animal models for the design and development of adjunctive therapies for HIV-1 dementia and other neuroinflammatory disorders. J Neuroimmune Pharmacol. 2007;2:8–13. doi: 10.1007/s11481-006-9050-2. [DOI] [PubMed] [Google Scholar]
- Genis P, Jett M, Bernton EW, Boyle T, Gelbard HA, Dzenko K, Keane RW, Resnick L, Mizrachi Y, Volsky DJ, Epstein LG, Gendelman HE. Cytokines and arachidonic metabolites produced during human immunodeficiency virus (HIV)-infected macrophage interactions: implications for the neuropathogenesis of HIV disease. J Exp Med. 1992;176:1703–1718. doi: 10.1084/jem.176.6.1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert GL, Kim HJ, Waataja JJ, Thayer SA. Delta9-tetrahydrocannabinol protects hippocampal neurons from excitotoxicity. Brain Res. 2007;1128:61–69. doi: 10.1016/j.brainres.2006.03.011. [DOI] [PubMed] [Google Scholar]
- Hasbani MJ, Schlief ML, Fisher DA, Goldberg MP. Dendritic spines lost during glutamate receptor activation reemerge at original sites of synaptic contact. J Neurosci. 2001;21:2393–2403. doi: 10.1523/JNEUROSCI.21-07-02393.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haughey NJ, Mattson MP. Calcium dysregulation and neuronal apoptosis by the HIV-1 proteins Tat and gp120. JAIDS. 2002;31:S55–S61. doi: 10.1097/00126334-200210012-00005. [DOI] [PubMed] [Google Scholar]
- Haughey NJ, Nath A, Mattson MP, Slevin JT, Geiger JD. HIV-1 Tat through phosphorylation of NMDA receptors potentiates glutamate excitotoxicity. J Neurochem. 2001;78:457–467. doi: 10.1046/j.1471-4159.2001.00396.x. [DOI] [PubMed] [Google Scholar]
- Herz J, Chen Y. Reelin, lipoprotein receptors and synaptic plasticity. Nat Rev Neurosci. 2006;7:850–859. doi: 10.1038/nrn2009. [DOI] [PubMed] [Google Scholar]
- Hudson L, Liu J, Nath A, Jones M, Raghavan R, Narayan O, Male D, Everall I. Detection of the human immunodeficiency virus regulatory protein Tat in CNS tissues. J Neurovirol. 2000;6:145–155. doi: 10.3109/13550280009013158. [DOI] [PubMed] [Google Scholar]
- Hult B, Chana G, Masliah E, Everall I. Neurobiology of HIV. Int Rev Psychiatry. 2008;20:3–13. doi: 10.1080/09540260701862086. [DOI] [PubMed] [Google Scholar]
- Kaul M, Lipton SA. Mechanisms of neuronal injury and death in HIV-1 associated dementia. Curr HIV Res. 2006;4:307–318. doi: 10.2174/157016206777709384. [DOI] [PubMed] [Google Scholar]
- Kim BO, Liu Y, Ruan Y, Xu ZC, Schantz L, He JJ. Neuropathologies in transgenic mice expressing human immunodeficiency virus type 1 Tat protein under the regulation of the astrocyte-specific glial fibrillary acidic protein promoter and doxycycline. Am J Pathol. 2003;162:1693–1707. doi: 10.1016/S0002-9440(10)64304-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HJ, Waataja JJ, Thayer SA. Cannabinoids inhibit network-driven synapse loss between hippocampal neurons in culture. J Pharmacol Exp Ther. 2008;325:850–858. doi: 10.1124/jpet.107.131607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King JE, Eugenin EA, Buckner CM, Berman JW. HIV Tat and neurotoxicity. Microbes Infect. 2006;8:1347–1357. doi: 10.1016/j.micinf.2005.11.014. [DOI] [PubMed] [Google Scholar]
- Kruman II, Nath A, Mattson MP. HIV-1 protein Tat induces apoptosis of hippocampal neurons by a mechanism involving caspase activation, calcium overload, and oxidative stress. Exp Neurol. 1998;154:276–288. doi: 10.1006/exnr.1998.6958. [DOI] [PubMed] [Google Scholar]
- Letendre SL, Woods SP, Ellis RJ, Atkinson JH, Masliah E, van den Brande G, Durelle J, Grant I, Everall I. Lithium improves HIV-associated neurocognitive impairment. AIDS. 2006;20:1885–1888. doi: 10.1097/01.aids.0000244208.49123.1b. [DOI] [PubMed] [Google Scholar]
- Li ST, Matsushita M, Moriwaki A, Saheki Y, Lu YF, Tomizawa K, Wu HY, Terada H, Matsui H. HIV-1 Tat inhibits long-term potentiation and attenuates spatial learning. Ann Neurol. 2004;55:362–371. doi: 10.1002/ana.10844. [DOI] [PubMed] [Google Scholar]
- Liu Y, Jones M, Hingtgen CM, Bu G, Laribee N, Tanzi RE, Moir RD, Nath A, He JJ. Uptake of HIV-1 Tat protein mediated by low-density lipoprotein receptor-related protein disrupts the neuronal metabolic balance of the receptor ligands. Nat Med. 2000;6:1380–1387. doi: 10.1038/82199. [DOI] [PubMed] [Google Scholar]
- Maragos WF, Tillman P, Jones M, Bruce-Keller AJ, Roth S, Bell JE, Nath A. Neuronal injury in hippocampus with human immunodeficiency virus transactivating protein, Tat. Neuroscience. 2003;117:43–53. doi: 10.1016/s0306-4522(02)00713-3. [DOI] [PubMed] [Google Scholar]
- Masliah E, Terry R. The role of synaptic proteins in the pathogenesis of disorders of the central nervous system. Brain Pathol. 1993;3:77–85. doi: 10.1111/j.1750-3639.1993.tb00728.x. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Haughey NJ, Nath A. Cell death in HIV dementia. Cell Death Differ. 2005;12(Suppl 1):893–904. doi: 10.1038/sj.cdd.4401577. [DOI] [PubMed] [Google Scholar]
- May P, Woldt E, Matz RL, Boucher P. The LDL receptor-related protein (LRP) family: an old family of proteins with new physiological functions. Ann Med. 2007;39:219–228. doi: 10.1080/07853890701214881. [DOI] [PubMed] [Google Scholar]
- Meller R, Thompson SJ, Lusardi TA, Ordonez AN, Ashley MD, Jessick V, Wang W, Torrey DJ, Henshall DC, Gafken PR, Saugstad JA, Xiong ZG, Simon RP. Ubiquitin Proteasome-Mediated Synaptic Reorganization: A Novel Mechanism Underlying Rapid Ischemic Tolerance. J Neurosci. 2008;28:50–59. doi: 10.1523/JNEUROSCI.3474-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minagar A, Commins D, Alexander JS, Hoque R, Chiappelli F, Singer EJ, Nikbin B, Shapshak P. NeuroAIDS: characteristics and diagnosis of the neurological complications of AIDS. Molecular diagnosis and therapy. 2008;12:25–43. doi: 10.1007/BF03256266. [DOI] [PubMed] [Google Scholar]
- Nath A, Psooy K, Martin C, Knudsen B, Magnuson DS, Haughey N, Geiger JD. Identification of a human immunodeficiency virus type 1 Tat epitope that is neuroexcitatory and neurotoxic. J Virol. 1996;70:1475–1480. doi: 10.1128/jvi.70.3.1475-1480.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nath A, Conant K, Chen P, Scott C, Major EO. Transient exposure to HIV-1 Tat protein results in cytokine production in macrophages and astrocytes. A hit and run phenomenon. J Biol Chem. 1999;274:17098–17102. doi: 10.1074/jbc.274.24.17098. [DOI] [PubMed] [Google Scholar]
- Perez A, Probert AW, Wang KK, Sharmeen L. Evaluation of HIV-1 Tat induced neurotoxicity in rat cortical cell culture. J Neurovirol. 2001;7:1–10. doi: 10.1080/135502801300069575. [DOI] [PubMed] [Google Scholar]
- Peterson C, Neal JH, Cotman CW. Development of N-methyl-D-aspartate excitotoxicity in cultured hippocampal neurons. Brain Res Dev Brain Res. 1989;48:187–195. doi: 10.1016/0165-3806(89)90075-8. [DOI] [PubMed] [Google Scholar]
- Prendergast MA, Rogers DT, Mulholland PJ, Littleton JM, Wilkins LH, Jr, Self RL, Nath A. Neurotoxic effects of the human immunodeficiency virus type-1 transcription factor Tat require function of a polyamine sensitive-site on the N-methyl–aspartate receptor. Brain Res. 2002;954:300–307. doi: 10.1016/s0006-8993(02)03360-7. [DOI] [PubMed] [Google Scholar]
- Price RW, Brew B, Sidtis J, Rosenblum M, Scheck AC, Cleary P. The brain in AIDS: central nervous system HIV-1 infection and AIDS dementia complex. Science. 1988;239:586–592. doi: 10.1126/science.3277272. [DOI] [PubMed] [Google Scholar]
- Rechsteiner M, Rogers SW. PEST sequences and regulation by proteolysis. Trends Biochem Sci. 1996;21:267–271. [PubMed] [Google Scholar]
- Sa MJ, Madeira MD, Ruela C, Volk B, Mota-Miranda A, Paula-Barbosa MM. Dendritic changes in the hippocampal formation of AIDS patients: a quantitative Golgi study. Acta Neuropathol (Berl) 2004;107:97–110. doi: 10.1007/s00401-003-0781-3. [DOI] [PubMed] [Google Scholar]
- Schellenberg GD, D'Souza I, Poorkaj P. The genetics of Alzheimer's disease. Current psychiatry reports. 2000;2:158–164. doi: 10.1007/s11920-000-0061-z. [DOI] [PubMed] [Google Scholar]
- Self RL, Mulholland PJ, Nath A, Harris BR, Prendergast MA. The human immunodeficiency virus type-1 transcription factor Tat produces elevations in intracellular is Ca2+ that require function of an N-methyl-D-aspartate receptor polyamine-sensitive site. Brain Res. 2004;995:39–45. doi: 10.1016/j.brainres.2003.09.052. [DOI] [PubMed] [Google Scholar]
- Shen M, Thayer SA. Cannabinoid receptor agonists protect cultured rat hippocampal neurons from excitotoxicity. Mol Pharmacol. 1998a;54:459–462. doi: 10.1124/mol.54.3.459. [DOI] [PubMed] [Google Scholar]
- Shen M, Thayer SA. The cannabinoid agonist Win55,212–2 inhibits calcium channels by receptor-mediated and direct pathways in cultured rat hippocampal neurons. Brain Res. 1998b;783:77–84. doi: 10.1016/s0006-8993(97)01195-5. [DOI] [PubMed] [Google Scholar]
- Song L, Nath A, Geiger JD, Moore A, Hochman S. Human immunodeficiency virus type 1 Tat protein directly activates neuronal N-methyl-D-aspartate receptors at an allosteric zinc-sensitive site. J Neurovirol. 2003;9:399–403. doi: 10.1080/13550280390201704. [DOI] [PubMed] [Google Scholar]
- Speth C, Stöckl G, Mohsenipour I, Würzner R, Stoiber H, Lass-Flörl C, Dierich MP. Human immunodeficiency virus type 1 induces expression of complement factors in human astrocytes. J Virol. 2001;75:2604–2615. doi: 10.1128/JVI.75.6.2604-2615.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swann JW, Al-Noori S, Jiang M, Lee CL. Spine loss and other dendritic abnormalities in epilepsy. Hippocampus. 2000;10:617–625. doi: 10.1002/1098-1063(2000)10:5<617::AID-HIPO13>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- Tymianski M, Spigelman I, Zhang L, Carlen PL, Tator CH, Charlton MP, Wallace MC. Mechanism of action and persistence of neuroprotection by cell-permeant Ca2+ chelators. J Cereb Blood Flow Metab. 1994;14:911–923. doi: 10.1038/jcbfm.1994.122. [DOI] [PubMed] [Google Scholar]
- Uo T, Kinoshita Y, Morrison RS. Apoptotic actions of p53 require transcriptional activation of PUMA and do not involve a direct mitochondrial/cytoplasmic site of action in postnatal cortical neurons. J Neurosci. 2007;27:12198–12210. doi: 10.1523/JNEUROSCI.3222-05.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, Fotouhi N, Liu EA. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303:844–848. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- Waataja JJ, Kim HJ, Roloff AM, Thayer SA. Excitotoxic loss of post-synaptic sites is distinct temporally and mechanistically from neuronal death. J Neurochem. 2008;104:364–375. doi: 10.1111/j.1471-4159.2007.04973.x. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Xiong Y, Yarbrough WG. ARF promotes MDM2 degradation and stabilizes p53: ARF-INK4a locus deletion impairs both the Rb and p53 tumor suppression pathways. Cell. 1998;92:725–734. doi: 10.1016/s0092-8674(00)81401-4. [DOI] [PubMed] [Google Scholar]
- Zink WE, Anderson E, Boyle J, Hock L, Rodriguez-Sierra J, Xiong H, Gendelman HE, Persidsky Y. Impaired spatial cognition and synaptic potentiation in a murine model of human immunodeficiency virus type 1 encephalitis. J Neurosci. 2002;22:2096–2105. doi: 10.1523/JNEUROSCI.22-06-02096.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]