

Abstract
A refined computational structural model of the oxygen-evolving complex (OEC) of photosystem II (PSII) is introduced. The model shows that the cuboidal core Mn3CaO4 with a “dangler” Mn ligated to a corner μ4-oxide ion is maximally consistent with the positioning of the amino acids around the metal cluster as characterized by XRD models and high-resolution spectroscopic data, including polarized-EXAFS of oriented single crystals and isotropic EXAFS. It is, therefore, natural to expect that the proposed structural model should be particularly useful to establish the structure of the OEC, consistently with high-resolution spectroscopic data, and for elucidating the mechanism of water-splitting in PSII as described by the intermediate oxidation states of the OEC along the catalytic cycle.
The oxygen-evolving complex (OEC) of photosystem II (PSII) is a Mn4Ca cofactor that catalyzes the oxidation of water to dioxygen in the thylakoid membrane of green plant chloroplasts and internal membranes of cyanobacteria during the light period of photosynthesis.1 Besides its biological significance, the OEC is a paradigm system for engineering direct solar fuel production catalysts since it is based on inexpensive metals and achieves an overall catalytic efficiency of water splitting that is yet to be matched by artificial systems. Elucidating its detailed molecular structure is, therefore, central for understanding the catalytic mechanism of photosynthetic water oxidation as well as for the development of biomimetic catalysts for oxidation chemistry.2 In this paper, we introduce for the first time a refined structural model of the OEC of PSII that is maximally consistent with both high-resolution polarized-Extended X-ray Absorption Fine Structure (polarized-EXAFS) spectroscopy of single crystals of PSII,3 and the positioning of the amino acids around the metal cluster as characterized by X-ray diffraction (XRD) data.4,5
The OEC has been extensively investigated by biochemical and spectroscopic studies, including mutagenesis work in combination with Fourier transform infrared (FTIR),6 fluorescence,7 and electron paramagnetic resonance (EPR) methods,8 time-resolved Mass Spectrometry (MS),9 XRD4,5 and X-ray absorption (XAS) spectroscopies,10 including both high-resolution EXAFS3 and X-ray absorption near-edge spectroscopy (XANES).3,10–12 However, the exact configuration of the Mn4Ca cluster and its ligation scheme have yet to be established.
Recent breakthroughs in XRD studies have resolved nearly all cofactors and most of the amino acid residues in PSII at 3.0 3.5 Å resolution.4,5 However, the precise positions of the Mn ions, substrate water molecules, and proteinaceous ligands could not be resolved since the coordinate error in the density maps of the OEC was as high as 1 Å 13 and the resolution of bridging ligands was typically out of reach.14 Nevertheless, the XRD models4,5 have tentatively proposed positions for the Mn ions consistently with the overall electronic density maps and the metal-metal distances determined by EXAFS studies.15 In addition, the XRD model 1S5L from the cyanobacterium Thermosynechococcus elongatus4 suggested μ-oxo bridges linking the metal centers, forming a Mn3CaO4 cuboidal core with a ‘dangler’ Mn ligated to a cornerμ4-oxide ion similar to earlier proposals,8,16,17 (Fig. 1, top left). ‘Dangler Mn’ models were originally proposed within a set of 11 possible structural motives, suggested by solution EXAFS studies, from which the ‘dimer-of-dimers’ model was extensively discussed.17 Similar ‘dangler Mn’ models were also preferred by 55Mn-electron nuclear double resonance (ENDOR) studies that strongly disfavored the ‘dimer-of-dimers’ motif over models with a trinuclear-Mn core and a fourth Mn set off from the core by a longer Mn-Mn internuclear distance.18 However, recent polarized-EXAFS studies of single crystals have disfavored ‘dangler’ models, and have reduced the original set of 11 possible models to four models (see models I, II, IIa and III from Ref. [3], also described in the Supporting Information document).
Figure 1.

Comparison of the Mn4Ca inorganic core of the OEC of PSII as described by the empirical model IIa from Ref. [3] (right) and the XRD model 1S5L4 (left).
Figure 1 compares the Mn4Ca inorganic core proposed by the XRD model 1S5L (top, left) and one of the polarized-EXAFS models from Ref. [3] (top right), a model that reproduces the experimental polarized-EXAFS data along the three axes of the PSII crystal (Fig. 2, top). In contrast, the polarized-EXAFS spectra calculated from the XRD model 1S5L show significant deviations when compared to experimental data (Fig. 2, bottom). The calculated EXAFS spectra (blue lines in Fig. 2) were obtained by using the ab initio real space Green function approach, as implemented in the program FEFF8 (version 8.2),19 using the coordinates for the Mn4Ca cluster provided in Ref. [3] (Fig. 2, top) and Ref. [4] (Fig. 2, bottom), respectively.
Figure 2.
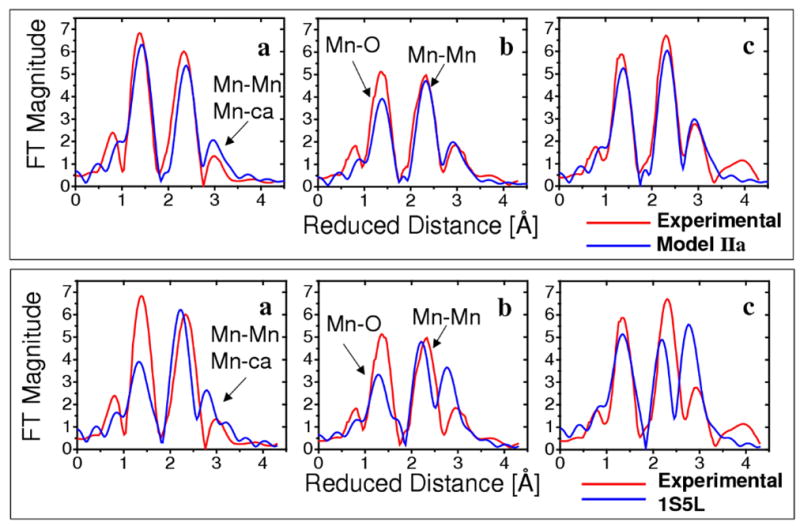
Comparison of the experimental polarized-EXAFS spectra (red),3 along the PSII crystal axes a, b and c, and the calculated spectra (blue) based on the polarized-EXAFS model IIa from Ref. [3] (top) and the XRD model 1S5L (bottom).4
The disagreement between the experimental EXAFS spectra and the simulated spectra based on the XRD model 1S5L is likely due to the intrinsic errors of the XRD model with moderate 3.5 Å resolution. In addition to structural disorder leading to XRD structures that might represent some sort of average of oxidized and photoreduced states, there is strong evidence for significant photoreduction of the Mn4Ca cluster due to the high doses of X-rays required during XRD data collection (where fully reduced Mn centers are expected to have lower coordination numbers than high-valent Mn).11,20 In contrast, EXAFS spectra3,10 are collected with much lower X-ray doses and therefore can provide metrical data of the OEC at a much higher (0.02 Å) resolution.
The four polarized-EXAFS models of the OEC, reported in Ref. [3], are currently considered the most ‘rigorous’ empirical models of the Mn4Ca metal cluster since they only empirical models that properly describe the dependence of high-resolution EXAFS amplitudes with the orientation of 3-dimensionally ordered single crystals of PSII. However, structural differences between some of them (e.g., Model I and Model II) are large. Furthermore, placing any of the four models into the XRD structures of PSII results in unsatisfactory metal-ligand distances, coordination numbers and geometries.21 Such inconsistencies might be due to the coordinate error in the X-ray crystal models, or the intrinsic limitations of polarized-EXAFS models built by neglecting the contributions of electron scattering paths from the ligands (i.e., assuming that the EXAFS amplitudes result solely from scattering paths associated with metal centers and oxo-bridges in the inorganic core).
In contrast to the empirical models, the recently developed DFT-QM/MM model of PSII22,23 is largely consistent with the Mn3CaO4 cuboidal core of the OEC and the distribution of proteinaceous ligands suggested by XRD measurements4,5 as well as with isotropic EXAFS spectra (see also supporting information).10 In addition, as shown in Fig. 3 (top panel), the calculated spectra obtained with the DFT-QM/MM model of the OEC reproduces the angular dependence of polarized-EXAFS amplitudes in much better agreement with experiments than the XRD model 1S5L (see Fig. 2, bottom panel). Nevertheless, the deviations shown in Fig. 3 (top panel) leave the OEC structure partially underdetermined until quantitative agreement between calculated and experimental high-resolution data is reached. Such agreement, however, is currently beyond the limitations of DFT methods, with typical errors in metal-metal and metal-ligand distances of the order of 0.05 Å.24 Therefore, a perfect match between the simulated EXAFS spectra based on DFT optimized structures and high-resolution EXAFS data (with an error of the order of 0.02 Å) is not expected.
Figure 3.
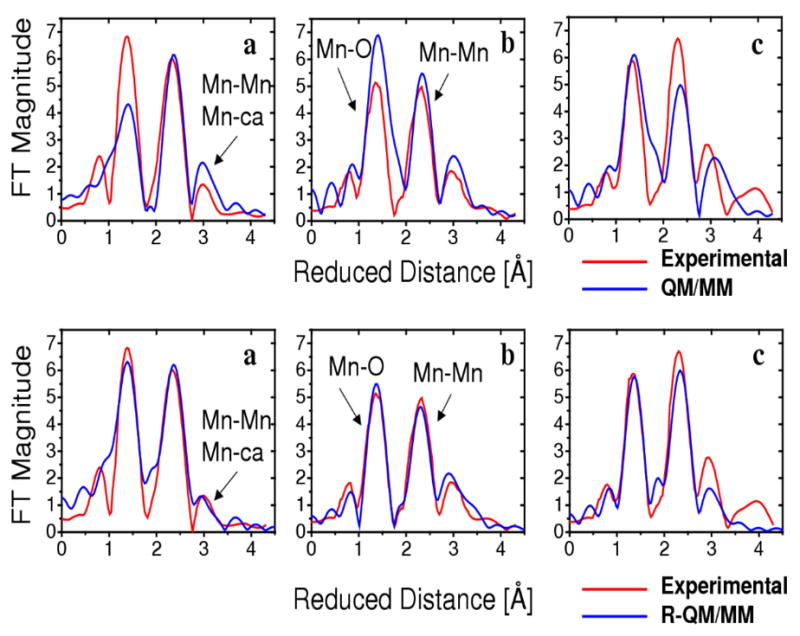
Comparison of the experimental polarized-EXAFS spectra (red),3 along the PSII crystal axes a, b and c, and the calculated spectra (blue) based on the DFT-QM/MM model (top)22 and the R-QM/MM model (bottom) introduced in this paper.
In order to obtain a model of the OEC in quantitative agreement with high-resolution spectroscopic data, beyond the limitations of current DFT methods, we have developed an EXAFS structural refinement method based on simulations of isotropic and polarized EXAFS spectra and direct comparisons with experimental data. The underlying computational procedure iteratively adjusts the molecular configuration of the system of interest, minimizing a scoring function defined in terms of the sum of squared deviations between calculated and experimental EXAFS spectra and a quadratic penalty factor that ensures minimum displacements of the nuclear positions relative to a reference configuration (i.e., the minimum energy DFT-QM/MM structure). Upon convergence of such a structural refinement procedure, we find that the isotropic and polarized EXAFS spectra of the resulting refined (R)-QM/MM model are in quantitative agreement with experimental data (see Fig. 3, lower panel, and the supporting information document for the comparison of isotropic EXAFS spectra). In spite of the agreement with high-resolution spectroscopic data, however, the R-QM/MM model does not rule out other possibilities since it is only a local solution (relative to the reference DFT-QM/MM structure) obtained by a refinement protocol based on umbrella sampling of configurations next to the reference DFT-QM/MM structure.
In contrast to the polarized-EXAFS models, the resulting R-QM/MM structure preserves intact the architecture of the cuboidal core Mn3CaO4 with a “dangler” Mn ligated to a corner μ4-oxide ion, as well as the DFT-QM/MM ligation scheme of the reference configuration (see Fig. 4). The refined model core also has good qualitative agreement with the analogous core in the 1S5L XRD structure and semiquantitative agreement with the reference DFT-QM/MM model. The main advantage of the refined model, relative to the earlier structures, is that the R-QM/MM model has metal-metal and metal-ligand distances and orientations corrected to ensure quantitative agreement with high-resolution spectroscopic data (see Table 1, Fig. 3, and extended table of interatomic distances and coordinates provided in the supporting information document).
Figure 4.
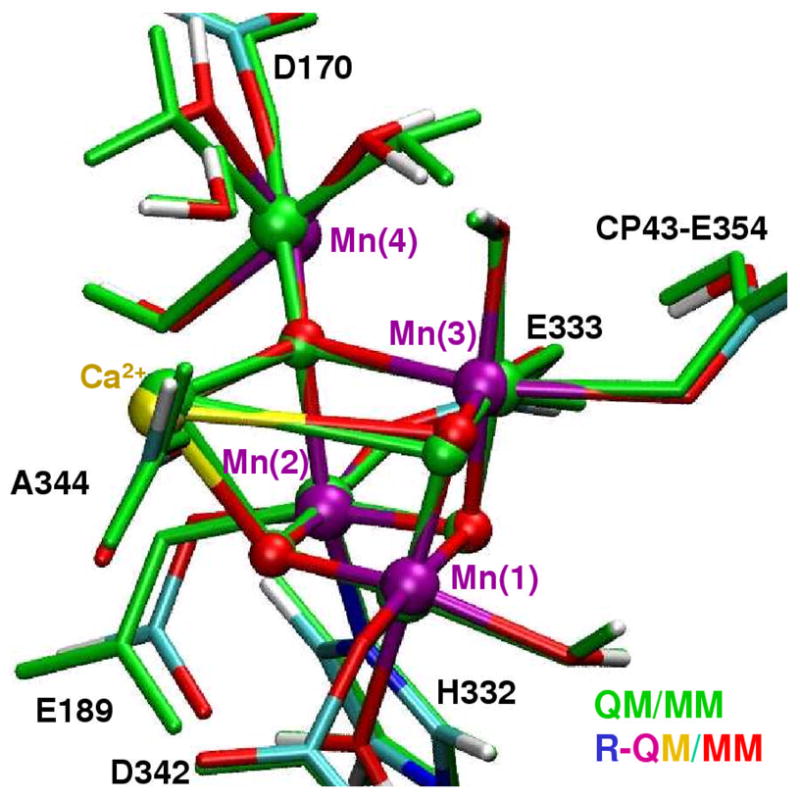
Comparison of structural models of the Mn4Ca metal cluster and ligands of the OEC of PSII, as described by the QM/MM (green) and the R-QM/MM (colored) models.
Table 1.
Metal-metal distances and bond-vector angles relative to the membrane normal in the QM/MM22 and R-QM/MM structural models of the OEC of PSII and the 1S5L XRD model.4
| Bond Vector | QM/MM | R-QM/MM | X-Ray | |||
|---|---|---|---|---|---|---|
| Å | ° | Å | ° | Å | ° | |
| Mn(1)Mn(2) | 2.76 | 57 | 2.74 | 58° | 2.65 | 59° |
| Mn(1)Mn(3) | 2.76 | 85 | 2.63 | 82° | 2.67 | 79° |
| Mn(2)Mn(3) | 2.82 | 63 | 2.77 | 67° | 2.72 | 71° |
| Mn(2)Mn(4) | 3.34 | 54 | 3.34 | 56° | 3.25 | 58° |
| Mn(3)Mn(4) | 3.72 | 29 | 3.68 | 37° | 3.26 | 38° |
| Ca-Mn(2) | 3.31 | 53 | 3.29 | 51° | 3.40 | 59° |
| Ca-Mn(3) | 3.95 | 35 | 3.88 | 36° | 3.38 | 39° |
In summary, we have developed a structural model of the OEC of PSII that is maximally consistent with both high-resolution polarized-EXAFS3 and isotropic-EXAFS10 as well as with the distribution of amino acid residues predicted by XRD.4,5 The R-QM/MM model involves a cuboidal core Mn3CaO4, with a “dangler” Mn ligated to a corner μ4-oxide ion, partially consistent with previous proposals,4,18,22,23 including empirical models,4,18 and DFT-QM/MM structures.22,23 Furthermore, the proteinaceous ligation of the Mn4Ca cluster provides metal-ligand distances, metal coordination numbers, and configurations of amino-acid residues surrounding the Mn4Ca cluster that are consistent with two XRD structures.4,5 It is, therefore, natural to expect that the R-QM/MM model should be particularly useful to establish the structure of the OEC consistently with high-resolution spectroscopic data, as well as for elucidating the water-splitting mechanism in PSII as described by the intermediate oxidation states of the OEC along the catalytic cycle.23
Supplementary Material
Acknowledgments
V.S.B. acknowledges supercomputer time from NERSC and financial support from the grants NSF CHE # 0345984, NSF ECCS # 0404191, NIH 2R01-GM043278-14, DOE DE-FG02-07ER15909 and the US-Israel BSF. G.W.B acknowledges support from the National Institutes of Health grant GM32715. E.M.S. thanks Dr. J. Yano for the numerical values of parameters associated with the simulations of polarized-EXAFS.
Footnotes
Supporting Information Available: Description of the methods used to prepare and analyze the computational structural models of the OEC of PSII, including figures and coordinates of the resulting models and a detailed comparative analysis of EXAFS spectra. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.McEvoy JP, Brudvig GW. Chem Rev. 2006;106:4455–4483. doi: 10.1021/cr0204294. [DOI] [PubMed] [Google Scholar]
- 2.Abuabara SG, Cady CW, Baxter JB, Schmuttenmaer CA, Crabtree RH, Brudvig GW, Batista VS. J Phys Chem C. 2007;111(32):11982–11990. [Google Scholar]; Limburg J, Vrettos JS, Liable-Sands LM, Rheingold AL, Crabtree RH, Brudvig GW. Science. 1999;283(5407):1524–1527. doi: 10.1126/science.283.5407.1524. [DOI] [PubMed] [Google Scholar]; Das S, Incarvito CD, Crabtree RH, Brudvig GW. Science. 2006;312(5782):1941–1943. doi: 10.1126/science.1127899. [DOI] [PubMed] [Google Scholar]
- 3.Yano J, Kern J, Sauer K, Latimer MJ, Pushkar Y, Biesiadka J, Loll B, Saenger W, Messinger J, Zouni A, Yachandra VK. Science. 2006;314:821–825. doi: 10.1126/science.1128186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ferreira KN, Iverson TM, Maghlaoui K, Barber J, Iwata S. Science. 2004;303(5665):1831–1838. doi: 10.1126/science.1093087. [DOI] [PubMed] [Google Scholar]
- 5.Loll B, Kern J, Saenger W, Zouni A, Biesiadka J. Nature. 2005;438:1040–1044. doi: 10.1038/nature04224. [DOI] [PubMed] [Google Scholar]
- 6.Debus RJ. In: Photosystem II: The Light-Driven Water:Plastoquinone Oxidoreductase. Wydrzynski TJ, Satoh K, editors. Vol. 22. Springer; Dordrecht: 2005. pp. 261–284. [Google Scholar]; Strickler MA, Walker LM, Hillier W, Britt RD, Debus RJ. Biochemistry. 2007;46:3151–3160. doi: 10.1021/bi062195e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bergmann U, Grush MM, Horne CR, DeMarois P, Penner-Hahn JE, Yocum CF, Wright DW, Dube CE, Armstrong WH, Christou G, Eppley HJ, Cramer SP. J Phys Chem B. 1998;102(42):8350–8352. [Google Scholar]
- 8.Britt RD, Campbell KA, Peloquin JM, Gilchrist ML, Aznar CP, Dicus MM, Robblee J, Messinger J. Biochim Biophys Acta. 2004;1655:158–171. doi: 10.1016/j.bbabio.2003.11.009. [DOI] [PubMed] [Google Scholar]
- 9.Hillier W, Wydrzynski T. Phys Chem Chem Phys. 2004;6(20):4882–4889. [Google Scholar]; Wydrzynski T, Hillier W, Messinger J. Physiologia Plantarum. 1996;96(2):342–350. [Google Scholar]
- 10.Haumann M, Muller C, Liebisch P, Iuzzolino L, Dittmer J, Grabolle M, Neisius T, Meyer-Klaucke W, Dau H. Biochemistry. 2005;44(6):1894–1908. doi: 10.1021/bi048697e. [DOI] [PubMed] [Google Scholar]
- 11.Yano J, Kern J, Irrgang KD, Latimer MJ, Bergmann U, Glatzel P, Pushkar Y, Biesiadka J, Loll B, Sauer K, Messinger J, Zouni A, Yachandra VK. Proc Natl Acad Sci USA. 2005;102(34):12047–12052. doi: 10.1073/pnas.0505207102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Haumann M, Liebisch P, Muller C, Barra M, Grabolle M, Dau H. Science. 2005;310:1019–1021. doi: 10.1126/science.1117551. [DOI] [PubMed] [Google Scholar]; Dau H, Liebisch P, Haumann M. Phys Chem Chem Phys. 2004;6(20):4781–4792. [Google Scholar]
- 13.Kamiya N, Shen JR. Proc Natl Acad Sci. 2003;100:98–103. doi: 10.1073/pnas.0135651100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dau H, Liebisch P, Haumann M. Phys Chem Chem Phys. 2004;6(20):4781–4792. [Google Scholar]
- 15.Robblee JH, Cinco RM, Yachandra VK. Biochim Biophys Acta. 2001;1503(1–2):7–23. doi: 10.1016/s0005-2728(00)00217-6. [DOI] [PMC free article] [PubMed] [Google Scholar]; Dau H, Iuzzolino L, Dittmer J. Biochim Biophys Acta. 2001;1503(1–2):24–39. doi: 10.1016/s0005-2728(00)00230-9. [DOI] [PubMed] [Google Scholar]
- 16.Peloquin JM, Campbell KA, Randall DW, Evanchik MA, Pecoraro VL, Armstrong WH, Britt RD. J Am Chem Soc. 2000;122(44):10926–10942. [Google Scholar]; Hasegawa K, Ono TA, Inoue Y, Kusunoki M. Chem Phys Lett. 1999;300(1–2):9–19. [Google Scholar]
- 17.Yachandra VK, Sauer K, Klein MP. Chemical Reviews. 1996;96(7):2927–2950. doi: 10.1021/cr950052k. [DOI] [PubMed] [Google Scholar]
- 18.Britt RD, Peloquin JM, Campbell KA. Ann Rev Biophys Biomol Struct. 2000;29:463–495. doi: 10.1146/annurev.biophys.29.1.463. [DOI] [PubMed] [Google Scholar]
- 19.Ankudinov AL, Bouldin CE, Rehr JJ, Sims J, Hung H. Physical Review. 2002;B 65(10) [Google Scholar]; Bouldin C, Sims J, Hung H, Rehr JJ, Ankudinov AL. X-Ray Spectrometry. 2001;30(6):431–434. [Google Scholar]
- 20.Grabolle M, Haumann M, Muller C, Liebisch P, Dau H. J Biolog Chem. 2006;281:4580–4588. doi: 10.1074/jbc.M509724200. [DOI] [PubMed] [Google Scholar]
- 21.Barber J, Murray JW. Phil Trans Royal Soc London Ser B-Biol Sci. 2008;363(1494):1129–1137. doi: 10.1098/rstb.2007.2208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sproviero EM, Gascon JA, McEvoy JP, Brudvig GW, Batista VS. J Chem Theor Comput. 2006;2:1119–1134. doi: 10.1021/ct060018l. [DOI] [PubMed] [Google Scholar]; Sproviero EM, Gascon JA, McEvoy JP, Brudvig GW, Batista VS. Curr Op Struct Biol. 2007;17:173–180. doi: 10.1016/j.sbi.2007.03.015. [DOI] [PubMed] [Google Scholar]; Sproviero EM, Gascon JA, McEvoy JP, Brudvig GW, Batista VS. Coord Chem Rev. 2008;252:395–415. doi: 10.1016/j.ccr.2007.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]; Sproviero EM, Shinopoulos K, Gascon JA, McEvoy JP, Brudvig GW, Batista VS. Phil Trans Royal Soc London Series B - Biol Sci. 2008;363:1149–1156. doi: 10.1098/rstb.2007.2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sproviero EM, Gascon JA, McEvoy JP, Brudvig GW, Batista VS. J Am Chem Soc. 2008;130:3428–3442. doi: 10.1021/ja076130q. [DOI] [PubMed] [Google Scholar]
- 24.Sproviero EM, Gascon JA, McEvoy JP, Brudvig GW, Batista VS. J Inorg Biochem. 2006;100:786–800. doi: 10.1016/j.jinorgbio.2006.01.017. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.