

Abstract
Estrogens are involved in the hypothalamic control of multiple homeostatic functions including reproduction, stress responses, energy metabolism, sleep cycles, temperature regulation, and motivated behaviors. The critical role of 17β-estradiol (E2) is evident in hypoestrogenic states (e.g., postmenopause) in which many of these functions go awry. The actions of E2 in the brain have been attributed to the activation of estrogen receptors α and β through nuclear, cytoplasmic, or membrane actions. However, we have identified a putative membrane-associated estrogen receptor that is coupled to desensitization of GABAB and μ-opioid receptors in guinea pig and mouse hypothalamic proopiomelanocortin neurons. We have synthesized a new nonsteroidal compound, STX, which selectively targets the Gαq-coupled phospholipase C–protein kinase C–protein kinase A pathway, and have established that STX is more potent than E2 in mediating this desensitization in an ICI 182, 780-sensitive manner in both guinea pig and mouse neurons. Both E2 and STX were fully efficacious in estrogen receptor α,β knock-out mice. Moreover, in vivo treatment with STX, similar to E2, attenuated the weight gain in hypoestrogenic female guinea pigs. Therefore, this membrane-delimited signaling pathway plays a critical role in the control of energy homeostasis and may provide a novel therapeutic target for treatment of postmenopausal symptoms and eating disorders in females.
Keywords: GPCR, body weight, GABAB receptor, intracellular signaling, potassium channels, POMC neurons
Introduction
The hypothalamus controls multiple homeostatic functions that change dramatically during the ovulatory cycle and especially during pregnancy and menopause. Fluctuations in circulating estrogens are thought to be responsible for these changes (Komesaroff et al., 1999; Milewicz et al., 2001; Sherwin, 2002; Stearns et al., 2002). The hypothalamic proopiomelanocortin (POMC) neurons are critical neurons in the hypothalamic circuits controlling energy homeostasis (Cone, 2005; Murphy and Bloom, 2005). POMC neurons through direct synaptic contacts modulate the excitability of hypothalamic neurons that control appetite, fluid balance, temperature, stress and reproduction (Horvath, 2005). In addition, POMC neurons project to other brain areas (e.g., midbrain) to control motivated behaviors such as sex and food consumption (Koob, 1992). Although many of the actions of estrogen are mediated at the level of the hypothalamus, the cellular targets and signaling cascades involved in the effects of estrogen on homeostatic functions remain unclear.
Until recently, the actions of estrogen in the hypothalamus and other brain areas have been attributed to the activation of estrogen receptor α (ERα) or ERβ (McEwen and Alves, 1999; Toran-Allerand et al., 1999; Kelly and Levin, 2001; Rønnekleiv and Kelly, 2005). However, we have identified a putative membrane-associated estrogen receptor (mER) that is Gq-coupled to a phospholipase C–protein kinase C–protein kinase A (PLC–PKC–PKA) pathway (Lagrange et al., 1997; Qiu et al., 2003). The activation of this pathway by E2 can rapidly disinhibit female POMC neurons through desensitizing μ-opioid and GABAB receptors in POMC neurons. We hypothesize that this mER signaling pathway is involved in heterologous desensitization of μ-opioid and GABAB in hypothalamic neurons similar to neurotransmitters like serotonin. Presently, we have characterized the signaling of a nonsteroidal compound, STX, that specifically targets the G-protein-coupled signaling pathway in both male and female hypothalamic POMC neurons. Furthermore, we have established that E2 and STX are fully efficacious in estrogen receptor knock-out (KO) mice. Finally, we show that STX, similar to estrogen, regulates homeostatic functions (e.g., energy metabolism) in ovariectomized (hypoestrogenic) females.
Materials and Methods
Animals and treatments.
All animal procedures described in this study are in accordance with institutional guidelines based on National Institutes of Health standards. Female Topeka guinea pigs (400–600 g), bred in our institutional breeding facility, and female multicolor guinea pigs (400–500 g; Elm Hill Breeding Labs, Chelmsford, MA) were used in these experiments. The guinea pigs were maintained under constant temperature (26°C) and light (on between 6:30 A.M. and 8:30 P.M.). Animals were housed individually, with food and water provided ad libitum. They were gonadectomized under ketamine–xylazine anesthesia (33 and 6 mg/kg, respectively, s.c.) 5–7 d before experimentation, and they were given sesame oil vehicle (0.1 ml, s.c.) 24 h before experimentation. Serum estrogen concentrations were measured in the ovariectomized females by radioimmunoassay (Oregon National Primate Research Center Radioimmunoassay Core, Portland, OR) from trunk blood collected on the day of experimentation and were <10 pg/ml. An additional group of animals were ovariectomized and, after 1 week, were injected with oil vehicle or estradiol benzoate (8 μg/kg in oil; n = 6) or STX (2 mg/kg in oil; n = 4) every other day, 3 times/week for 4 weeks. On completion of the experiments, the uterine weights were obtained.
Wild-type (WT) C57BL/6 mice in these studies were obtained from The Jackson Laboratory (Bar Harbor, ME). ERαKO (from P. Chambon; C57BL/6) mice were generated from breeding pairs heterozygous for the disrupted ERα gene, and offspring were screened by PCR amplification of tail DNA using primers as described previously (Dupont et al., 2000). The ERαβ (double) knock-out (from P. Chambon; C57BL/6) mice were generated from breeding pairs heterozygous for the disrupted ERα and ERβ genes as described previously (Dupont et al., 2000). ERβKO (from K. S. Korach; C57BL/6) mice were generated from breeding pairs heterozygous for the disrupted ERβ gene as described previously (Krege et al., 1998). Offspring were screened with two sets of primers: the first primer pair (5′ACATCCATACACCCCCACTCAACC 3′ and 5′AAAGAAACATGTCCTGGCAAATCA 3′) produced a 600 bp product for the WT allele and no product for the KO allele. The second primer pair (5′ TTCTGAGGGATCCGCTGTAAG 3′ and 5′AGGCTGCTGATCTC-GTTCT 3′) produced a 450 bp product for the KO allele and no product for the WT allele. In addition, hypothalamic slices from WT and KO mice were tested for immunoreactive ERα using the C1355 antibody kindly provided by Dr. Margaret Shupnik (University of Virginia, Charlottesville, VA). With this antibody we found a high concentration of ERα-immunoreactive neurons in the hypothalamus from WT and ERβKO, but no stain in the ERαKO or the ERαβ KO animals. ERβ expression was tested using reverse transcription (RT)-PCR of hypothalamic RNA with primer pairs designed to flank the neomycin-resistance gene insert in exon 3 of the ERβKO mice (Krege et al., 1998) (forward primer 830–848 bp and reverse primer 950–968 bp; GenBank accession number NM_207707). These primers produced a PCR product in WT, heterozygous, and ERαKO mice but not in ERβKO mice. All animals were maintained under controlled temperature (25°C) and photoperiod conditions (12 h light/dark cycle; lights on between 6:00 A.M. and 6:00 P.M.) with food and water ad libitum. Adult female mice were ovariectomized under ketamine–xylazine anesthesia (10 mg/kg, each, i.p.) and allowed to recover for 1 week. Adult male ERαβKO mice were only available for these studies, and they were not gonadectomized because we found that gonad-intact males responded to E2 and STX. Therefore, we decided not to do the invasive surgery in these extremely valuable animals.
Drugs.
The preparation and concentrations of the drugs for this study have been published (Qiu et al., 2003), with the exception of STX, which was prepared by chemical synthesis via a procedure that has been reported previously (Tobias et al., 2006).
Electrophysiology.
Female adult Topeka guinea pigs or adult mice were gonadectomized 6–10 d before each experiment. Hypothalamic slices were prepared and whole-cell recordings were done as described previously (Qiu et al., 2003). The mean resting membrane potential (RMP) for female guinea pig neurons was −55.3 ± 0.7 mV (n = 97). For the C57BL/6 background mice, the RMP was −53.0 ± 1.0 mV (n = 37) for wild-type; −53.4 ± 1.2 mV (n = 25) for ERαKO; −52.2 ± 1.5 mV (n = 17) for ERβKO; and −55.9 ± 1.2 mV (n = 24) for intact male ERαβKO mice. The mean input resistance (Rin) for the female guinea pig arcuate neurons was 610 ± 50 MΩ but was greater for the smaller mouse arcuate neurons (860 ± 60 MΩ). There was no difference in the baclofen (EC50)-induced outward K+ current between the guinea pig (24.5 ± 1.1 pA; n = 97) and mouse (wild-type, 23.4 ± 4.0 pA, n = 20; ERαKO, 25.1 ± 2.4 pA, n = 14; ERβKO, 23.4 ± 3.5 pA, n = 15; ERαβKO, 21.6 ± 1.9 pA, n = 22) experimental groups, and only cells that gave a reversible baclofen response of at least 10 pA were included in the study.
Composite dose–response curves were generated from the following logistic equation fitted by computer (Origin 4.1; Microcal) to the data: ΔImax = 100 × ([agonist]n/([agonist]n + EC50n)), where ΔImax is the maximum outward current for a given agonist, EC50 represents the agonist potency, and n is the Hill slope.
Immunocytochemistry.
After electrical recording, the slices were prepared for fluorescence immunocytochemistry as described previously (Qiu et al., 2003).
Statistical analyses.
Comparisons between groups were performed using a one-way or two-way ANOVA (with post hoc Newman–Keuls paired analysis). Differences were considered statistically significant if the probability of error was <5%.
Results
STX is more potent than E2 to attenuate the GABAB response
Estrogen rapidly reduces the potency of the GABAB receptor agonist baclofen to activate G-protein-coupled, inwardly rectifying K+ (GIRK) channels in hypothalamic neurons (Lagrange et al., 1994, 1996, 1997; Kelly et al., 1999; Qiu et al., 2003). These effects are mimicked by the membrane impermeant E2-BSA, suggesting that E2 signals through a membrane receptor (Qiu et al., 2003). Presently, we compared the potency and efficacy of E2 and a new diphenylacrylamide compound STX that does not bind to ERα or ERβ (Qiu et al., 2003) using whole-cell patch recording in hypothalamic arcuate neurons in slices prepared from ovariectomized female guinea pigs. Seventy-five percent of these neurons responded to E2 or STX (n = 67), including all of the β-endorphin identified (POMC) neurons (n = 27). For measuring E2 or STX modulation of the GABAB response, we used an EC50 concentration (5 μm) of baclofen and a protocol in which a densensitization of the response with multiple applications of baclofen was not observed (Qiu et al., 2003). Baclofen induced a robust outward current that subsided after washout, and the application of baclofen ∼20 min later elicited the same robust response. However, if E2 or STX was applied during the interim period (i.e., after the washout of the first application of baclofen), there was a significant (p < 0.005) decrease of >40% in the response to a second application of baclofen (Fig. 1). Concentration–response curves showed that E2 and STX rapidly attenuate the GABAB response in a concentration-dependent manner with 50% inhibition at 46.0 nm for E2 and at 2.6 nm for STX, which was equally as efficacious as estrogen. Therefore, STX is ∼20 times more potent than E2 in desensitizing the GABAB response in hypothalamic arcuate neurons.
Figure 1.

Composite dose–response curves for E2 and STX illustrating the difference in potency between E2 and STX in arcuate POMC neurons. a, Experiments were conducted as described in Materials and Methods. Cells were perfused with different concentrations of E2 (0, 10, 50, 100, and 1000 nm) or STX (0, 1, 10, 50, and 100 nm). Data are presented as mean ± SEM (n = 4–11 cells/data point). Based on a logistics equation fit to the data points (see Materials and Methods), the EC50 for STX was 2.6 nm, which is 17-fold lower than that found for E2 (46.0 nm). The Hill slope for E2 and STX was 0.80 and 1.24, respectively. b, A, Biocytin–streptavidin–Cy2 labeling of a small pyramidal arcuate neuron that responded to STX. B, Immunocytochemical staining of β-endorphin in the same neuron (arrow). C, Overlay of A and B. Forty percent of the neurons (n = 49) were identified as POMC neurons. Scale bar: A–C, 20 μm. c, The chemical structures of 17-β-estradiol (E2) and the diphenylacrylamide STX.
To determine whether the response to STX could be blocked with an estrogen receptor antagonist, we used ICI 182,780 (ICI) (1 μm) to antagonize the inhibitory effects of STX on the baclofen response. Indeed, ICI 182,780 when coperfused with STX blocked the effects of STX (Fig. 2). Treatment with ICI 182,780 alone had no effect on the baclofen response (Fig. 2). Therefore, these data are consistent with a high-affinity membrane receptor that can bind both estrogen and STX.
Figure 2.

STX attenuation of the GABAB response depends on activation of PKC and PKA activation in hypothalamic arcuate (POMC) neurons. a, Three representative traces of the baclofen response in the presence of STX (100 nm), STX and Rp-cAMPs (200 μm), or STX and rottlerin (5 μm) in arcuate neurons from ovariectomized guinea pigs are shown. b, Bar graphs summarizing the effects of inhibitors of the effects of STX. Veh, Vehicle. Experiments were conducted as described in Materials and Methods. Cells were dialyzed for 15 min before baclofen application with BIS (100 nm) or Rp-cAMPs (200 μm), which were included in the patch-pipette solution. STX (100 nm) rapidly attenuated the outward (GIRK) current induced by the GABAB receptor agonist baclofen in arcuate (POMC) neurons, which was reversed by the estrogen receptor antagonist ICI 182,780 (1 μm). ICI alone had no effect. BIS (an inhibitor of all PKC isoforms), the selective PKCδ inhibitor rottlerin (5 μm), and the PKA inhibitor Rp-cAMPs could reverse, but the selective inhibitor of conventional PKC isoforms, Gö6976 (2 μm), could not block the attenuation of the baclofen response by STX in arcuate neurons including POMC neurons. BIS (n = 7), RpcAMPs (n = 4), and rottlerin (n = 3) alone had no effect on the baclofen response (data not shown). Bars represent the mean ± SEM. Thirty-nine percent of the neurons were β-endorphin (POMC) neurons (n = 54). **p < 0.01 and ***p < 0.001 versus vehicle control.
Desensitization of the GABAB response by STX involves protein kinase A and PKCδ
Previously, we reported that the membrane estrogen receptor in arcuate (POMC) neurons is Gαq-coupled to activation of phospholipase C, leading to the upregulation of protein kinase Cδ and protein kinase A activity in arcuate (POMC) neurons (Qiu et al., 2003). Therefore, we explored whether STX utilizes the same pathway to desensitize the GABAB receptor-mediated response. If activation of the PKA or PKC pathway is involved, inhibition of PKA and/or PKC activity should abrogate the effects of STX on GABAB responses. As shown in Figure 2, when we dialyzed neurons with a selective PKA inhibitor, the nonhydrolyzable cAMP analog Rp-cAMPS, the STX-induced reduction of the GABAB response was abolished. These results indicate that the suppression of the GABAB response by STX requires the activation of PKA. Furthermore, activation of PKC is also critical for STX modulation of the GABAB response. Treatment of neurons with bisindolymaleimide (BIS), a broad-spectrum inhibitor of PKC, eliminated the effects of STX (Fig. 2). In addition, the selective PKCδ inhibitor rottlerin completely blocked the ability of STX to inhibit the GABAB response in hypothalamic neurons, whereas a selective inhibitor of the conventional PKC isoforms, Gö6976, did not attenuate the effects of STX. Altogether, these data indicate that STX and E2 are signaling via the same mER that is coupled to a Gαq–PKC–PKA pathway. In fact, the effects of estrogen and STX were nonadditive such that coperfusion of both drugs did not generate a greater response (R2/R1 = 58.1 ± 2.7%; n = 4).
E2 and STX are fully efficacious in ERαKO, ERβKO, and ERαβKO mice
As described previously by Blaustein (1994), ERα is abundantly expressed in guinea pig arcuate neurons. Also, ERβ mRNA and protein are expressed in the rodent arcuate nucleus but to a much lesser extent than ERα (Shughrue et al., 1997; Fodor and Delemarre-van de Waal, 2001). Therefore, to verify that neither ERα nor ERβ are involved in the rapid desensitization of the GABAB response by estrogen, we compared the effects of E2 and STX on the GABAB response in arcuate neurons including POMC neurons from ERα knock-out, ERβ knock-out, and wild-type (C57BL/6) mice (Fig. 3). As observed in ovariectomized female guinea pigs, we found that both STX and E2 significantly attenuated the GABAB response in arcuate neurons from ovariectomized ERαKO and ERβKO female and ERαβKO male mice. Moreover, the efficacy of the response was equivalent in wild-type (C57BL/6) and KO mice, indicating that the effects of STX and E2 are mediated by a membrane ER (mER) distinct from ERα or ERβ (Fig. 3).
Figure 3.

E2 and STX rapidly attenuated the baclofen response in hypothalamic arcuate (POMC) neurons in wild-type and ERαKO, ERβKO and ERαβKO mice. a, Experiments were conducted as described in the Materials and Methods. Four representative traces of the baclofen response in the presence of vehicle, E2 (100 nm), or STX (100 nm) in arcuate neurons from wild-type (A, B) and ERαKO (C, D) mice are shown. b, Bar graphs summarizing the effects of E2 or STX in arcuate (POMC) neurons from ovariectomized, wild-type, ERαKO, and ERβKO mice. E2 and STX (100 nm) rapidly attenuated the outward (GIRK) current induced by the GABAB receptor agonist baclofen in arcuate (POMC) neurons from ovariectomized wild-type (C57BL/6), ERαKO, and ERβKO mice. Bars represent the mean ± SEM. ###p < 0.001, E2-treated wild-type group versus vehicle (Veh) wild-type control; ***p < 0.001, E2- or STX-treated ERαKO or ERβKO groups versus vehicle ERKO control. The data for the ERαKO and ERβKO control responses were combined in one bar graph. c, Double labeling of POMC neurons that responded to STX from ERαKO (A–C) and ERβKO (D–F) mice. Arcuate neurons were filled with biocytin during the whole-cell recording. A, D, Biocytin–streptavidin–Cy2 labeling of two small pyramidal arcuate neurons. B, E, Immunocytochemical staining of β-endorphin in the same neurons (arrow). C, overlay of A and B; F, overlay of D and E. Nineteen of the 46 responsive neurons (41%) were identified as β-endorphin neurons. Scale bar: A–F, 20 μm. d, Bar graphs summarizing the effects of E2, STX, or ICI 182,780 in arcuate (POMC) neurons from male ERαβKO mice. E2 and STX (100 nm) rapidly attenuated the outward (GIRK) current induced by the GABAB receptor agonist baclofen, which was antagonized by ICI. ICI alone had no effect. Twelve of the 20 (60%) neurons were identified as POMC neurons. Bars represent the mean ± SEM. †††p < 0.001, E2- or STX- versus vehicle-treated cells from ERαβKO mice.
E2 and STX prevent excess body weight gain after ovariectomy in guinea pigs
Evidence from several species indicates that food intake and body weight are influenced both by changes in endogenous estrogens and by exogenous estrogenic treatments (Czaja and Goy, 1975). Butera and Czaja (1984) have shown that the anorexigenic effects of estrogen are attributable to the direct actions of the steroid in the arcuate-ventromedial hypothalamic nuclei. Therefore, we compared the efficacy of STX and E2 in attenuating the weight gain in guinea pigs after ovariectomy (Fig. 4). Female guinea pigs were ovariectomized and allowed to recover for 7 d, at which time they received subcutaneous injections of estradiol benzoate (8 μg/kg), STX (2 mg/kg), or a sesame oil vehicle (0.1 ml) every other day, three times per week for 4 weeks. The body weights were measured before each injection. We found that E2 treatment significantly decreased body weight, which is similar to previous reports on the effect of subcutaneous E2 treatment in ovariectiomized females (Fig. 4a) (Czaja and Goy, 1975). As expected, estradiol benzoate significantly increased uterine weight (Fig. 4c) (EB: 2.87 ± 0.25 g vs oil: 0.37 ± 0.08 g; p < 0.001). STX also attenuated the body weight gain after ovariectomy (Fig. 4b). However, unlike estradiol benzoate, STX, as shown previously in the mouse (Qiu et al., 2003), did not increase uterine weight (STX: 0.23 ± 0.02 g vs oil: 0.28 ± 0.03 g; p > 0.05). Therefore, the nonsteroidal STX, which is selective for the G-protein-coupled estrogen receptor, can mimic the effects of estrogen to alter energy metabolism in females but not the proliferative effects on peripheral reproductive organs.
Figure 4.
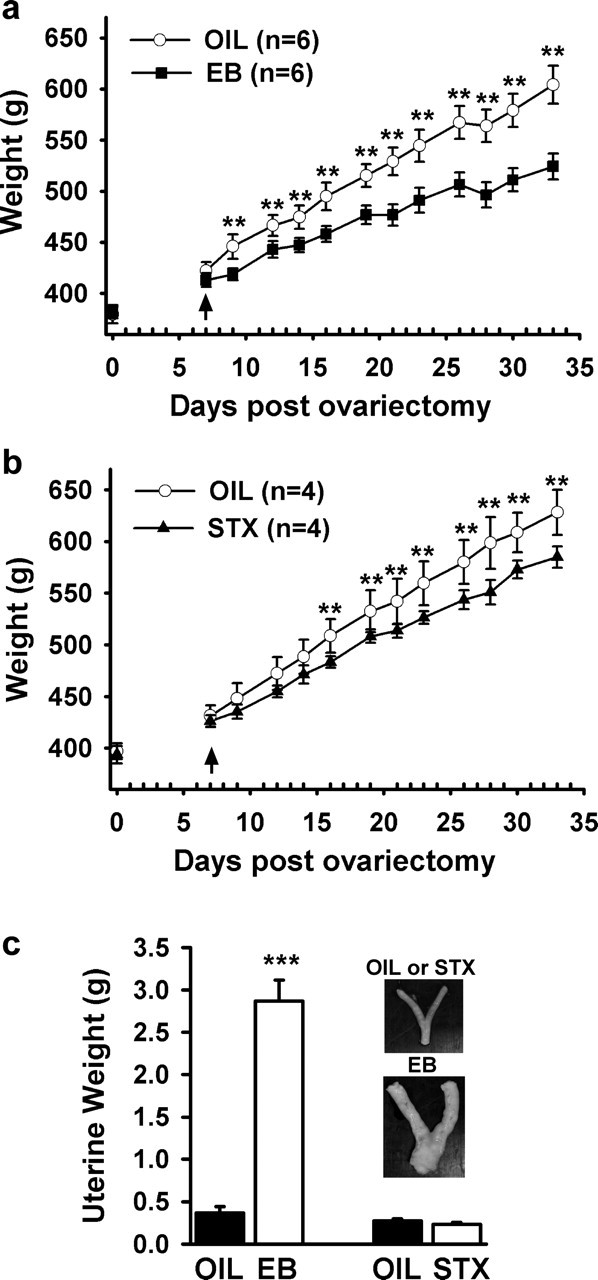
Estrogen (a) and STX (b) significantly attenuate the body weight gain in female guinea pigs after ovariectomy. The female guinea pigs were ovariectomized (on day 0) and allowed to recover for 1 week before being given bi-daily subcutaneous injections of oil (OIL), estradiol benzoate (EB), or STX (see Materials and Methods). A two-way ANOVA (repeated measures) revealed an overall significant effect of both estrogen and STX (p < 0.001), and post hoc Newman–Keuls analysis revealed daily significant differences between estrogen and oil-treated, and STX and oil-treated groups (**p < 0.01). Bars represent the mean ± SEM of six and four animals per group for estrogen and STX treatment, respectively. c, Uteri are enlarged after estradiol, but not after STX or oil-vehicle treatment (inset). After the treatment period, the uteri of the guinea pigs were harvested and examined. There was a significant increase in uterine size after EB, compared with oil-vehicle or STX treatment. Bar graphs represent mean uterine weights. ***p < 0.001, EB versus oil-treated females; n = 6 guinea pigs/group.
Discussion
For the first time we have identified a nonsteroidal compound, STX, which selectively targets the mER and is more potent than E2 in activating a membrane delimited pathway in hypothalamic arcuate neurons. STX is fully efficacious in ERα and ERβ knock-out mice and has no proliferative effects on reproductive organs, yet it is effective in reducing the weight gain associated with hypoestrogenic states. Although we found that other arcuate neurons express this mER signaling pathway (Qiu et al., 2003), this pathway is involved in heterologous desensitization of the μ-opioid (autoreceptor) and GABAB receptors in POMC neurons and therefore would play a critical role in dictating the level of excitability POMC neurons. These neurons appear to be the critical “relay” neurons in the melanocortin pathway, and their activity ultimately dictates the level of excitability in feeding circuits (Saper et al., 2002; Jobst et al., 2004; Cone, 2005).
Presently, we have characterized an mER that is Gq-coupled to a PLC–PKC–PKA pathway using STX as a ligand. This mER has subnanomolar affinity for estrogen based on our previous pharmacological (Schild) analysis (Lagrange et al., 1997). However, STX is a more selective ligand with even greater affinity (∼20-fold) for the mER. The activation of this pathway by E2 and STX disinhibits female arcuate neurons by uncoupling GABAB receptors from downstream K+ channels. Also, E2 attenuates the coupling of μ-opioid receptors, which are autoreceptors in POMC neurons, to the same population of K+ channels (Kelly et al., 1992; Lagrange et al., 1994, 1997). Therefore, the desensitization of both GABAB and μ-opioid receptors by activation of this mER signaling pathway has the potential to affect synaptic transmission not only in POMC neurons but also in other hypothalamic neurons. Indeed, we also identified this mER in dopamine neurons (Qiu et al., 2003). In dopamine neurons, this mER signaling pathway may be involved in dopamine-mediated inhibition of prolactin secretion (Frawley and Neill, 1980). Although we targeted the POMC- and dopamine-rich areas of the mediobasal hypothalamus, this mER signaling pathway could be involved in numerous other homeostatic pathways yet to be determined.
The pharmacological characteristics of the hypothalamic mER appear to be different from estrogen receptor “X,” which is expressed in the cortex during development and after ischemic injury, is activated by 17α-estradiol, and is not antagonized by ICI 182,780 (Toran-Allerand et al., 2002). However, the molecular identity of the hypothalamic mER is not known. Recently, an orphan GPCR (GPR 30) has been identified in breast carcinoma cell lines that binds estrogen with relatively high affinity, but ICI 182,780 stimulates adenylyl cyclase activity in SKBR3 cells that express GPR 30, GPR 30-transfected MDA-MB231 cells, and GPR 30-transfected HEK (human embryonic kidney) cells (Filardo et al., 2002; Revankar et al., 2005; Thomas et al., 2005). GPR 30 mRNA has been localized in the hypothalamus by in situ hybridization, but the pharmacology has not been characterized in neurons (O’Dowd et al., 1998). In preliminary experiments, we have measured mRNA expression of GPR30 in arcuate neurons using single-cell RT-PCR, but we do not know whether GPR30 is coupled to the phospholipase C–protein kinase C–protein kinase A signaling pathway in these native neurons.
The physiological importance of the hypothalamic mER is highlighted by the fact that systemically administered STX was effective, albeit less than the natural hormone estrogen, in attenuating the weight gain after ovariectomy. Although STX is more potent at the cellular level (in vitro), the pharmacokinetics of this drug are presumably similar to 4-hydroxytamoxifen because of the strong structural similarity with this SERM (selective estrogen receptor modulator). Hydroxytamoxifen does not cross the blood–brain barrier as well as estrogen does, and therefore higher doses are needed to have the same physiological effects in monkeys and in women (Stearns et al., 2003; Wilson et al., 2003). Based on the early studies of Czaja and colleagues (Czaja and Goy, 1975; Butera and Czaja, 1984), we know that estrogen modulates energy metabolism via its direct actions on the hypothalamus, and the present findings suggest that this is mediated, in part, via the mER. The critical role of estrogen in controlling energy metabolism is highlighted by the high incidence of obesity in women after menopause (Milewicz et al., 2000; American College of Obstetricians and Gynecologists, 2005). It is possible that the mER is responsible in part for sex differences in the control of feeding and eating disorders, which are much more prevalent (90–95%) in young women as compared with men (Sodersten et al., 2003).
Our original hypothesis was that this hypothalamic mER signaling pathway was involved in heterologous desensitization of μ-opioid and GABAB receptors in POMC and dopamine neurons similar to neurotransmitters like serotonin, which affects a number of homeostatic processes. However, we do not think that E2 (STX) is allosterically interacting with the receptors based on the lack of an effect of the steroid on the μ-opioid and GABAB agonist-stimulated GTPγS binding in arcuate neurons (Cunningham et al., 1998). In addition, the slope conductance of the G-protein-gated inwardly rectifying K+ channel (GIRK) current is not altered, indicating that E2 (STX) is not acting directly on the channel pore (Lagrange et al., 1997; Qiu et al., 2003). However, we know that the signaling pathways of E2 (STX) and serotonin are in parallel but convergent pathways in these same neurons (Qiu et al., 2005). Indeed, the serotonin 5-HT2C receptor is also Gαq-coupled and impinges on the GABAB signaling pathway in POMC neurons (Qiu et al., 2005), and the expression of 5-HT2C receptors is critical for maintaining energy homeostasis (Tecott et al., 1995; Heisler et al., 2002). Interestingly, serotoninergic drugs (selective serotonin reuptake inhibitors) and E2 are effective in alleviating postmenopausal symptoms in women (Stearns et al., 2002). It may be that E2 and serotonin (via mER and 5-HT2C receptors, respectively) synergize to regulate energy metabolism in postpubertal females.
In summary, our findings indicate that the mER in hypothalamic arcuate (POMC) neurons is distinct from ERα or ERβ. Moreover, we have identified a nonsteroidal compound, STX, as a selective ligand for the mER. STX is significantly more potent then estrogen in activating cellular cascades that lead to desensitization of GABAB and μ-opioid receptors in hypothalamic arcuate neurons that are involved in the control of homeostasis.
The fact that E2 and STX are fully efficacious in activating this signaling pathway in double-estrogen receptor knock-out mice is further proof for the existence of a novel mER that is involved in critical physiological processes. In addition to its role in energy homeostasis, this hypothalamic mER may also be involved in temperature regulation, circadian rhythms, stress responses, and motivated behaviors.
Footnotes
This work was supported by United States Public Health Service Grants NS43330, NS38809, NS49210, DK68098, and DK57574. We recognize the help of Dr. Emilie F. Rissman (University of Virginia, Charlottesville, VA) in establishing the transgenic mouse colonies.
References
- American College of Obstetricians and Gynecologists. (2005). Body mass index and insulin resistance. Obstet Gynecol 104:5s–10s. [DOI] [PubMed] [Google Scholar]
- Blaustein JD (1994). Estrogen receptors in neurons: new subcellular locations and functional implications. Endocr J 2:249–258. [Google Scholar]
- Butera PC, Czaja JA (1984). Intracranial estradiol in ovariectomized guinea pigs: effects on ingestive behaviors and body weight. Brain Res 322:41–48. [DOI] [PubMed] [Google Scholar]
- Cone RD (2005). Anatomy and regulation of the central melanocortin system. Nat Neurosci 8:571–578. [DOI] [PubMed] [Google Scholar]
- Cunningham MJ, Fang Y, Selley DE, Kelly MJ (1998). μ-Opioid agonist-stimulated [35S]GTPgammaS binding in guinea pig hypothalamus: effects of estrogen. Brain Res 791:341–346. [DOI] [PubMed] [Google Scholar]
- Czaja JA, Goy RW (1975). Ovarian hormones and food intake in female guinea pigs and rhesus monkeys. Horm Behav 6:329–349. [DOI] [PubMed] [Google Scholar]
- Dupont S, Krust A, Gansmuller A, Dierich A, Chambon P, Mark M (2000). Effect of single and compound knockouts of estrogen receptors α (ERα) and β (ERβ) on mouse reproductive phenotypes. Development 127:4277–4291. [DOI] [PubMed] [Google Scholar]
- Filardo EJ, Quinn JA, Frackelton AR Jr, Bland KI (2002). Estrogen action via the G protein-coupled receptor, GPR30: stimulation of adenylyl cyclase and cAMP-mediated attenuation of the epidermal growth factor receptor-to-MAPK signaling axis. Mol Endocrinol 16:70–84. [DOI] [PubMed] [Google Scholar]
- Fodor M, Delemarre-van de Waal HA (2001). Are POMC neurons targets for sex steroids in the arcuate nucleus of the rat? NeuroReport 12:3989–3991. [DOI] [PubMed] [Google Scholar]
- Frawley LS, Neill JD (1980). Effect of estrogen on serum prolactin levels in rhesus monkeys after hypophyseal stalk transection. Biol Reprod 22:1089–1093. [DOI] [PubMed] [Google Scholar]
- Heisler LK, Cowley MA, Tecott LH, Fan W, Low MJ, Smart JL, Rubinstein M, Tatro JB, Marcus JN, Holstege H, Lee CE, Cone RD, Elmquist JK (2002). Activation of central melanocortin pathways by fenfluramine. Science 297:609–611. [DOI] [PubMed] [Google Scholar]
- Horvath TL (2005). The hardship of obesity: a soft-wired hypothalamus. Nat Neurosci 8:561–565. [DOI] [PubMed] [Google Scholar]
- Jobst EE, Enriori PJ, Cowley MA (2004). The electrophysiology of feeding circuits. Trends Endocrinol Metab 15:488–499. [DOI] [PubMed] [Google Scholar]
- Kelly MJ, Levin ER (2001). Rapid actions of plasma membrane estrogen receptors. Trends Endocrinol Metab 12:152–156. [DOI] [PubMed] [Google Scholar]
- Kelly MJ, Loose MD, Rønnekleiv OK (1992). Estrogen suppresses μ-opioid and GABAB-mediated hyperpolarization of hypothalamic arcuate neurons. J Neurosci 12:2745–2750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly MJ, Lagrange AH, Wagner EJ, Rønnekleiv OK (1999). Rapid effects of estrogen to modulate G protein-coupled receptors via activation of protein kinase A and protein kinase C pathways. Steroids 64:64–75. [DOI] [PubMed] [Google Scholar]
- Komesaroff PA, Sudhir K, Esler MD (1999). Effects of estrogen on stress responses in women. J Clin Endocrinol Metab 84:4292–4293. [DOI] [PubMed] [Google Scholar]
- Koob GF (1992). Drugs of abuse: anatomy, pharmacology and function of reward pathways. Trends Pharmacol Sci 13:177–184. [DOI] [PubMed] [Google Scholar]
- Krege JH, Hodgin JB, Couse JF, Enmark E, Warner M, Mahler JF, Sar M, Korach KS, Gustafsson JÅ, Smithies O (1998). Generation and reproductive phenotypes of mice lacking estrogen receptor beta. Proc Natl Acad Sci USA 95:15677–15682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagrange AH, Rønnekleiv OK, Kelly MJ (1994). The potency of μ-opioid hyperpolarization of hypothalamic arcuate neurons is rapidly attenuated by 17β-estradiol. J Neurosci 14:6196–6204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagrange AH, Wagner EJ, Rønnekleiv OK, Kelly MJ (1996). Estrogen rapidly attenuates a GABAB response in hypothalamic neurons. Neuroendocrinology 64:114–123. [DOI] [PubMed] [Google Scholar]
- Lagrange AH, Rønnekleiv OK, Kelly MJ (1997). Modulation of G protein-coupled receptors by an estrogen receptor that activates protein kinase A. Mol Pharmacol 51:605–612. [DOI] [PubMed] [Google Scholar]
- McEwen BS, Alves SE (1999). Estrogen actions in the central nervous system. Endocr Rev 20:279–307. [DOI] [PubMed] [Google Scholar]
- Milewicz A, Bidzinska B, Mikulski E, Demissie M, Tworowska U (2000). Influence of obesity and menopausal status on serum leptin, cholecystokinin, galanin and neuropeptide Y levels. Gynecol Endocrinol 14:196–203. [DOI] [PubMed] [Google Scholar]
- Milewicz A, Tworowska U, Demissie M (2001). Menopausal obesity–myth or fact? Climacteric 4:273–283. [PubMed] [Google Scholar]
- Murphy KG, Bloom SR (2005). Peripheral influences on central melanocortin neurons. Peptides 26:1744–1752. [DOI] [PubMed] [Google Scholar]
- O’Dowd BF, Nguyen T, Marchese A, Cheng R, Lynch KR, Heng HH, Kolakowski LFJ, George SR (1998). Discovery of three novel G-protein-coupled receptor genes. Genomics 47:310–313. [DOI] [PubMed] [Google Scholar]
- Qiu J, Bosch MA, Tobias SC, Grandy DK, Scanlan TS, Rønnekleiv OK, Kelly MJ (2003). Rapid signaling of estrogen in hypothalamic neurons involves a novel G protein-coupled estrogen receptor that activates protein kinase C. J Neurosci 23:9529–9540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu J, Bosch MA, Rønnekleiv OK, Kelly MJ (2005). Convergence of membrane estrogen receptor and serotonin 5 HT2A/C receptor signaling in hypothalamic neurons. Soc Neurosci Abstr 31632.5.
- Revankar CM, Cimino DF, Sklar LA, Arterburn JB, Prossnitz ER (2005). A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science 307:1625–1630. [DOI] [PubMed] [Google Scholar]
- Rønnekleiv OK, Kelly MJ (2005). Diversity of ovarian steroid signaling in the hypothalamus. Front Neuroendocrinol 26:65–84. [DOI] [PubMed] [Google Scholar]
- Saper CB, Chou TC, Elmquist JK (2002). The need to feed: homeostatic and hedonic control of eating. Neuron 36:199–211. [DOI] [PubMed] [Google Scholar]
- Sherwin BB (2002). Estrogen and cognitive aging in women. Trends Pharmacol Sci 23:527–534. [DOI] [PubMed] [Google Scholar]
- Shughrue PJ, Lane MV, Merchenthaler I (1997). Comparative distribution of estrogen receptor-α and -β mRNA in the rat central nervous system. J Comp Neurol 388:507–525. [DOI] [PubMed] [Google Scholar]
- Sodersten P, Bergh C, Ammar C (2003). Anorexia nervosa: towards a neurobiologically based therapy. Eur J Pharmacol 480:67–74. [DOI] [PubMed] [Google Scholar]
- Stearns V, Ullmer L, Lopez JF, Smith Y, Isaacs C, Hayes DF (2002). Hot flushes. Lancet 360:1851–1861. [DOI] [PubMed] [Google Scholar]
- Stearns V, Johnson MD, Rae JM, Morocho A, Novielli A, Bhargava P, Hayes DF, Desta Z, Flockhart DA (2003). Active tamoxifen metabolite plasma concentrations after coadministration of tamoxifen and the selective serotonin reuptake inhibitor paroxetine. J Natl Cancer Inst 95:1758–1764. [DOI] [PubMed] [Google Scholar]
- Tecott LH, Sun LM, Akana SF, Strack AM, Lowenstein DH, Dallman MF, Julius D (1995). Eating disorder and epilepsy in mice lacking 5-HT2c serotonin receptors. Nature 374:542–546. [DOI] [PubMed] [Google Scholar]
- Thomas P, Pang Y, Filardo EJ, Dong J (2005). Identity of an estrogen membrane receptor coupled to a G-protein in human breast cancer cells. Endocrinology 146:624–632. [DOI] [PubMed] [Google Scholar]
- Tobias SC, Qiu J, Kelly MJ, Scanlan TS (2006). Synthesis and biological evaluation of SERMs with potent nongenomic estrogenic activity. Chem Med Chem 1:565–571. [DOI] [PubMed] [Google Scholar]
- Toran-Allerand CD, Singh M, Setalo G Jr (1999). Novel mechanisms of estrogen action in the brain: New players in an old story. Front Neuroendocrinol 20:97–121. [DOI] [PubMed] [Google Scholar]
- Toran-Allerand CD, Guan X, MacLusky NJ, Horvath TL, Diano S, Singh M, Connolly ES Jr, Nethrapalli IS, Tinnikov AA (2002). ER-X: a novel, plasma membrane-associated, putative estrogen receptor that is regulated during development and after ischemic brain injury. J Neurosci 22:8391–8401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson ME, Mook D, Graves F, Felger J, Bielsky IF, Wallen K (2003). Tamoxifen is an estrogen antagonist on gonadotropin secretion and responsiveness of the hypothalamic-pituitary-adrenal axis in female monkeys. Endocrine 22:305–315. [DOI] [PubMed] [Google Scholar]