

Abstract
Three free base porphyrins have been prepared that bear a polar and facially encumbering 2,4,6-tris(carboxymethoxy)phenyl motif at one meso (5-) position. The only other substituent (15-position) comprises phenyl, formyl, or p-aminophenyl. The porphyrins exhibit solubility in water (or aqueous buffer solutions) at pH ≥7 and concentrations >1 mM at room temperature. The concise syntheses, water-solubility, and bioconjugatable handle make these porphyrin constructs suitable for biological applications.
Keywords: Porphyrin, Water, Hydrocarbon, Water-soluble, Facial encumbrance, Bioconjugation
1. Introduction
Tetrapyrrolic molecules have distinct chemical and photophysical properties that are attractive for a wide range of diagnostic and therapeutic applications in the field of photomedicine. The applications encompass optical imaging of diseased tissue,1–3 fluorescent labeling in flow cytometry,4–7 photodynamic inactivation of microbial infections,8–10 and photodynamic therapy of solid tumors.11–15 A key challenge to the implementation of tetrapyrrolic molecules in photomedicine entails tailoring the molecules to exhibit appropriate solubility. In particular, synthetic motifs that impart water solubility are important for a number of biological studies. Tetrapyrrolic macrocycles (porphyrins, chlorins, and bacteriochlorins) present a particular challenge in this regard owing to the large size of the planar hydrocarbon macrocycle. In recent years, approaches for solubilizing related carbon-rich molecules such as fullerenes and nanotubes have relied on grafting polar substituents to create a polar layer or sheath that encompasses the hydrophobic molecule.16–20 For biological applications of tetrapyrrolic macrocycles, a typical requirement is to introduce substituents to achieve both water-solubility and facility for conjugation to biomolecules.21
Water solubilization of porphyrins has traditionally been achieved by use of uroporphyrin, which bears eight β-substituted alkylcarboxylic acids,22–26 or by attachment of small aryl motifs such as N-alkyl pyridinium or phenylsulfonic acid units to the meso sites of synthetic porphyrins27 (Chart 1). In the latter case, four polar substituents (one at each of the meso sites) traditionally were introduced to impart water solubility. The use of four substituents stemmed from the synthetic methods of the day,28 which only readily supported the synthesis of such A4-porphyrins. Regardless, the most easily synthesized members – the pyridinium- or sulfoaryl-derivatized porphyrins – exhibit only limited solubility in aqueous media. Moreover, the sense that four such groups were essential also presented an impediment to further functionalization of the macrocycle, as required for applications where bioconjugation is required. The advent of versatile and rational synthetic routes to compact porphyrins that bear less than four meso substituents29,30 (e.g., trans-AB-porphyrins) has opened the door to consideration of new molecular designs for water solubilization that are both potent and compatible with features required for bioconjugation.
Chart 1.

In parallel with the development of new synthetic methods, new water-solubilization motifs have been developed to serve a variety of objectives. Representative examples of such solubilization motifs include oligoethylene glycol monomers31 and dendrimers,32 alkyl polyamines,33 and polycarboxy chains.34 While each such group has merit, most are quite large and are not well suited for our goals in developing compact bioconjugatable molecules for use in photomedical applications.
We initiated a program to develop water-solubilization motifs that ideally adhere to the following design guidelines: (1) ionic groups are more potent than non-ionic groups for solubilization, (2) anionic groups are generally preferred over cationic groups to minimize non-specific binding to cellular structures, (3) the projection of polar substituents above and below the plane of the macrocycle is expected to suppress cofacial aggregation of the porphyrins, and (4) solubilization is desired with a single group so as to achieve a compact molecular architecture. The emphasis on groups that project over the porphyrin macrocycle stemmed from our finding that the mesityl group and homologues (e.g., 2,4,6-triethylphenyl) impart increased organic solubility (versus phenyl) to porphyrins.35,36 The results from this program include a 1,5-diphosphonopent-3-yl group (swallowtail moiety, I),7,37 a N,N-diethylimidazolium-2-yl unit (II),38 and a 2,6-bis(phosphonomethoxy)phenyl moiety (III)39 (Chart 2). The first two solubilization moieties (I, II) have been incorporated as one substituent in trans-AB-porphyrins wherein the distal substituent constituted a bioconjugatable handle (carboxylic acid, iodoacetamide).7,37,39 The diphosphonate solubilization moieties (I, III) have been incorporated in a synthetic chlorin.39 Jux also has developed facially encumbering motifs for porphyrins by reaction of a 2,6-bis(bromomethyl)aryl group located at the porphyrin meso-position with 4-tert-butylpyridine or with diethyl malonate.40–43
Chart 2.

Here we describe an additional water-solubilization motif that provides facial encumbrance, the 2,4,6-tris(carboxymethoxy)phenyl unit. This motif is incorporated as one substituent in a trans-AB-porphyrin. The other substituent is a handle suitable for bioconjugation. Typical groups for bioconjugation include isothiocyanate and imidate esters (for amines) and iodoacetamide (for sulfhydryls).44 To perform bioconjugation in aqueous solution, and therefore avoid the solubility problems that can arise with mixed aqueous-organic media, the carboxy moieties must be deprotected. Hence, the conjugatable groups should be compatible with the carboxylic motif to allow for selective bioconjugation. We have elected to focus on aldehyde and amine groups, which are suitable for reductive amination in aqueous media. Reductive amination has been used successfully for derivatization of tetrapyrrolic macrocycles.45–48 This design complements prior examples of trans-AB-porphyrins that bear a bioconjugatable handle including isothiocyanate,21 carboxylic acid/ester,7,38 and iodoacetamide.37 Taken together, the work described herein provides an additional approach for the design and synthesis of compact, water-soluble, bioconjugatable porphyrins.
2. Results and discussion
2.1. Molecular design and synthesis strategy
Each target molecule (H2P-1–3) is a free base trans-AB-porphyrin (Chart 3). The trans-AB-porphyrin architecture, wherein the porphyrin contains only two meso substituents, was chosen to maintain a compact size. Each of the porphyrins is tailored with a water-solubilizing 2,4,6-tris(carboxymethoxy)phenyl substituent at one meso position. The 2,4,6-tris(carboxymethoxy)phenyl unit is relatively compact. The mass of this water-solubilizing moiety (C12H11O9, 299 Da) is comparable to that of the core porphine unit (C20H12N4, 308 Da) in the trans-AB-porphyrin.
Chart 3.

The 2,4,6-tris(carboxymethoxy)phenyl unit is an o,o′-disubstituted meso-aryl motif, whereby the polar groups are (i) in close proximity to the porphyrin macrocycle and (ii) are thrust in an orientation that encumbers the faces of porphyrin, thereby impeding aggregation. The introduction of 2,6-disubstituted aryl units to porphyrins has presented challenges over the years; however, the o-benzyloxy motif is compatible with the steric constraints encountered upon formation of the pyrromethane intermediate (e.g., dipyrromethane, porphyrinogen) on the path to the porphyrin.49 Indeed, we previously synthesized porphyrins that bear 2,6-bis(benzyloxy) and 2,4,6-tris(benzyloxy) substituents at the meso positions wherein a number of substituents were introduced on the arene moiety of the benzyloxy group.36,50,51
The conjugatable groups should be compatible with carboxylic acids to allow for selectivity during the bioconjugation step. Hence, conjugatable groups include a formyl group for porphyrin H2P-2 and a p-aminophenyl group for H2P-3. Porphyrin H2P-1, which has a meso-phenyl substituent in lieu of a bioconjugatable handle, was prepared as a benchmark for the solubility studies.
The synthesis relied on two methods for the preparation of trans-AB-porphyrins. One method employs condensation of a 1,9-bis(N,N-dimethylaminomethyl)dipyrromethane with a dipyrromethane in refluxing ethanol containing zinc acetate followed by DDQ oxidation.29 The second method requires the 1,9-diformylation of a dipyrromethane and subsequent imination of the 1,9-diformyldipyrromethane by treatment with n-propylamine. Reaction of the resulting 1,9-bis(imino)dipyrromethane with a dipyrromethane in the presence of zinc acetate and air leads to the corresponding trans-AB-porphyrin.30 In both routes the zinc porphyrin is obtained. Both routes typically afford a single porphyrin product, albeit in rather low yield (5–30%).
The tert-butyl group was chosen as the protective moiety for each of the three carboxylic acids of the water-solubilization motif. The water-solubilization motif to be introduced thus contains three phenolic ethers and three esters. Accordingly, the 1,9-functionalization (imination or aminoalkylation) was performed on the complementary dipyrromethane.
2.2. Synthesis
2.2.1. Porphyrin H2P-1
The O-alkylation51,52 of 2,4,6-trihydroxybenzaldehyde with tert-butyl bromoacetate in the presence of K2CO3 gave the alkylated product 1 in 82% yield. Condensation53 of aldehyde 1 with pyrrole (40 equiv) in the presence of InCl3 followed by crystallization gave dipyrromethane 2a in 35% yield (Scheme 1). Condensation of dipyrromethane 2a with 1,9-bis(imino)dipyrromethane 3a30 in refluxing ethanol containing excess Zn(OAc)2 with exposure to air gave the corresponding protected zinc ester-porphyrin ZnP-1′ in 18% yield. Treatment of ZnP-1′ with aqueous KOH (2 M) in THF at 60 °C for 8 h afforded cleavage of the esters. The porphyrin was extracted in the aqueous layer and concentrated to give the zinc porphyrin with free carboxylic acids. Upon adjusting an aqueous solution of the latter compound to pH = 3 (by addition of 2 M aqueous HCl), the zinc porphyrin was demetalated and the resulting free base porphyrin H2P-1 was isolated as a precipitate in 82% yield (overall from ZnP-1′). The porphyrins were characterized by 1H NMR spectroscopy, ESI-MS or LD-MS (laser-desorption mass spectrometry in the absence of a matrix),54 absorption spectroscopy, and fluorescence spectroscopy.
Scheme 1.

2.2.2. Porphyrin H2P-2
Condensation of the 1,9-bis(N,N-dimethylaminomethyl)-5-acetaldipyrromethane 429 and dipyrromethane 2a in refluxing ethanol containing Zn(OAc)2 followed by oxidation with DDQ gave the corresponding zinc porphyrin ZnP-2′ in 10% yield (Scheme 2). Attempts to simultaneously cleave both the tert-butyl and the acetal protective groups with a biphasic solution55 of TFA/H2O/CH2Cl2 gave a mixture of porphyrins. Treatment of ZnP-2′ with neat TFA at room temperature for 3 days unveiled the carboxylic and formyl groups, giving H2P-2 in 77% yield. The structure of H2P-2 was confirmed by LD-MS, ESI-MS and 1H NMR analyses. The purity of the porphyrin H2P-2 was estimated to be 90% according to the 1H NMR analysis. Attempts to purify porphyrin H2P-2 employing reversed-phase (RP) chromatography were not successful.
Scheme 2.

To achieve greater purity, stepwise deprotection conditions were employed. The carboxylic acid units were unveiled by treatment of porphyrin ZnP-2′ with aqueous KOH (2 M) in THF at 60 °C for 12 h. Aqueous-organic workup and subsequent treatment of the crude product with neat TFA at 50 °C for 24 h resulted in deprotection of the formyl group. The crude reaction mixture was diluted with H2O to pH 3 to induce precipitation of the porphyrin. Sonication, centrifugation, decanting and drying of the resulting solid pellet gave porphyrin H2P-2 (overall 65% yield) in pure form without chromatography. Porphyrin H2P-2 was characterized by 1H NMR and 13C NMR spectroscopy, absorption and fluorescence spectroscopy, and LD-MS and ESI-MS analyses.
2.2.3. Porphyrin H2P-3
Dipyrromethane 2b56 was subjected to Vilsmeier formylation to give the corresponding 1,9-diformyldipyrromethane 5 in 68% yield. Condensation of 5 with excess n-propylamine gave the corresponding 1,9-bis(imino)dipyrromethane 3b quantitatively. The resulting 1,9-bis(imino)dipyrromethane 3b was employed promptly for porphyrin formation without further purification. The condensation of 1,9-bis(imino)dipyrromethane 3b and dipyrromethane 2a in refluxing toluene for 12 h gave porphyrin ZnP-3′ in 28% yield (Scheme 3). No other porphyrin products (e.g., derived from scrambling) were detectable by LD-MS analysis.
Scheme 3.

The deprotection of ZnP-3′ was examined under various conditions, including TFA in CH2Cl2 (6 days), neat TFA (2 days), concentrated aqueous HCl/CH2Cl2 (5 days), TMSBr/triethylamine in CHCl3 (2 days), and LiOH in aqueous THF (1 day), all of which gave incomplete reaction or mixtures of products. Treatment with KOH (2 M) and THF for 28 h at 60 °C gave a porphyrin that lacked the tert-butyl esters but retained the tert-BOC protective group on the amino moiety, as indicated by LD-MS and ESI-MS data. Treatment with TFA at room temperature for 11 h gave the target porphyrin H2P-3 in 83% overall yield. Porphyrin H2P-3 was characterized by 1H NMR and 13C NMR spectroscopy, absorption and fluorescence spectroscopy, and LD-MS and ESI-MS analyses.
2.3. Characterization
2.3.1. Molecular modeling
A molecular model of porphyrin H2P-1 is shown in Figure 1. The figure shows the projection of the carboxymethoxy substituents above and below the plane of the porphyrin macrocycle. A considerable range of motion is available to the 2,6-carboxymethoxy units owing to rotation about the single bonds of the carboxymethoxy moiety, and the torsion of the aryl unit with respect to the plane of the porphyrin (by rotation about the single bond between the aryl carbon and the porphyrin meso carbon). On account of the combined motions, the hydroxy moiety of the carboxylic acid can extend as far as the pyrrolic NH and pyrrolenyl units (and contact the π-chromophore of the porphyrin), project almost normal to the plane of the porphyrin macrocycle, or project back over the aryl unit.
Figure 1.
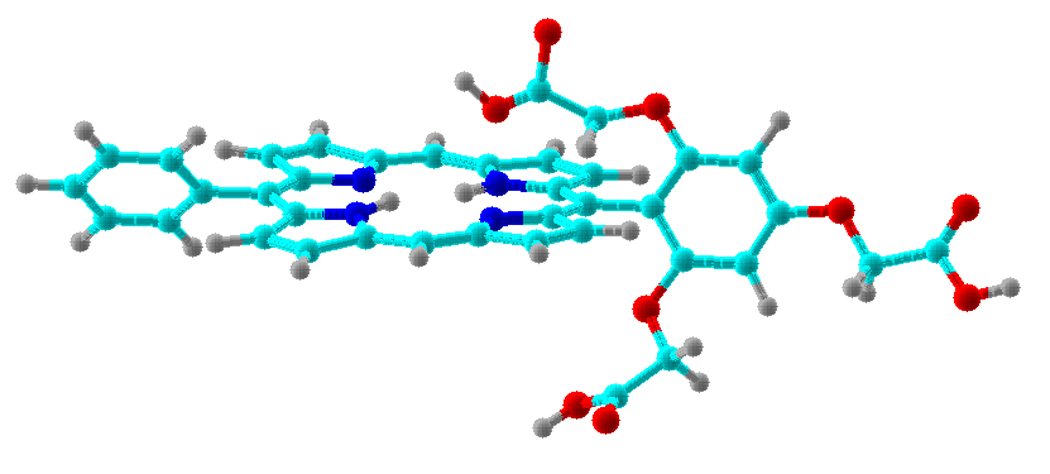
Molecular model of porphyrin H2P-1 showing conformations (not energy-minimized) of the 2,6-carboxymethoxy units attached to the aryl moiety.
2.3.2. Chemical characterization
All porphyrins were characterized by 1H NMR and 13C NMR spectroscopy, mass spectrometry (LD-MS, ESI-MS and/or FAB-MS), and absorption and fluorescence spectroscopy. Analytical RP-HPLC was also employed to confirm the purity of the hydrophilic porphyrins (H2P-1–3). The hydrophobic porphyrins (ZnP-1′–3′) were examined by 1H NMR spectroscopy in CDCl3, and by absorption and emission spectroscopy in toluene. The hydrophilic porphyrins (H2P-1–3) were examined by 1H NMR spectroscopy in DMSO, and by absorption and emission spectroscopy in water.
The hydrophilic porphyrins did not give observable NH and COOH signals upon 1H NMR spectroscopic analysis in DMSO-d6 due to broadening/exchange with solvent. The 1H NMR spectra were typically characteristic of meso-substituted porphyrins, with the exception of a splitting in the peak attributed to the inner NH of the free base porphyrins; this phenomenon occurred with each of H2P-1–3. A variable temperature experiment with H2P-2 over the range 20 °C to 80 °C showed that the peaks coalesced upon heating to 80 °C, with a coalescence temperature estimated to be ~60 °C (Figure 2). The origin of the splitting at room temperature is not known but likely stems from different conformations of the 2,6-bis(carboxymethoxy)phenyl unit (vide supra).
Figure 2.

Variable temperature 1H NMR spectra for porphyrin H2P-2 in DMSO-d6.
The hydrophilic porphyrins were examined by absorption spectroscopy in a range of solvents, including THF, DMSO, water, and aqueous buffer solutions. In each case, the Soret band (~410 nm) was broadened in all solvents examined versus that of a benchmark trans-A2-porphyrin (5,15-diphenylporphyrin,57 H2P-4) in toluene or DMSO. The absorption spectrum of H2P-3 is displayed in three solvents (water, THF, DMSO) in Figure 3.
Figure 3.

Absorption spectra of porphyrin H2P-3 at room temperature in water (solid line), THF (dashed line) and DMSO (dotted line).
The breadth of the Soret band was assessed by the full width at half maximum (fwhm) for the hydrophilic porphyrins in a range of solvents (THF, MeOH, DMSO, water, aqueous buffer solutions of pH 7 and 10) and for the hydrophobic porphyrins in toluene and DMSO. Regardless of solvent, the hydrophilic porphyrins (H2P-1–3) exhibited broader Soret bands (~20–30 nm or greater versus ~11–14 nm) than those of the hydrophobic porphyrins (ZnP-1′–3′). This difference does not stem from the different metalation states (the former are free base porphyrins whereas the latter are zinc porphyrins) but must be due to the different conformations of the polar, ionizable carboxylic acid moieties (at the 2,6-positions of the meso-aryl unit) and resulting range of electronic interaction with the π-chromophore of the porphyrin. Indeed, the benchmark porphyrin (H2P-4) lacks the solubilizing feature and exhibits a sharp Soret band (fwhm = 13 nm). The results from an extensive study are summarized in Table 1.
Table 1.
Absorption spectra of porphyrins in various solvents
| Entry | Cmpd | M | Solvent | λSoret (nm) | fwhm (nm) | λQ (nm) |
|---|---|---|---|---|---|---|
| 1 | ZnP-1′ | Zn | toluene | 413 | 13 | 539, 573 |
| 2 | ZnP-1′ | Zn | MeOH | 410 | 10 | 544, 579 |
| 3 | ZnP-1′ | Zn | DMSO | 417 | 10 | 548, 583 |
| 4 | ZnP-2′ | Zn | toluene | 411 | 14 | 502, 539, 574 |
| 5 | ZnP-2′ | Zn | MeOH | 408 | 10 | 542, 577 |
| 6 | ZnP-2′ | Zn | DMSO | 414 | 11 | 502, 545, 580 |
| 7 | ZnP-3′ | Zn | toluene | 414 | 13 | 539, 575 |
| 8 | ZnP-3′ | Zn | THF | 414 | 11 | 545, 580 |
| 9 | ZnP-3′ | Zn | DMSO | 418 | 11 | 548, 583 |
| 10 | H2P-1 | H, H | DMSO | 416 | 27 | 506, 547, 577, 632 |
| 11 | H2P-1 | H, H | MeOH | 406 | 21 | 501, 542, 574, 629 |
| 12 | H2P-1 | H, H | H2O | 402 | 30 | 505, 564, 620 |
| 13 | H2P-1 | H, H | pH 7 | 402 | 20 | 504, 541, 617 |
| 14 | H2P-1 | H, H | pH 10 | 403 | 20 | 504, 541, 617 |
| 15 | H2P-2 | H, H | DMSO | 415 | 31 | 518, 560, 641 |
| 16 | H2P-2 | H, H | MeOH | 411 | 33 | 516, 559, 642 |
| 17 | H2P-2 | H, H | H2O | 407 | 33 | 519, 561, 637 |
| 18 | H2P-2 | H, H | pH 7 | 408 | 41 | 518, 562, 633 |
| 19 | H2P-2 | H, H | pH 10 | 395 | 63 | 519, 565, 637 |
| 20 | H2P-3 | H, H | DMSO | 414 | 34 | 507, 548, 579, 647 |
| 21 | H2P-3 | H, H | THF | 409 | 22 | 504, 541, 579, 636 |
| 22 | H2P-3 | H, H | H2O | 404 | 24 | 505, 543, 568, 623 |
| 23 | H2P-3 | H, H | pH 7 | 405 | 23 | 506, 544, 567, 622 |
| 24 | H2P-3 | H, H | pH 10 | 405 | 24 | 506, 544, 567, 622 |
| 25 | H2P-4 | H, H | toluene | 408 | 13 | 503, 535, 576, 633 |
| 26 | H2P-4 | H, H | DMSO | 407 | 13 | 502, 537, 574, 629 |
The only notable absorption spectral distinction observed in water versus other solvents among the hydrophilic porphyrins occurred for H2P-2, the porphyrin bearing a meso-formyl group. In DMSO, examination of the visible region showed peaks (declining order of intensity) at 561, 641, 584 (shoulder) and 518 nm. In water the peaks in the visible region were substantially broader, which remained unchanged upon dilution (vide infra), suggesting that these features are characteristic of the meso-formyl porphyrin in aqueous media rather than due to aggregation. Finally, upon increasing the pH to 10, the Soret band broadened to a fwhm of 63 nm (entry 19, Table 1).
The porphyrins also were characterized by fluorescence emission spectroscopy with excitation into the Soret band. The hydrophobic, zinc ester-porphyrins were analyzed in toluene and exhibited emission characteristic of zinc trans-AB-porphyrins, which consisted of two bands at ~578 and 632 nm. The hydrophilic, deprotected free base porphyrin analogues were analyzed in water and also exhibited two emission bands. The emission maxima were typical for free base trans-AB-porphyrins bearing meso-aryl substituents for H2P-1 (625 and 686 nm) and H2P-3 (629 and 688 nm), but were shifted bathochromically for H2P-2 (641 and 707 nm). The shift in the emission spectrum of H2P-2 stems from the presence of the meso-formyl substituent, which is known to be a potent auxochrome.58,59
2.3.3. Water solubility
The hydrophilic porphyrins (H2P-1–3) were examined for solubility at room temperature in aqueous media of physiologic pH 7.4 and higher. Solubility was assessed by visual inspection using pure water or aqueous buffer solutions (pH 7, 7.4, 8, and 10) at porphyrin concentrations of 10 to 14 mM (using short-pathlength cells). Each of the porphyrins H2P-1–3 gave an optically clear solution in an aqueous buffer solution at pH 7 or higher for porphyrin concentrations in this regime.
One method to detect the aggregation of porphyrins entails the comparison of absorption spectra of various concentrations. In the case of no aggregation, the shape of the absorption spectra remain constant upon change of concentration of the sample. For this determination, stock solutions of porphyrins H2P-1 (14 mM), H2P-2 (10 mM) and H2P-3 (14 mM) were prepared in an aqueous buffer at pH 7. The absorption spectra were collected in the same buffer solution at concentrations obtained by 1, 5 and 10 fold-dilution using cuvettes of different path lengths (l = 1, 5 and 10 mm), thereby in principle affording equal absorbancies. The spectra of porphyrins H2P-1–3 are shown in Figure 4. A very slight change was observed in the spectra of H2P-1 (over the concentration of 2.8, 0.56 and 0.28 mM) and H2P-2 (2.5, 0.5 and 0.25 mM), whereas the three spectra of porphyrin H2P-3 (3, 0.6 and 0.3 mM) were superimposable with each other. The spectra suggest that any aggregation across this concentration regime is at most very slight.
Figure 4.

Absorption spectra of porphyrins in aqueous buffer solution (pH = 7) at room temperature as a function of concentration. (A) Porphyrin H2P-1: (solid line) c = 2.8 mM, l = 1 mm; (dotted line) c = 0.56 mM, l = 5 mm; (dashed line) c = 0.28 mM, l = 10 mm. (B) Porphyrin H2P-2: (solid line) c = 2.5 mM, l = 1 mm; (dotted line) c = 0.5 mM, l = 5 mm; (dashed line) c = 0.25 mM, l = 10 mm. (C) Porphyrin H2P-3: (solid line) c = 3 mM, l = 1 mm; (dotted line) c = 0.6 mM, l = 5 mm; (dashed line) c = 0.3 mM, l = 10 mm.
3. Outlook
The molecular design of water-soluble tetrapyrrolic molecules generally begins with a non-polar, carbon-rich macrocycle that must be functionalized to achieve water solubility (and bioconjugatability). The use of tetrapyrrolic molecules in aqueous media might seem unnecessarily complex, given the availability of numerous organic dyes that are soluble in water. However, such dyes typically bear an intrinsic charge; consequently, the synthetic chemistry and purification of elaborate analogues of such dyes can be quite cumbersome.60 The tetrapyrrole family presents the opportunity to carry out all synthetic manipulations on neutral, non-ionic compounds in organic solution, and then unveil the groups for water-solubilization and bioconjugation in the last step of the synthetic process. In addition, few organic dyes present the opportunities for molecular tuning that are available with the tetrapyrrolic macrocycles by virtue of chelation of different metals, reduction of the macrocycle (porphyrin, chlorin, bacteriochlorin), and introduction of diverse substituents.
The trans-AB-porphyrins described herein provide a compact architecture with controlled substitution pattern. The presence of a single conjugatable group enables formation of a single product upon bioconjugation, as opposed to a mixture that results with porphyrins that have multiple reactive functional groups. It is noteworthy that chromatography was not employed in the purification of the water-soluble porphyrins upon unveiling the water-solubilizing carboxylic acid moieties. As anticipated, introduction of the polar and facially encumbering 2,4,6-tris(carboxymethoxy)phenyl motif on the macrocyclic perimeter increased the hydrophilicity of the porphyrins. The hydrophilic porphyrins proved to be soluble in aqueous buffer solutions at pH ≥ 7 and concentrations of at least 1 mM at room temperature. The target molecules are amenable to conjugation with biologically active molecules.
We now have described four motifs for imparting water solubility to tetrapyrrolic macrocycles,7,37–39 all of which rely on facial encumbrance of polar groups. The potency of water-solubilization appears to decline in the order of diphosphonate-bearing groups (swallowtail, 2,6-aryl) > 2,4,6-tris(carboxymethoxy)phenyl > N,N-dialkylimidazolium-2-yl. One distinction between the three types of groups is that the diphosphonate-derivatized group is expected to be ionized (and hence to impart solubility) at essentially all physiological pH ranges, the carboxy-derivatized group would be expected to be ionized only at neutral to basic pH ranges, and the imidazolium group is permanently charged. Accordingly, the use of the carboxy-containing motif described herein may be expected to enable ready penetration of lipid membranes and also selective precipitation upon exposure to acidic environments. The former feature may be essential for bioavailability whereas the latter may be attractive for localization in sub-cellular domains, as required for staining or for targeted molecular brachytherapy.61
4. Experimental section
4.1. General methods
All 1H NMR spectra (400 MHz) and 13C NMR spectra (100 MHz) were collected in CDCl3 unless noted otherwise. The DMSO-d6 employed was 99.9% pure. Absorption spectra were collected in toluene at room temperature unless noted otherwise. Mass spectra of porphyrins were obtained via laser desorption mass spectrometry (LD-MS) without a matrix,54 by high-resolution fast atom bombardment mass spectrometry (FAB-MS) using a matrix of nitrobenzyl alcohol and polyethylene glycol, and by electrospray ionization mass spectrometry (ESI-MS). Positive ions were detected unless noted otherwise. Silica gel (40 µm average particle size) was used for column chromatography. Molecular modeling was carried out using Pcmodel version 7.50.00 (Serena Software).
The aqueous buffer solutions employed were the following: (i) potassium phosphate (0.05 M, pH 7); (ii) potassium phosphate monobasic (0.05 M, pH 7) was titrated with sodium hydroxide to give pH 7.4 or pH 8; and (iii) potassium carbonate/potassium borate/potassium hydroxide (0.05 M, pH 10).
Analytical RP-HPLC in all cases was carried out using a reversed-phase, C-18 column (5 um, 125 mm × 4 mm) with the following elution program: flow rate = 1.5 mL/min; 0–2 min, 0% B; 2–40 min 95% B; 40–50 min 100% A; A = water (0.1% TFA), B = methanol (0.1% TFA); detection at 405 nm. In all cases, the void volume corresponded to t = 2 min.
4.2. Noncommercial compounds
Dipyrromethanes 2b,56 3a,30 and 429 were prepared following reported procedures.
4.3. Synthetic procedures
4.3.1. 2,4,6-Tris(tert-butoxycarbonylmethoxy)benzaldehyde (1)
Following the K2CO3 alkylation method,51,52 a solution of 2,4,6-trihydroxybenzaldehyde (2.0 g, 13 mmol) in anhydrous DMF (43 mL, 0.3 M) was treated with K2CO3 (17.8 g, 129 mmol, dried in vacuo at 140 °C overnight), and the mixture was heated at 60 °C. After 30 min, tert-butyl bromoacetate (7.70 mL, 51.6 mmol) was added dropwise to the reaction mixture, and the temperature was set at 80 °C. After stirring for 1 h at 80 °C, the reaction mixture was allowed to reach ambient temperature. The reaction mixture was treated with water (20 mL) and extracted with ethyl acetate. The organic layer was washed (brine and water), separated, dried (Na2SO4) and concentrated to a red oil. Purification by column chromatography [silica, hexanes/ethyl acetate (1:1)] gave a red-brown viscous oil that solidified upon standing for 1 day at room temperature (5.2 g, 82%): mp 55–60 °C; 1H NMR δ 1.48 (s, 18H), 1.49 (s, 9H), 4.49 (s, 2H), 4.57 (s, 4H), 6.01 (s, 2H), 10.43 (s, 1H); 13C NMR δ 28.2, 65.8, 66.8, 82.9, 83.2, 93.5, 110.6, 162.3, 163.7, 166.9, 167.1, 187.5; FAB-MS obsd 497.2396, calcd 497.2387 (C25H36O10); Anal. Calcd for C25H36O10: C, 60.47; H, 7.31; Found: C, 60.03; H, 7.33.
4.3.2. 5-(2,4,6-Tris(tert-butoxycarbonylmethoxyphenyl)dipyrromethane (2a)
Following a procedure for dipyrromethane formation,53 a sample of aldehyde 1 (1.53 g, 3.08 mmol) and pyrrole (8.4 mL, 0.12 mol) was purged with argon for 10 min. Then InCl3 (66 mg, 0.30 mmol) was added, and the mixture was stirred at room temperature under argon. After 1 h, the reaction mixture was treated with powdered NaOH (360 mg, 6.00 mmol), and stirring was continued for 30 min. The reaction mixture was filtered through a Buchner funnel, and the filtrate was concentrated. The resulting solid was treated with hexanes, and the suspension was sonicated. The volatile components were evaporated. This sequence of operations was repeated thrice in order to remove traces of pyrrole. Crystallization from ethanol-water afforded an ash-grey solid (659 mg, 35%): mp 107–112 °C; 1H NMR δ 1.45 (s, 18H), 1.50 (s, 9H), 4.39 (s, 6H), 5.94–5.97 (m, 2H), 6.01–6.06 (m, 2H), 6.09 (s, 2H), 6.23 (s, 1H), 6.64–6.68 (m, 2H), 9.50 (br, 2H); 13C NMR δ 28.26, 28.29, 32.8, 66.1, 66.8, 82.8, 106.3, 107.4, 116.2, 116.6, 132.7, 158.1, 167.8, 168.3; FAB-MS obsd 612.3082, calcd 612.3047 (C33H44N2O9).
4.3.3. Zn(II)-5-Phenyl-15-[2,4,6-tris(tert-butoxycarbonylmethoxy)phenyl]porphyrin (ZnP-1′)
Following a reported procedure,30 a solution of 2a (150 mg, 0.245 mmol) and 3a (88.0 mg, 0.245 mmol) in EtOH (25 mL, 0.01 M) was treated with Zn(OAc)2 (450 mg, 2.45 mmol) and heated to reflux for 16 h. The heat source was removed, and the reaction mixture was concentrated. Purification by column chromatography [alumina, hexanes/ethyl acetate (1:3)] gave a pink solid (40 mg, 18%): 1H NMR δ 1.24 (s, 18H), 1.25 (s, 9H), 4.11 (s, 4H), 4.75(s, 2H), 6.51 (s, 2H), 7.77–7.80 (m, 3H), 8.25–8.28 (m, 2H), 9.11 (d, J = 4.4 Hz, 2H), 9.21 (d, J = 4.7 Hz, 2H), 9.40 (d, J = 4.4, 4H), 10.26 (s, 2H); 13C NMR δ 28.1, 28.4, 66.4, 66.7, 82.2, 82.9, 93.7, 105.9, 126.8, 127.6, 131.5, 132.26, 132.39, 132.54, 134.9, 143.0, 149.5, 149.8, 151.4, 159.83, 159.99, 167.9; LD-MS obsd 914.5; ESI-MS obsd 915.2927, calcd 915.2942 [(M + H)+; M = C50H51N4O9Zn]; λabs 412, 538 nm; λem (λex = 412 nm) 578, 632 nm.
4.3.4. 5-Phenyl-15-[2,4,6-tris(carboxymethoxy)phenyl]porphyrin (H2P-1)
A solution of ZnP-1′ (20 mg, 0.022 mmol) in THF (4 mL, 5 mM) was treated with 1 mL of aqueous KOH (2 M) and heated at 60 °C. After 8 h, CH2Cl2 (15 mL) and H2O (15 mL) were added to the reaction mixture. The aqueous layer, which contained the porphyrin, was separated and concentrated to a pink solid. The solid was dissolved in H2O (3 mL) and treated with aqueous HCl (2 M) to obtain pH = 3, whereupon the porphyrin precipitated. The mixture was sonicated, centrifuged, and decanted to afford a solid pellet. Water was added followed by sonication, centrifugation, and decanting; this treatment was performed four times, whereupon the supernatant was colorless. The pellet was dried in vacuo to afford a red-brown solid (12 mg, 80% yield): 1H NMR (DMSO-d6) δ –3.32 (s, 1H), −3.24 (s, 1H), 4.46 (s, 4H), 4.95 (s, 2H), 6.66 (s, 2H), 7.87–7.90 (m, 3H), 8.28–8.31 (m, 2H), 9.01 (d, J = 4.9 Hz, 2H), 9.10 (d, J = 4.4 Hz, 2H), 9.53 (d, J = 4.4, 2H), 9.63 (d, J = 4.9, 2H), 10.53 (s, 2H); 13C NMR δ 64.7, 65.2, 92.8, 105.1, 110.9, 127.3, 130.5, 131.3, 132.1, 134.6, 159.1, 169.7, 170.1; ESI-MS obsd 684.1866, calcd 684.1856 [(M − H)+; M = C38H28N4O9]; λabs (H2O) 401, 504, 563 nm; λem (λex = 401 nm) 625, 686 nm; RP-HPLC tR = 30.05 min. In one case, the deprotection was carried out with isolation of the zinc ester-porphyrin: A solution of ZnP-1′ (9 mg, 0.009 mmol) in THF (2.0 mL, 4.5 mM) was treated with 0.5 mL of aqueous KOH (2 M) and heated at 60 °C. After 8 h, CH2Cl2 (10 mL) and H2O (10 mL) were added to the reaction mixture. The aqueous layer, which contained the porphyrin, was separated and concentrated to give Zn(II)-5-phenyl-15-[2,4,6-tris(carboxymethoxy)phenyl]porphyrin as a pink solid: 1H NMR (methanol-d4) δ 4.09 (s, 4H), 4.69 (s, 2H), 6.78 (s, 2H), 7.80–7.82 (m, 3H), 8.22–8.25 (m, 2H), 8.98 (d, J = 4.4 Hz, 2H), 9.17 (d, J = 4.4 Hz, 2H), 9.36–9.40 (m, 4H), 10.23 (s, 2H); ESI-MS obsd 745.0917, calcd 745.0918 [(M − H)+; M = C38H26N4O9Zn; λabs (H2O) 409, 543 nm; λem (λex = 409 nm) 587, 638 nm.
4.3.5. Zn(II)-5-(5,5-Dimethyl-1,3-dioxan-2-yl)-15-[2,4,6-tris(tert-butoxycarbonylmethoxy)phenyl]-porphyrin (ZnP-2′)
Following a reported procedure,29 a solution of 2a (0.163 g, 0.267 mmol) and 4 (0.100 g, 0.267 mmol) in ethanol (26 mL, 10 mM) was treated with anhydrous Zn(OAc)2 (0.489 mg, 2.67 mmol) and heated to reflux. After 16 h, DDQ (0.181 g, 0.801 mmol) was added, and the mixture was stirred for 15 min. Triethylamine (187 µL, 1.33 mmol) was added, and the reaction mixture was concentrated to dryness. Column chromatography [alumina, hexanes/ethyl acetate, (1:3)] afforded a purple solid (26 mg, 10%): 1H NMR δ 1.23 (s, 27H), 1.64 (s, 6H), 4.11 (s, 4H), 4.38–4.39 (m, 4H), 4.74 (s, 2H), 6.49 (s, 2H), 8.10 (s, 1H), 9.19 (d, J = 4.3 Hz, 2H), 9.36 (d, J = 4.3 Hz, 2H), 9.50 (d, J = 4.3 Hz, 2H), 10.16 (d, J = 4.7 Hz, 2H), 10.24 (s, 2H); 13C NMR δ 22.9, 25.4, 28.0, 28.2, 29.9, 31.2, 66.4, 66.6, 67.6, 80.5, 82.2, 82.9, 93.5, 106.0, 107.0, 130.4, 131.8, 132.43, 132.58, 149.3, 149.5, 149.7, 150.7, 159.7, 159.9, 167.8; LD-MS obsd 952.3; FAB-MS obsd 952.3222; calcd 952.3237 (C50H56N4O11Zn); λabs 411, 543, 578 nm; λem (λex = 411 nm) 578, 632 nm.
4.3.6. 5-Formyl-15-[2,4,6-tris(carboxymethoxy)phenyl]porphyrin (H2P-2)
A sample of porphyrin ZnP-2′ (15 mg, 0.016 mmol) in THF (1.6 mL, 0.01M) was treated with aqueous KOH (0.75 mL, 2 M) at 60 °C for 10 h. The reaction mixture was allowed to reach ambient temperature. The reaction mixture was treated with H2O (10 mL) and CH2Cl2 (10 mL). The aqueous layer, which contained the porphyrin, was separated and concentrated to a pink solid. The solid was treated with neat TFA (1 mL) and stirred at 50 °C for 24 h. The mixture was diluted with H2O to adjust the pH to 3. The resulting suspension was sonicated and centrifuged. Decanting afforded a solid pellet. Water was added followed by sonication, centrifugation, and decanting; this treatment was performed three times, whereupon the supernatant was colorless. The pellet was dried in vacuo to a green solid (6.5 mg, 65%): 1H NMR (DMSO-d6) δ –2.58 (s, 1H), −2.52 (s, 1H), 4.44 (s, 4H), 4.91 (s, 2H), 6.65 (s, 2H), 9.09 (d, J = 4.7 Hz, 2H), 9.46 (d, J = 4.3 Hz, 2H), 9.78 (d, J = 4.7 Hz, 2H), 10.32 (d, J = 4.7 Hz, 2H), 10.59 (s, 2H), 12.52 (s, 1H); 13C NMR δ 64.6, 65.1, 92.7, 104.9, 116.6, 128.8, 131.87, 132.04, 134.9, 143.7, 145.6, 147.4, 148.9, 158.8, 160.1, 170.0; LD-MS obsd 637.0; ESI-MS obsd 636.1479, calcd 636.1492 (C33H24N4O10); λabs (H2O) 409, 562, 633 nm; λem (H2O, λex = 409 nm) 641, 707; RP-HPLC tR = 28.94 min.
4.3.7. 1,9-Diformyl-5-{4-[N-(tert-butoxycarbonyl)amino]phenyl}dipyrromethane (5)
Following a reported procedure,30 a solution of 2b (500 mg, 1.48 mmol) in anhydrous DMF (1.50 mL) at 0 °C under argon was treated dropwise with POCl3 (0.28 mL, 3.1 mmol). The mixture was stirred at room temperature. After 1 h the reaction mixture was poured into a saturated aqueous solution of NaOAc (50 mL). The milky reaction mixture was extracted with CH2Cl2. The organic extract was washed with water, dried (Na2SO4), and filtered. The filtrate was concentrated to dryness. The crude residue was chromatographed [silica, CH2Cl2/ethyl acetate (3:1)] to afford a beige solid (400 mg, 68%): mp 196–202 °C; 1H NMR δ 1.51 (s, 9H), 5.51 (s, 1H), 6.05–6.08 (m, 2H), 6.84–6.90 (m, 2H), 7.15–7.18 (m, 2H), 7.34–7.32 (m, 2H), 9.24 (s, 2H), 10.94 (br, 2H); 13C NMR δ 28.5, 43.9, 111.7, 119.2, 122.7, 129.2, 132.8, 133.8, 138.1, 142.2, 179.3; FAB-MS obsd 394.1773, calcd 394.1767 [(M + H)+; M = C22H23N3O4].
4.3.8. 1,9-Bis(propylimino)-5-{4-[N-(tert-butoxycarbonyl)amino]phenyl}dipyrromethane (3b)
Following a reported procedure,30 a solution of 5 (250 mg, 0.630 mmol) and n-propylamine (1.00 mL, 12.7 mmol) in THF (2 mL) was stirred at room temperature for 1 h. Removal of the solvent and excess n-propylamine gave a beige solid (300 mg, quantitative): mp 65–68 °C; 1H NMR δ 0.88 (t, J = 7.2 Hz, 6H), 1.49 (s, 9H), 1.57–1.63 (m, 4H), 1.99 (s, 2H), 3.38 (t, J = 7.2 Hz, 4H), 5.40 (s, 1H), 5.92 (d, J = 3.6 Hz, 2H), 6.41 (d, J = 3.6 Hz, 2H), 7.07–7.10 (m, 2H), 7.23–7.25 (m, 2H), 7.82 (s, 2H); 13C NMR δ 11.9, 24.3, 28.5, 43.9, 61.5, 109.8, 118.81, 118.84, 129.14, 129.20, 135.2, 137.7, 139.0, 151.6; FAB-MS obsd 476.3026, calcd 476.3056 [(M + H)+; M = C28H37N5O2].
4.3.9. Zn(II)-5-{4-[N-(tert-butyloxycarbonyl)amino]phenyl}-15-[2,4,6-tris(tert-butoxycarbonylmethoxy) phenyl]porphyrin (ZnP-3′)
Following a reported procedure,30 a solution of 2a (123 mg, 0.200 mmol) and 3b (95 mg, 0.20 mmol) in toluene (20 mL, 0.01 M) was treated with anhydrous Zn(OAc)2 (367 mg, 2.00 mmol) and refluxed for 12 h with exposure to air. The reaction mixture was concentrated to dryness. Column chromatography [alumina, hexanes/ethyl acetate (1:1)] afforded a purple solid (58 mg, 28%): 1H NMR δ 1.15 (s, 9H), 1.62 (s, 27H), 4.15 (s, 4H), 4.79 (s, 2H), 6.49 (s, 2H), 7.76–7.78 (m, 2H), 8.17 (d, J = 8.0 Hz, 2H), 9.18 (d, J = 4.0 Hz, 2H), 9.10 (d, J = 4.0 Hz, 2H), 9.16 (d, J = 4.0 Hz, 2H), 9.37 (dd, J = 4.3 Hz, J = 4.3 Hz, 2H), 10.20 (s, 2H); 13C NMR δ 28.0, 28.4, 28.7, 66.4, 66.6, 82.2, 82.9, 90.7, 105.9, 110.3, 114.9, 116.8, 119.7, 131.4, 132.21, 132.34, 135.4, 137.84, 137.96, 149.4, 149.7, 149.9, 151.3, 153.3, 159.7, 159.9, 163.2, 167.8, 168.0, 204.21, 204.25, 211.1; LD-MS obsd 1029.2; FAB-MS obsd 1029.3558, calcd 1029.3503 (C55H59N5O11Zn); λabs 414, 545, 580 nm; λem (λex = 414 nm) 579, 634 nm.
4.3.10. 5-(4-Aminophenyl)-15-[2,4,6-tris(carboxymethoxy)phenyl]porphyrin (H2P-3)
A solution of ZnP-3′ (20 mg, 0.019 mmol) in THF (1.9 mL, 0.01 M) was treated with aqueous KOH (0.5 mL, 2 M) at 60 °C for 28 h. The reaction mixture was treated with water and CH2Cl2. The aqueous layer, which contained the porphyrin, was separated and concentrated to a pink solid. The solid was diluted with water. The solution was acidified with aqueous HCl (2 M) to pH 3, whereupon the porphyrin precipitated. The suspension was sonicated, centrifuged, and decanted to afford a solid pellet; this procedure was repeated four times, whereupon the supernatant was colorless. The solid pellet was dried and treated with neat TFA (1 mL). The resulting solution was stirred at room temperature for 11 h. The mixture was diluted with water (1 mL). The suspension was sonicated and centrifuged. Decanting afforded a solid pellet. Water was added followed by sonication, centrifugation, and decanting; this treatment was performed three times, whereupon the supernatant was colorless. The resulting solid was dried in vacuo to afford a dark-purple solid (11 mg, 83%): 1H NMR (DMSO-d6) δ –3.21 (s, 1H), −3.11 (s, 1H), 4.45 (s, 4H), 4.94 (s, 2H), 6.65 (s, 2H), 7.07 (d, J = 8.0 Hz, 2H), 7.95 (d, J = 8.4 Hz, 2H), 9.06 (d, J = 4.4 Hz, 2H), 9.13 (d, J = 4.4 Hz, 2H), 9.49 (d, J = 4.8 Hz, 2H), 9.58 (d, J = 4.4 Hz, 2H), 10.46 (s, 2H); 13C NMR δ 64.8, 65.2, 92.7, 104.8, 110.1, 110.8, 112.9, 120.4, 127.66, 127.68, 130.8, 131.0, 131.6, 132.0, 135.9, 144.4, 146.5, 148.1, 148.7, 159.1, 159.8, 169.8, 170.1; LD-MS obsd 700.3, ESI-MS obsd 700.2032, calcd 700.2038 [(M + H)+; M = C38H29N5O9]; λabs (H2O) 404, 505, 544, 567, 622 nm; λem (H2O, λex = 407 nm) 629, 688; RP-HPLC tR = 22.25 min.
Acknowledgements
This research was supported by a grant from the NIH (GM36238). Mass spectra were obtained at the Mass Spectrometry Laboratory for Biotechnology at North Carolina State University. Partial funding for the Facility was obtained from the North Carolina Biotechnology Center and the National Science Foundation. We thank NingNing Xu, Ibrahim Bori, and Dhanalekshmi Savithri for early work on this project.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Licha K. Top. Curr. Chem. 2002;222:1–29. [Google Scholar]
- 2.Berg K, Selbo PK, Weyergang A, Dietze A, Prasmickaite L, Bonsted A, Engesaeter BØ, Angell-Petersen E, Warloe T, Frandsen N, Høgset A. J. Microscopy. 2005;218:133–147. doi: 10.1111/j.1365-2818.2005.01471.x. [DOI] [PubMed] [Google Scholar]
- 3.Kee HL, Nothdurft R, Muthiah C, Diers JR, Fan D, Ptaszek M, Bocian DF, Lindsey JS, Culver JP, Holten D. Photochem. Photobiol. 2008;84 doi: 10.1111/j.1751-1097.2008.00409.x. [DOI] [PubMed] [Google Scholar]
- 4.De Rosa SC, Brenchley JM, Roederer M. Nature Med. 2003;9:112–117. doi: 10.1038/nm0103-112. [DOI] [PubMed] [Google Scholar]
- 5.Perfetto SP, Chattopadhyay PK, Roederer M. Nature Rev.: Immun. 2004;4:648–655. doi: 10.1038/nri1416. [DOI] [PubMed] [Google Scholar]
- 6.Chattopadhyay PK, Price DA, Harper TF, Betts MR, Yu J, Gostick E, Perfetto SP, Goepfert P, Koup RA, De Rosa SC, Bruchez MP, Roederer M. Nature Med. 2006;12:972–977. doi: 10.1038/nm1371. [DOI] [PubMed] [Google Scholar]
- 7.Borbas KE, Mroz P, Hamblin MR, Lindsey JS. Bioconjugate Chem. 2006;17:638–653. doi: 10.1021/bc050337w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wainright M. J. Antimicrob. Chemo. 1998;42:13–28. doi: 10.1093/jac/42.1.13. [DOI] [PubMed] [Google Scholar]
- 9.Maisch T, Szeimies R-M, Jori G, Abels C. Photochem. Photobiol. Sci. 2004;3:907–917. doi: 10.1039/b407622b. [DOI] [PubMed] [Google Scholar]
- 10.Hamblin MR, Hasan T. Photochem. Photobiol. Sci. 2004;3:436–450. doi: 10.1039/b311900a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sternberg ED, Dolphin D, Brückner C. Tetrahedron. 1998;54:4151–4202. [Google Scholar]
- 12.Pandey RK, Zheng G. In: The Porphyrin Handbook. Kadish KM, Smith KM, Guilard R, editors. Vol. 6. San Diego, CA: Academic Press; 2000. pp. 157–230. [Google Scholar]
- 13.Bonnett R. Chemical Aspects of Photodynamic Therapy. Amsterdam: Gordon and Breach Science Publishers; 2000. [Google Scholar]
- 14.Detty MR, Gibson SL, Wagner SJ. J. Med. Chem. 2004;47:3897–3915. doi: 10.1021/jm040074b. [DOI] [PubMed] [Google Scholar]
- 15.Nyman ES, Hynninen PH. J. Photochem. Photobiol. B: Biol. 2004;73:1–28. doi: 10.1016/j.jphotobiol.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 16.Da Ros T, Prato M. J. Org. Chem. 1996;61:9070–9072. doi: 10.1021/jo961522t. [DOI] [PubMed] [Google Scholar]
- 17.Hirsch A. Tetrahedron Lett. 1998;39:2731–2734. [Google Scholar]
- 18.Georgakilas V, Tagmatarchis N, Pantarotto D, Bianco A, Briand J-P, Prato M. Chem. Commun. 2002:3050–3051. doi: 10.1039/b209843a. [DOI] [PubMed] [Google Scholar]
- 19.Zhao B, Hu H, Haddon RC. Adv. Funct. Mater. 2004;14:71–76. [Google Scholar]
- 20.Guldi DM, Rahman GMA, Jux N, Balbinot D, Hartnagel U, Tagmatarchis N, Prato M. J. Am. Chem. Soc. 2005;127:9830–9838. doi: 10.1021/ja050930o. [DOI] [PubMed] [Google Scholar]
- 21.Sutton JM, Clarke OJ, Fernandez N, Boyle RW. Bioconjugate Chem. 2002;13:249–263. doi: 10.1021/bc015547x. [DOI] [PubMed] [Google Scholar]
- 22.Mauzerall D. J. Am. Chem. Soc. 1962;84:2437–2445. [Google Scholar]
- 23.Mauzerall D. J. Phys. Chem. 1962;66:2531–2533. [Google Scholar]
- 24.Mauzerall D. Biochemistry. 1965;4:1801–1810. [Google Scholar]
- 25.Carapellucci PA, Mauzerall D. Ann. New York Acad. Sci. 1975;244:214–238. doi: 10.1111/j.1749-6632.1975.tb41533.x. [DOI] [PubMed] [Google Scholar]
- 26.Feitelson J, Mauzerall D. J. Phys. Chem. B. 2002;106:9674–9678. [Google Scholar]
- 27.Hambright P. In: The Porphyrin Handbook. Kadish KM, Smith KM, Guilard R, editors. Vol. 3. San Diego, CA: Academic Press; 2000. pp. 129–210. [Google Scholar]
- 28.Lindsey JS. In: The Porphyrin Handbook. Kadish KM, Smith KM, Guilard R, editors. Vol. 1. San Diego, CA: Academic Press; 2000. pp. 45–118. [Google Scholar]
- 29.Fan D, Taniguchi M, Yao Z, Dhanalekshmi S, Lindsey JS. Tetrahedron. 2005;61:10291–10302. [Google Scholar]
- 30.Taniguchi M, Balakumar A, Fan D, McDowell BE, Lindsey JS. J. Porphyrins Phthalocyanines. 2005;9:554–574. [Google Scholar]
- 31.Hornung R, Fehr MK, Walt H, Wyss P, Berns MW, Tadir Y. Photochem. Photobiol. 2000;72:696–700. doi: 10.1562/0031-8655(2000)072<0696:pmtmpe>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 32.Zingg A, Felber B, Gramlich V, Fu L, Collman JP, Diederich F. Helv. Chim. Acta. 2002;85:333–351. [Google Scholar]
- 33.Lamarche F, Sol V, Huang Y-M, Granet R, Guilloton M, Krausz P. J. Porphyrins Phthalocyanines. 2002;6:130–134. [Google Scholar]
- 34.Subbarayan M, Shetty SJ, Srivastava TS, Noronha OPD, Samuel AM, Mukhtar H. Biochem. Biophys. Res. Commun. 2001;281:32–36. doi: 10.1006/bbrc.2001.4289. [DOI] [PubMed] [Google Scholar]
- 35.Lindsey JS, Prathapan S, Johnson TE, Wagner RW. Tetrahedron. 1994;50:8941–8968. [Google Scholar]
- 36.Loewe RS, Tomizaki K-Y, Youngblood WJ, Bo Z, Lindsey JS. J. Mater. Chem. 2002;12:3438–3451. [Google Scholar]
- 37.Borbas KE, Kee HL, Holten D, Lindsey JS. Org. Biomol. Chem. 2008;6:187–194. doi: 10.1039/b715072e. [DOI] [PubMed] [Google Scholar]
- 38.Bhaumik J, Yao Z, Borbas KE, Taniguchi M, Lindsey JS. J. Org. Chem. 2006;71:8807–8817. doi: 10.1021/jo061461r. [DOI] [PubMed] [Google Scholar]
- 39.Borbas KE, Chandrashaker V, Muthiah C, Kee HL, Holten D, Lindsey JS. J. Org. Chem. 2008;73:3145–3158. doi: 10.1021/jo7026728. [DOI] [PubMed] [Google Scholar]
- 40.Jux N. Org. Lett. 2000;2:2129–2132. doi: 10.1021/ol006028x. [DOI] [PubMed] [Google Scholar]
- 41.Guldi DM, Zilbermann I, Anderson G, Li A, Balbinot D, Jux N, Hatzimarinaki M, Hirsch A, Prato M. Chem. Commun. 2004:726–727. doi: 10.1039/b400027g. [DOI] [PubMed] [Google Scholar]
- 42.Jee J-E, Eigler S, Hampel F, Jux N, Wolak M, Zahl A, Stochel G, van Eldik R. Inorg. Chem. 2005;44:7717–7731. doi: 10.1021/ic050924t. [DOI] [PubMed] [Google Scholar]
- 43.Jee J-E, Eigler S, Jux N, Zahl A, van Eldik R. Inorg. Chem. 2007;46:3336–3352. doi: 10.1021/ic061732g. [DOI] [PubMed] [Google Scholar]
- 44.Hermanson GT. Bioconjugate Techniques. San Diego, CA: Academic Press; 1996. [Google Scholar]
- 45.Lindsey JS, Mauzerall DC. J. Am. Chem. Soc. 1982;104:4498–4450. [Google Scholar]
- 46.Lindsey JS, Delaney JK, Mauzerall DC, Linschitz H. J. Am. Chem. Soc. 1988;110:3610–3621. [Google Scholar]
- 47.Fan D, Taniguchi M, Lindsey JS. J. Org. Chem. 2007;72:5350–5357. doi: 10.1021/jo070785s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ruzié C, Krayer M, Balasubramanian T, Lindsey JS. J. Org. Chem. 2008;73 doi: 10.1021/jo800736c. [DOI] [PubMed] [Google Scholar]
- 49.Lindsey JS, Wagner RW. J. Org. Chem. 1989;54:828–836. [Google Scholar]
- 50.Wagner RW, Breakwell BV, Ruffing J, Lindsey JS. Tetrahedron Lett. 1991;32:1703–1706. [Google Scholar]
- 51.Wagner RW, Lindsey JS, Turowska-Tyrk I, Scheidt WR. Tetrahedron. 1994;50:11097–11112. [Google Scholar]
- 52.Wagner RW, Johnson TE, Lindsey JS. Tetrahedron. 1997;53:6755–6790. [Google Scholar]
- 53.Laha JK, Dhanalekshmi S, Taniguchi M, Ambroise A, Lindsey JS. Org. Process Res. Dev. 2003;7:799–812. [Google Scholar]
- 54.Srinivasan N, Haney CA, Lindsey JS, Zhang W, Chait BT. J. Porphyrins Phthalocyanines. 1999;3:283–291. [Google Scholar]
- 55.Lindsey JS, Brown PA, Siesel DA. Tetrahedron. 1989;45:4845–4866. [Google Scholar]
- 56.Schmidt I, Jiao J, Thamyongkit P, Sharada DS, Bocian DF, Lindsey JS. J. Org. Chem. 2006;71:3033–3050. doi: 10.1021/jo052650x. [DOI] [PubMed] [Google Scholar]
- 57.Manka JS, Lawrence DS. Tetrahedron Lett. 1989;30:6989–6992. [Google Scholar]
- 58.Balakumar A, Muthukumaran K, Lindsey JS. J. Org. Chem. 2004;69:5112–5115. doi: 10.1021/jo049819b. [DOI] [PubMed] [Google Scholar]
- 59.Yao Z, Bhaumik J, Dhanalekshmi S, Ptaszek M, Rodriguez PA, Lindsey JS. Tetrahedron. 2007;63:10657–10670. doi: 10.1016/j.tet.2007.07.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wagner RW, Lindsey JS. Pure Appl. Chem. 1996;68:1373–1380. (b) Corrigendum: Wagner RW, Lindsey JS. Pure Appl. Chem. 1998;70(8):i.
- 61.Yao Z, Borbas KE, Lindsey JS. New J. Chem. 2008;32:436–451. [Google Scholar]