

The gross structure of a streptomycete derived bisanthraquinone designated as BE-43472B was first claimed in a Japanese patent in 1996 as an antitumor agent.[1] Ten years later, Rowley and co-workers reported the isolation of a bisanthraquinone antibiotic to which they assigned the relative configuration of structure (−)-1 (Figure 1) from streptomyces strain #N1-78-1, isolated from cultured cells of an unidentified unicellular green alga (URI strain #N36-11-10), which, in turn, was isolated from the ascidian Ecteinascidia turbinata, collected from La Parguera, Puerto Rico.[2a] Although the structure (−)-1 was depicted in that[2a] and a subsequent publication,[2b] these studies left the absolute stereo-chemistry of this natural product unassigned. It was speculated[2a] on the basis of spectroscopic data comparisons that this antibiotic was most likely the same as the one previously discovered by the Japanese group.[1] In addition to antitumor properties, this bisanthraquinone antibiotic exhibited potent inhibitory activity against clinically derived isolates of methicillin-susceptible, methicillin-resistant, and tetracyclin-resistant Staphylococcus aureus (MSSA, MRSA, and TRSA, respectively), and vancomycin-resistant Enterococcus faecium (VRE).[2] Most impressively, this compound also demonstrated, in a time-kill study, significant bactericidal activity (>99.9 % kill) against MSSA, MRSA, and VRE.[2b] Given the urgency for new antibiotics to combat drug-resistant bacteria,[3] the novelty of the molecular structure of (−)-1 and the uncertainty about its absolute stereo-chemistry, this molecule became an attractive target for synthesis in our group.[4] In this communication, we report the first total synthesis of both enantiomers of this novel antibiotic and the assignment of its absolute configuration as that depicted by structure (+)-1 (Figure 1).
Figure 1.
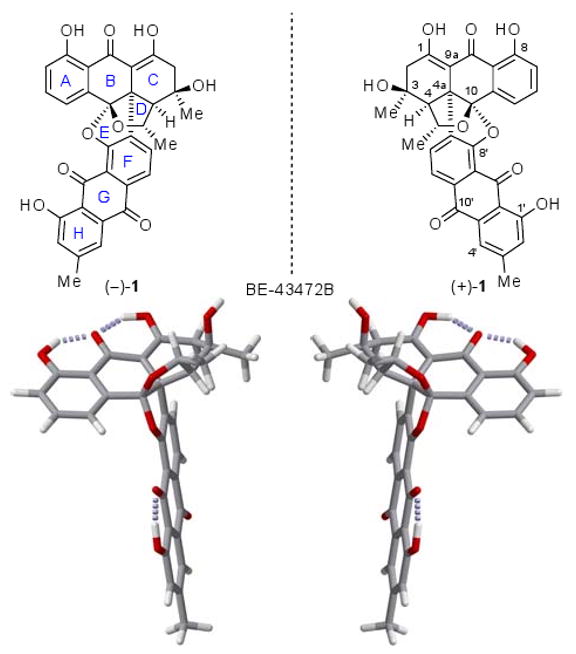
Enantiomers of the bisanthraquinone antibiotic BE-43472B [(−)-1 and (+)-1] and their computer-generated models (fully optimized on B3LYB/6-31G* level) (carbon: gray, hydrogen: white, oxygen: red, H-bonds: dotted gray).
The unprecedented and unique structure of bisanthraquinone natural product BE-43472B [(+)-1] is a composition of two different anthraquinone-type molecules joined together by a sterically hindered carbon-carbon bond and an oxygen bridge in an assembly containing no less than eight rings. Its five stereogenic centers are clustered on and around the DE ring system that contains an oxygen atom in each of its rings, forming an internal ketal. Its overall shape is that of a T, with its two sides more or less flat and with its junction where the stereocenters reside slightly distorted (see calculated structures, Figure 1).
Scheme 1 presents, in retrosynthetic format, the blueprint of the devised synthesis of bisanthraquinone antibiotic BE-43472B [(+)-1] (and its enantiomer [(−)-1]). Thus, disconnection of the ether bridge between the two anthraquinone moieties of the molecule by rupture of the aryl-oxygen bond (opening of ring E) and appropriate modifications of ring C led to the potential precursor 2. Dismantling the hemiketal ring of 2 then generated cyclohexene derivative 3, opening the way for the Diels–Alder reaction as the key step of the strategy.[5] That disconnection defined diene 4 and dienophile 5 as the required building blocks for our projected synthesis. The precise structures of these Diels–Alder partners were crucial for the desired regio- and stereochemical outcome of the [4+2] cycloaddition, as will be discussed below.
Scheme 1.

Retrosynthetic analysis of BE-43472B [(+)-1].
Scheme 2 summarizes the synthesis of the required diene 4 containing the first stereocenter of the molecule that would control the remaining four. Our starting material for this construction was an aldehyde derived from lactic acid methyl ester. Since the absolute configuration of our target was not known at the time, we chose the less expensive of the two enantiomers of this aldehyde [(S)-(−)-7] derived from natural, (S)-(−)-lactic acid ethyl ester which also happens to correspond to the structure of the natural product as depicted in the original structural elucidation report.[2] As events transpired, this choice led to (−)-1, the enantiomer of BE-43472B. Starting with (R)-(+)-7 (derived from unnatural (R)-(+)-lactic acid methyl ester)[6] we then synthesized (+)-1, the naturally occurring enantiomeric form of antibiotic BE-43472B; it is this synthesis we describe herein. Wittig reaction[7] of aldehyde (+)-7 with phosphorane 6 gave α,β-unsaturated ester 8 (95 % yield, E/Z >95:5), which was converted to ketone 10 (57 % yield over two steps) via addition of MeLi to the intermediate Weinreb amide 9.[8] Desilylation of 10 (TBAF, quantitative yield) led to allyl alcohol 11, which was converted to diene 4 by treatment with 2.3 equiv of LDA followed by trapping the resulting dianion with one equiv of TBSOTf (33 % yield, 93 % ee as determined by HPLC).
Scheme 2.

Synthesis of diene 4. Reagents and conditions: a) 7 (1.0 equiv), 6 (1.1 equiv), CH2Cl2, 25 °C, 6 h, 95 %; b) HNMe(OMe)•HCl (5.0 equiv), Me2AlCl (5.0 equiv), CH2Cl2, 0 → 25 °C, 16 h; c) MeLi, (2.0 equiv), Et2O, −78 °C, 1 h, 57 % over the two steps; d) TBAF (1.1 equiv), THF, 25 °C, 2.5 h, 100 %; e) LDA (2.3 equiv), THF, −78 °C; then TBSOTf (1.0 equiv), −78 °C, 1.5 h, −78 → 25 °C, 30 min, 33 % (93% ee, 70% yield brsm). brsm = based on recovered starting material; LDA = lithium diisopropylamide; TBAF = tetra-n-butyl-ammonium fluoride; TBSOTf = tert-butyldimethylsilyl triflate.
The synthesis of the dienophile 5 commenced with known tricyclic system 12 and proceeded as shown in Scheme 3.[9] Dibromination of 12 (LHMDS, NBS) yielded dibromoketone 13 (96 % yield), which upon sequential exposure to DBU and CsCO3-Me2SO4 resulted in the formation of anthracene derivative 15, through phenol 14, in 85 % overall yield for the two steps. Subsequent CAN oxidation furnished anthraquinone 16, in 77 % yield. Stannylation of 16 with Me3SnSnMe3 in the presence of Pd(PPh3)4 cat. led to stannane 17 (81 % yield), whose Pd(PPh3)4-catalyzed Stille coupling with bromoquinone 18 furnished pentacycle 19 in 65 % yield. Finally, removal of the MOM protecting group from 19 under acidic conditions (1 % HCl in MeOH) led to the desired dienophile bisquinone 5, in 86 % yield.
Scheme 3.

Synthesis of dienophile 5. Reagents and conditions: a) LHMDS (2.2 equiv), NBS (2.2 equiv), THF, 0 → 25 °C, 1 h, 98 %; b) DBU (1.0 equiv), CH2Cl2, 25 °C, 12 h; c) Me2SO4 (1.2 equiv), Cs2CO3 (1.2 equiv), acetone, reflux, 5 h, 74 % over the two steps; d) CAN (2.0 equiv), CH2Cl2, MeCN, H2O, 0 °C, 30 min, 77 %; e) Pd(PPh3)4 (0.1 equiv), Me3SnSnMe3 (1.5 equiv), toluene, 110 °C, 2 h, 95 %; f) 17 (1.0 equiv), 18 (1.5 equiv), Pd(PPh3)4 (0.1 equiv), CuI (0.2 equiv), THF, 70 °C, 22 h, 65 %; g) 1 % HCl in MeOH, CH2Cl2, 25 °C, 16 h, 85 %. CAN = cerium(IV) ammonium nitrate; DBU = 1,8-diazabicyclo-[5.4.0]undec-7-ene; LHMDS = lithium hexamethyldisilazide; MOM = methoxymethyl; NBS = N-bromosuccinimide.
With the two components 4 and 5 in hand, the stage was now set for their much anticipated union through a Diels–Alder reaction.[5] As seen in Scheme 4, heating 4 and 5 in CH2Cl2 at 85 °C in a sealed tube for 48 h, followed by addition of toluene and refluxing with a Dean-Stark apparatus for another 48 h, led to a remarkable series of transformations to afford octacyclic product 22 and its C-9a epimer epi-22 (epi-22:22 ca. 2:1) in 98 % overall yield. The only structural element not controlled in this fascinating cascade sequence was the easily epimerizable stereogenic center at C-9a, an element of little consequence, if any, since this stereocenter had to be subsequently erased. The success of this cascade depended crucially on special features encoded in the two reacting partners, diene 4 and dienophile 5. To explain the control of the high diastereoselectivity in forming the challenging quaternary stereocenter at C-4a, it is assumed that in the endo transition state TS-20 (Scheme 4) the free hydroxy group within 4 hydrogen bonds with the more Lewis basic carbonyl oxygen of the quinone moiety of dienophile 5 (the other carbonyl group is already engaged in H-bonding), orienting the reactive species in the indicated facial arrangement due to 1,3-allylic strain[10] and steric reasons.[11,12] The exquisite regiocontrol leading to the exclusive formation of Diels–Alder adduct 3 can also be explained by intuitive analysis of the molecular orbital coefficients of the reactants, if the bisanthraquinone substituent on the quinone dienophile is construed to be an electron withdrawing group and thereby enhancing the MO coefficient at C-9a (for the diene a larger coefficient is expected at C-1). The regioselective H–bonding may amplify the required orbital interactions.[13] Adduct 3 was observed by NMR spectroscopic analysis to exist in equilibrium with its hemiketal form 2 (2:3 ca. 4:1, benzene-d6). Manual molecular modelling indicated that 2 finds itself in a favourable conformation to undergo an intramolecular nucleophilic ipso substitution (2 → 22),[14] expelling a molecule of MeOH to afford the desired octacycle 22. This novel cyclization is apparently facilitated by the presence of the adjacent phenolic quinone moiety, which, being self-activated through H–bonding, invites the attack by the initiating hemiketal hydroxyl group as shown in structures 2 and 21.[15] The product 22 of this cascade reaction apparently epimerizes partially at C-9a under the toluene refluxing conditions, which also ensures the removal of the departing MeOH, as well as on silica gel to furnish epi-22. In refluxing benzene we also observed partial epimerization of hemiketal 2 yielding an equilibrating mixture of 2 and epi-2. With compounds epi-22 and 22 in hand, the stage was now set for the final drive to the target molecule (+)-1.
Scheme 4.

Diels–Alder reaction / hemiketal formation / nucleophilic aromatic substitution cascade sequence to form octacyclic structure 22. Reagents and conditions: 5 (1.0 equiv), 4 (2.0 equiv), CH2Cl2, 85 °C, sealed tube, 2 d; then toluene, 135°C, dean stark, 1 d, epi-22:22 (ca. 2:1 mixture of epimers) 98 % overall yield based on 5.
Scheme 5 depicts the final sequence that led to the completion of the total synthesis of antibiotic BE-43472B [(+)-1]. Thus, mCPBA epoxidation of epi-22/22 (2:1)[16] furnished epoxides 23 (major epimers, ca. 4:1 mixture with its α-epimers, not shown), which were desilylated (HF•py) and then oxidized with SeO2 in AcOH to afford enone 25 (49 % yield over the three steps) through hydroxy ketone intermediates 24, together with its chromatographically separable 3-epi-stereoisomer (epi-25, not shown, 39 % yield).
Scheme 5.

Completion of the total synthesis of BE-43472B [(+)-1]. Reagents and conditions: a) mCPBA (1.2 equiv), CH2Cl2, −20 °C, 2 h; b) HF•pyr (10.0 equiv), THF, 25 °C, 20 min, then TMSOMe; c) SeO2 (5.0 equiv), AcOH, 120 °C, 15 h, 25, 49 % and epi-25, 39 % over the three steps; d) HSCH2CH2SH, as solvent, BF3•OEt2 (10.0 equiv), 25 °C, 30 min, 63 %; e) Raney-Nickel 2400, MeOH, air, 25 °C, 1.5 h, then aq. HCl, 27, 45 % (56 % brsm); f) tBuOOH (5.0 equiv), DBU (3.0 equiv), CH2Cl2, −25 °C, 28, 84 % and β-28, 7 % ; g) hν (mercury lamp), benzene, 25 °C, 7 h, 77 % (83 % yield brsm). mCPBA = meta-chloroperbenzoic acid; py = pyridine.
An X-ray crystallographic analysis (see ORTEP drawing, Figure 2)[17,18] of single crystals of 25 (m. p. >250 °C, CH2Cl2) proved its relative configuration and confirmed the outcome of both the Diels–Alder and the epoxidation reactions. With this pleasing advance, the removal of the superfluous oxygen atom at C-2 and the introduction of the hydroxy group at C-1 became the next tasks. In a notably regioselective manner, tetracarbonyl compound 25 was reacted with ethane-1,2-dithiol (as solvent) in the presence of excess BF3•OEt2 to afford the desired dithioketal 26 (63 % yield), a substrate that underwent reductive desulfurization with Raney-Ni to generate enone 27 (45 % yield). All that was now left to reach the target molecule was the insertion of the missing oxygen atom in the C-H bond of the enone moiety of the latter compound. This was achieved through a remarkably smooth procedure involving, first, epoxidation of 27 to give epoxide 28 (tBuOOH, 84 % yield, plus the β-epimer of 28, 7% yield, separable by preparative TLC, silica), and then, reorganization of the epoxide structural motif to the required enol moiety through photolysis[19] of 28 to afford bisanthraquinone antibiotic BE-43472B [(+)-1] in 77 % yield (83 % yield brsm, 92 % conversion).[20] Synthetic (+)-1 exhibited essentially identical physical properties to those reported for the natural product,[1,2a] and so did (−)-1, except for its optical rotation which was of the opposite sign.[21]
Figure 2.
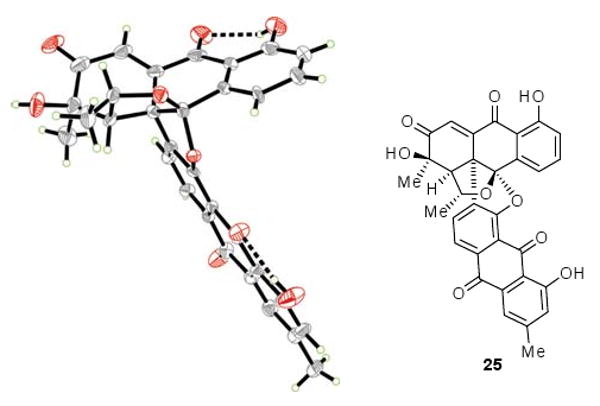
X-ray derived ORTEP drawing of 25 (Thermal ellipsoids are shown at 30 % probability).[17]
In addition to assigning the absolute configuration of bisanthraquinone antibiotic BE-43472B [(+)-1], the described total synthesis demonstrates the increasing power of cascade reactions in chemical synthesis[22] and opens the way for the construction of designed analogues of this intriguing antitumor antibiotic for biological investigations.
Supplementary Material
Footnotes
We gratefully acknowledge grants from the National Science Foundation (CHE-0603217) and National Institutes of Health (USA) and fellowships from the Alexander von Humboldt Foundation (J.B.), A*STAR (Y.H.L.), and the Skaggs-Oxford Scholarship program (Y.H.L.). We thank Prof. D. C. Rowley and A. Socha for a sample of BE-43472B [(+)-1].
Supporting information for this article is available on the WWW under http://www.angewandte.org or from authors.
References
- 1.Kushida H, Nakajima S, Koyama T, Suzuki H, Ojiri K, Suda H. Antitumoric BE-43472 manufacture with streptomyces. JP 08143569. 1996 [Google Scholar]
- 2.a) Socha AM, Garcia D, Sheffer R, Rowley DC. J Nat Prod. 2006;69:1070. doi: 10.1021/np050449b. [DOI] [PubMed] [Google Scholar]; b) Socha AM, LaPlante KL, Rowley DC. Bioorg Med Chem. 2006;14:8664. doi: 10.1016/j.bmc.2006.08.038. [DOI] [PubMed] [Google Scholar]
- 3.For recent reviews on this topic, see: Taubes G. Science. 2008;321:356. doi: 10.1126/science.321.5887.356.Nicolaou KC, Chen JS, Edmonds DJ, Estrada AA. Angew Chem. 2009;121:670. doi: 10.1002/anie.200801695.Angew Chem Int Ed. 2009;48:660.
- 4.For previous synthetic studies inspired by BE-43472B, see: Suzuki K, Takikawa H, Hachisu Y, Bode JW. Angew Chem. 2007;119:3316. doi: 10.1002/anie.200605138.Angew Chem Int Ed. 2007;46:3252.Takikawa H, Hikita K, Suzuki K. Angew Chem. 2008;120:10035. doi: 10.1002/anie.200801577.Angew Chem Int Ed. 2008;47:9887.
- 5.Diels O, Alder K. Justus Liebigs Ann Chem. 1926;450:237.Reviews: Corey EJ. Angew Chem. 2002;114:1724.Angew Chem Int Ed. 2002;41:1650.Nicolaou KC. Angew Chem. 2002;114:1742.Angew Chem Int Ed. 2002;41:1668.
- 6.Marshall JA, Yanik MM, Adams ND, Ellis KC, Chobanian HR. Org Synth. 2005;81:157. [Google Scholar]
- 7.Wittig G, Schöllkopf U. Chem Ber. 1954;87:1318. [Google Scholar]
- 8.Nahm S, Weinreb SM. Tetrahedron Lett. 1981;22:3815. [Google Scholar]
- 9.a) Nicolaou KC, Lim YH, Papageorgiou CD, Piper JL. Angew Chem. 2005;117:8131. [Google Scholar]; Angew Chem Int Ed. 2005;44:7917. [Google Scholar]; b) Nicolaou KC, Lim YH, Piper JL, Papageorgiou CD. J Am Chem Soc. 2007;129:4001. doi: 10.1021/ja0685708. [DOI] [PubMed] [Google Scholar]
- 10.Hoffmann RW. Chem Rev. 1989;89:1841. [Google Scholar]
- 11.For Diels–Alder reactions with substituted naphthoquinones, see: Trost BM, Ippen J, Vladuchick WC. J Am Chem Soc. 1977;99:8116.Boeckman RK, Jr, Dolak TM, Culos KO. J Am Chem Soc. 1978;100:7098.Kelly TR, Montury M. Tetrahedron Lett. 1978;45:4311.Rozeboom MD, Tegmo-Larsson IM, Houk KN. J Org Chem. 1981;46Tietze LF, Gericke KM, Singidi RR, Schuberth I. Org Biomol Chem. 2007;5:1191. doi: 10.1039/b700838d.
- 12.For a discussion of transition states of Diels–Alder reactions with chiral 1,3-dienes, see: McDougal PG, Jump JM, Rojas C, Rico JG. Tetrahedron Lett. 1989;30:3897.Carreño MC, Garciá-Cerrada S, Urbano A, Di Vitta C. J Org Chem. 2000;65:4355. doi: 10.1021/jo000210u.Barriault L, Thomas JDO, Clément R. J Org Chem. 2003;68:2317. doi: 10.1021/jo020664m.
- 13.The rather puzzling exquisite regiochemical outcome of this Diels–Alder reaction cannot be fully explained at this time and studies directed towards its further understanding are continuing. It should be noted however, that that a diene corresponding to 4 with a MEM-O group at its C-1 terminus (rather than a TBS-O group at its C-2 position) reacted with dienophile 5 to give exclusively the opposite regioisomeric Diels–Alder product. Further details will be reported in the full account of this work.
- 14.An alternative, but less likely, mechanism for the conversion of 2 to 22:epi-22 may involve initial oxonium ion formation followed by intramolecular attack from the nearby MeO group and extrusion of MeOH.
- 15.For examples of intermolecular SN(Ar) ipso substitution of a MeO group by carbon nucleophiles, see: Fuson RC, Speck SB. J Am Chem Soc. 1942;64:2446.Meyers AI, Mihelich ED. J Am Chem Soc. 1975;97:7383.Aki S, Haraguchi Y, Sakikawa H, Ishigami M, Fujioka T, Furuta T, Minikawa JI. Org Proc Res Dev. 2001;5:535.Reviews: Gaertner R. Chem Rev. 1949;45:493.Burnett JF, Zahler RE. Chem Rev. 1951;49:273.To the best of our knowledge, the present case is the first example of an intramolecular SN(Ar) ipso substitution of a MeO group by an oxygen nucleophile. For the introduction of the term ipso substitution see: Perrin CL, Skinner GA. J Am Chem Soc. 1971;93:3389.
- 16.Since epimers 22 and epi-22 are hardly separable and because the subsequent intermediates 23 and 24 were also shown to epimerize under the reaction conditions employed for their generation, it was more expedient and efficient to carry the mixture of epi-22 and 22 through the three-step sequence and separate the obtained enone 25 and its C-3 epimer epi-25 by chromatography.
- 17.CCDC-714178 contains the supplementary crystallographic data for 25 and is available free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
- 18.In the initial synthesis of (−)-1, we also obtained the X-ray crystal structure of ent-25. CCDC-711967 contains the supplementary crystallographic data for ent-25.
- 19.a) Bodforss S. Ber deut chem Ges. 1918;51:214. [Google Scholar]; b) Jeger O, Schaffner K. Pure Appl Chem. 1970;21:247. doi: 10.1351/pac197021020247. [DOI] [PubMed] [Google Scholar]
- 20.Interestingly, the minor epimeric oxirane epi-28 remained unchanged under the same photolytic conditions.
- 21.Since the spectroscopic data and optical rotation of our synthetic BE-43472B [(+)-1] are essentially the same as those reported in the Japanese patent[2] and ref. [2a] for naturally occurring BE-43472B [(+)-1], we assume that the two latter compounds are one and the same.
- 22.Reviews: Nicolaou KC, Montagnon T, Snyder SA. Chem Commun. 2003:551. doi: 10.1039/b209440c.Tietze LF, Brasche G, Gericke K. Domino Reactions in Organic Synthesis. Wiley-VCH; Weinheim: 2006. p. 672.Nicolaou KC, Edmonds DJ, Bulger PG. Angew Chem. 2006;118:7292. doi: 10.1002/anie.200601872.Angew Chem Int Ed. 2006;45:7134.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.