

Abstract
A route for the rapid assembly of the carbon framework of several resveratrol natural products is presented. A palladium-catalyzed domino reaction of bromostilbene derivative 6 and tolane 7, involving two sequential Heck coupling reactions, provides access to the benzofulvene-based core of various resveratrol-derived natural products. The carbon skeleton of pallidol and its congeners is achieved by a Lewis acid-induced Nazarov-type oxidative cyclization of 9.
Palladium-catalyzed domino reactions, coupled with carefully designed substrates, allow for rapid establishment of molecular complexity in the total synthesis of complex natural products.1 The addition of an aryl- or alkenylpalladium species to a triple or double bond provides an intermediate poised to participate in subsequent reactions. The reactive organopalladium intermediate may be trapped in various ways, e.g., by reaction with an additional equivalent of a triple or double bond. Scheme 1 depicts one such scenario, involving a sequential Heck/intramolecular cyclization cascade.
SCHEME 1.

A Palladium-Catalyzed Cascade
Exploiting the potential for domino sequences, multiple carbon-carbon bonds can be formed in a single synthetic operation through a palladium-catalyzed cascade. Furthermore, the substrates and reaction conditions may be tailored to produce a desired functional group or structural motif. For example, depending on the nature of the diene (A, Scheme 1)—cyclic, acyclic or aryl—a palladium-catalyzed cascade could give rise to either differentially substituted fulvene-based compounds or the analogous benzofulvene-containing structures (see E).
We recognized the potential utility of these palladium-catalyzed domino reactions in accessing the benzofulvene-based core of several biologically important resveratrol derivatives. Resveratrol (1, Figure 1) was first isolated from the roots of Veratrum grandiflorum in 1940.2 It was later classified as a phytoalexin (i.e., a toxin produced by plants in response to infection or stress).3 In further biological studies, resveratrol displayed both anti-inflammatory and anti-carcinogenic properties in vivo,4 and significantly extended the lifespan of vertebrates.5 To date, the specific mechanism of action of resveratrol remains elusive due to its rapid metabolism. Several resveratrol-derived natural products have shown biological activities comparable to their parent monomer, e.g., in vitro stimulation of osteoblast proliferation at concentrations as low as 10−10 g/mL.6d However, a thorough exploration of the properties and uses of resveratrol congeners requires efficient, facile and versatile syntheses of these molecules.
FIGURE 1.

Selected resveratrol-derived natural products.
In 2007, Snyder et al. reported a chemoselective approach to the synthesis of several polyphenolic natural products believed to result from resveratrol dimerizations.7a,b Snyder employed an easily accessible building block containing three aromatic rings, which was subjected to a series of electrophile-promoted cascades to complete the total syntheses of five resveratrol-based natural products, including quadrangularin A (2a) and pallidol (5).7a This strategy was recently extended to the synthesis of other members of the resveratrol class.7b A previous synthesis of quadrangularin based on a biomimetic dimerization of a resveratrol derivative has also been reported.7c In this Communication, we report our progress to date on a strategy for the synthesis of a subset of dimeric resveratrol-based natural products.
We sought to probe the synthetic versatility of a palladium-catalyzed cascade that would provide rapid access to several resveratrol-based natural products. Specifically, we envisioned a tandem Heck/intramolecular cyclization cascade between bromostilbene derivative 6 and tolane 7 (Scheme 2). In accord with our proposed mechanism, we expected initial regioselective addition of the arylpalladium species 6a to 7, which we anticipated would be governed by the electronics of the aromatic ring substituents of 7. Proximity-driven insertion of the resultant alkenylpalladium intermediate into the pendant double bond (see 8) should afford 9 following β-hydride elimination. Such a cascade would form two carbon-carbon bonds in a single step to provide an intermediate containing all of the carbon atoms present in several important resveratrol derivatives.
SCHEME 2.

Proposed Cascade
From common intermediate 9, we anticipated selective reduction of the exocyclic double bond could likely be achieved using the method employed by Katzenellenbogen et al. to reduce tri-substituted arylindenes.8 Indene 11 could then serve as a precursor to either quadrangularin A (2a) or parthenocissin A (2b).
To access the carbon skeleton present in pallidol (5), we expected a Nazarov-type cyclization of 9 would provide 10.9 The methyl ethers could presumably be cleaved with BBr3. Reduction of the tetrasubstituted double bond could then be achieved following the precedent of Brand et al. (Scheme 2).10
Our synthetic investigations commenced with the preparation of bromide 6. Following the procedure employed by Snyder et al., 6 was synthesized from commercially available 3,5-dimethoxybenzaldehyde (12) in five steps (75% overall yield).7a Tolane 711 was also assembled from 12. Under standard Ramirez olefination conditions, 12 was converted to dibromide 13 in 60% yield (Scheme 3).12 Subsequent treatment with n-BuLi for 10 h, followed by quenching with water, afforded terminal alkyne 14 in 94% yield, completing a Corey-Fuchs sequence.13, 14 Finally, alkyne 14 was cross-coupled to p-iodoanisole under standard Sonogashira conditions to afford tolane 7 in 99% yield (56% overall yield from 12).15
SCHEME 3.

Synthesis of Tolane 7
a) CBr4 (2.0 equiv), PPh3 (4.0 equiv), CH2Cl2, 0 °C, 1 h, 60%; b) n- BuLi (2.8 equiv), THF, −78 °C→25 °C, 10 h, 94%; c) CuI (0.1 equiv), Pd(PPh3)2Cl2 (0.05 equiv), Et3N (0.1 M), 25 °C, 18 h, 99%.
Preliminary investigations confirmed the viability of our proposed cascade—when 6 and 7 were subjected to standard Heck conditions (Pd(OAc)2, PPh3, DIPEA, DMF, 135 °C, 15 h), 916 was isolated as a bright red solid in 53% yield (Scheme 4). Slow diffusion of pentane into a solution of 9 in CH2Cl2 resulted in the formation of crystals suitable for X-ray diffraction. The ORTEP representation of 9 (Figure 2) revealed an E orientation of the exocyclic double bond, as in parthenocissin A (2b).
SCHEME 4.

Synthesis of 9 and 15
FIGURE 2.
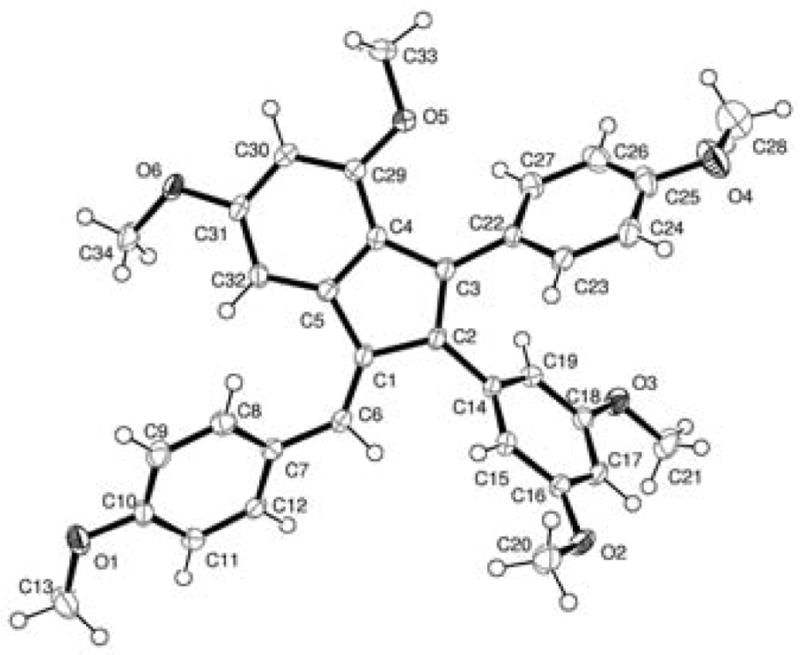
Molecular structure of 9; ellipsoids drawn at the 50% probability level.
With 9 in hand, a variety of Lewis and Bronsted acids were tested in an effort to form 10 via our proposed Nazarov-type cyclization (Scheme 2). We found success with the treatment of 9 with FeCl3•6H2O (1 equiv) in CH2Cl2 to yield diene 1517 in 38% yield as a maroon solid (Scheme 4). Although 10 was not observed, pentalene 15 could presumably have formed via the intermediacy of 10, which was likely oxidized under the reaction conditions. Alternatively, 15 may have resulted directly from 9 via a cyclodehydrogenation in keeping with the precedent of Kovacic.18 Slow evaporation of a CH2Cl2 solution of 15 yielded X-ray quality crystals, which confirmed the connectivity of 15 (Figure 3). Attempts to reduce 15, as well as the exo- and endocyclic double bonds of 9, are ongoing.
FIGURE 3.
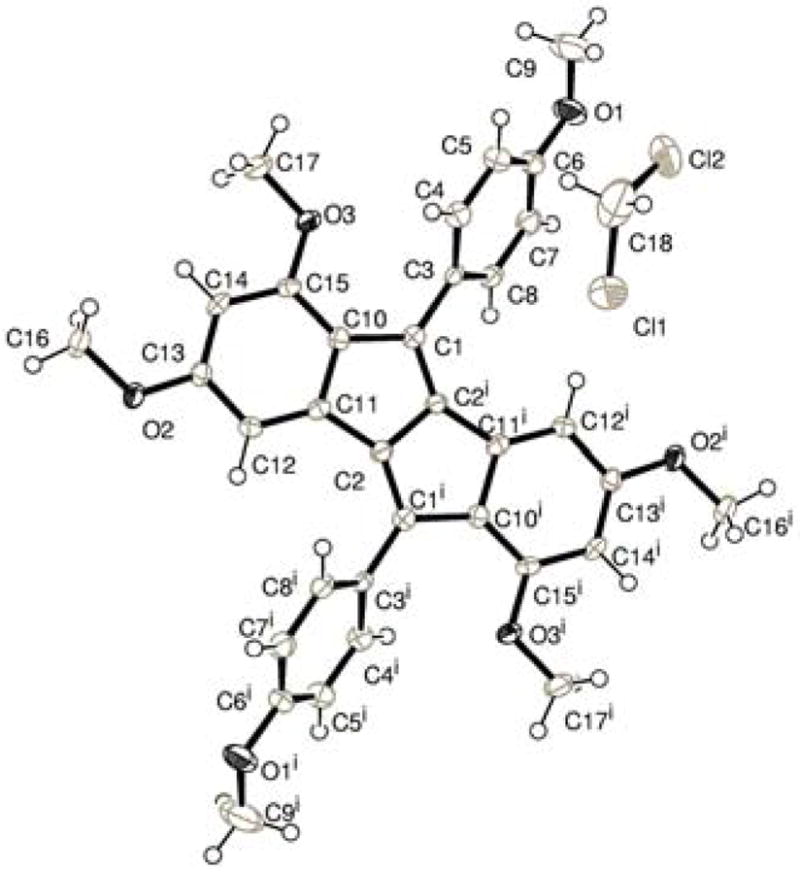
ORTEP representation of 15; ellipsoids drawn at the 50% probability level.
In conclusion, we have demonstrated a novel palladium-catalyzed cascade reaction, which assembles the carbon framework of several resveratrol-derived natural products. Starting from two readily accessible building blocks, we have synthesized potential precursors to a large family of natural products, including quadrangularin A, parthenocissin A and pallidol. Studies on the functionalization of these compounds and completion of the total syntheses of a series of resveratrol-based natural products are underway and will be reported in due course.
Acknowledgments
The authors are grateful to UC Berkeley, GlaxoSmithKline, Amgen, Dupont, and Abbott laboratories for generous, unrestricted financial support. We would also like to thank Dr. Antonio DiPasquale for obtaining the X-ray structures of 9 and 15.
Footnotes
Supplementary Material
Experimental details and characterization data for all new compounds; X-ray structures of 9 and 15, structure refinement details, tables of atomic coordinates, thermal parameters, bond lengths, and bond angles. This material is available upon request from the authors. Crystallographic data (excluding structure factors) for the structures in this paper have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication nos. 718629 and 718630. Copies of the data can be obtained, free of charge, on application to CCDC, 12 Union Road, Cambridge CB2 1EZ, UK, (fax: +44-(0)1223-336033 or e-mail: deposit@ccdc.cam.ac.Uk).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.For a review of domino reactions in organic synthesis, see: Tietze LF. Chem Rev. 1996;96:115–136. doi: 10.1021/cr950027e.For a review of the Heck reaction in modern synthesis and a list of several examples of palladium-catalyzed cascade reactions, see: De Meijere A, Meyer FE. Angew Chem Int Ed Engl. 1994;33:2379–2411.
- 2.Takaoka M. Proc Imp Acad Tokyo. 1940;16:405–407. [Google Scholar]
- 3.Langcake P, Pryce RJ. Physiol Plant Pathol. 1976;9:77–86. [Google Scholar]
- 4.Jang M, Cai L, Udeani GO, Slowing KV, Thomas CF, Beecher CWW, Fong HHS, Farnsworth NR, Kinghorn AD, Mehta RG, Moon RC, Pezzuto JM. Science. 1997;275:218–220. doi: 10.1126/science.275.5297.218. [DOI] [PubMed] [Google Scholar]
- 5.Valenzano DR, Terzibasi E, Genade T, Cattaneo A, Domenici L, Cellerino A. Curr Biol. 2006;16:296–300. doi: 10.1016/j.cub.2005.12.038. [DOI] [PubMed] [Google Scholar]
- 6.(a) Ohyama M, Tanaka T, Iinuma M. Phytochemistry. 1995;38:733–740. [Google Scholar]; (b) Ohyama M, Tanaka T, Iinuma M. Chem Pharm Bull. 1994;42:2117–2120. [Google Scholar]; (c) Adesanya SA, Nia R, Martin TM, Boukamcha N, Montagnac A, Pais M. J Nat Prod. 1999;62:1694–1695. [Google Scholar]; (d) Luo HF, Zhang LP, Hu CQ. Tetrahedron. 2001;57:4849–4854. [Google Scholar]; (e) Li WW, Ding LS, Li BG, Chien YZ. Phytochemistry. 1996;42:1163–1165. [Google Scholar]; (f) Tanaka T, Ito T, Nakaya K, Iinuma M, Riswan S. Phytochemistry. 2000;54:63–69. doi: 10.1016/s0031-9422(00)00026-1. [DOI] [PubMed] [Google Scholar]; (g) Coggon P, Janes NF, King TJ, Wallwork SC. J Chem Soc. 1965:406–408. [Google Scholar]; (h) Ito T, Tanaka T, Iinuma M, Iliya I, Nakaya K, Ali Z, Takahashi Y, Sawa R, Shirataki Y, Murata J, Darnaedi D. Tetrahedron. 2003;59:5347–5363. [Google Scholar]; (i) Supudompol B, Likhitwitayawuid K, Houghton PJ. Phytochemistry. 2004;65:2589–2594. doi: 10.1016/j.phytochem.2004.08.003. [DOI] [PubMed] [Google Scholar]; (j) Sahadin Hakim EH, Juliawaty LD, Syah YM, bin Din L, Ghisalberti EL, Latip J, Said IM, Achmad SAZ. Naturforsch, C: Biosci. 2005;60:723–727. doi: 10.1515/znc-2005-9-1011. [DOI] [PubMed] [Google Scholar]; (k) Ito T, Tanaka T, Iinuma M, Nakaya K, Takahashi Y, Sawa R, Murata J, Darnaedi D. J Nat Prod. 2004;67:932–937. doi: 10.1021/np030236r. [DOI] [PubMed] [Google Scholar]
- 7.(a) Snyder SA, Zografos AL, Lin Y. Angew Chem Int Ed. 2007;46:8186–8191. doi: 10.1002/anie.200703333. [DOI] [PubMed] [Google Scholar]; (b) Snyder SA, Breazzano SP, Ross AG, Lin Y, Zografos AL. J Am Chem Soc. 2009 doi: 10.1021/ja806183r. [DOI] [PubMed] [Google Scholar]; (c) Li W, Li H, Li Y, Hou Z. Angew Chem Int Ed. 2006;45:7609–7611. doi: 10.1002/anie.200603097. [DOI] [PubMed] [Google Scholar]
- 8.Anstead GM, Ensign JL, Peterson CS, Katzenellenbogen JA. J Org Chem. 1989;54:1485–1491. [Google Scholar]
- 9.Nazarov IN, Zaretskaya II. Bull Acad Sci URSS, Classe sci chim. 1942:200–209.For a review of the Nazarov cyclization, see: Habermas KL, Denmark SE, Jones TK. Org React. 1994;45:1–158.
- 10.Brand K, Krey W. J Prakt Chem. 1925;110:10–25. [Google Scholar]
- 11.7: To a solution of p-iodoanisole (0.100 g, 0.427 mmol), Pd(PPh3)2Cl2 (0.015 g, 0.0214 mmol), and CuI (0.0081 g, 0.0427 mmol) in Et3N (5 mL) under N2 was added ethynylbenzene 14 (0.0970 g, 0.598 mmol). The solution was stirred at rt for 18 h. The solvent was removed in vacuo and the crude brown residue was purified by flash chromatography (6:1 hexanes/EtOAc) to give 0.113 g (99% yield) of 7 as a colorless oil. Rf 0.52 (2:1 hexanes/EtOAc); 1H NMR (400 MHz, CDCl3) δ 7.46 (d, J = 8.8 Hz, 2H), 6.86 (d, J = 8.8 Hz, 2H), 6.67 (s, 2H), 6.43 (s, 1H), 3.80 (s, 3H), 3.77 (m, 6H); 13C NMR (125 MHz, CDCl3) δ 160.72, 159.88, 133.32, 125.07, 115.36, 114.20, 110.37, 109.37, 103.12, 101.74, 89.18, 88.28, 81.78, 55.62; IR (film) νmax 3002, 2957, 2935, 2837, 2216, 1650, 1588, 1511, 1452, 1419, 1357, 1347, 1290, 1248, 1205, 1156, 1121, 1064, 1031 cm−1; HRMS (ESI) m/z 269.1178 [(M+H)+; calculated for [C17H17O3]+: 269.1172].
- 12.Ramirez F, Desai NB, McKelvie N. J Am Chem Soc. 1962;84:1745–1747. [Google Scholar]
- 13.Corey EJ, Fuchs PL. Tetrahedron Lett. 1972;13:3769–3772.For a review of alkyne synthesis using phosphorous compounds, see: Eymery F, Iorga B, Savignac P. Synthesis. 2000:185–213.
- 14.Nakamura H, Kuroda H, Saito H, Suzuki R, Yamori T, Maruyama K, Haga T. Chem Med Chem. 2006;1:729–740. doi: 10.1002/cmdc.200600068. [DOI] [PubMed] [Google Scholar]
- 15.Sonogashira K, Tohda Y, Hagihara N. Tetrahedron Lett. 1975:4467–4470.For a review of organopalladium chemistry in organic synthesis, see: Sonogashira K. Synthesis. 2002;1:493–529.
- 16.9: To a flame-dried Schlenk tube containing Pd(OAc)2 (0.007 g, 0.031 mmol) and triphenylphosphine (0.016 g, 0.062 mmol) was added a solution of bromide 6 (0.050 g, 0.154 mmol), tolane 7 (0.081 g, 0.301 mmol) and N,N-diisopropylethylamine (DIPEA) (0.08 mL, 0.461 mmol) in DMF (1 mL) via syringe. The combined reaction mixture was evacuated, backfilled with N2 (x 3), sealed and heated in an oil bath at 135 °C for 15 h. The reaction mixture was allowed to cool to rt, diluted with Et2O (10 mL) and washed with 1% aqueous HCl (3 × 5 mL). The combined aqueous washings were reextracted with Et2O (2 × 5 mL). The combined organic extracts were washed with 1% aqueous HCl (5 mL), brine (2 × 5 mL), dried over MgSO4, filtered and concentrated in vacuo to afford a dark red residue. The crude product was purified by flash chromatography (12:1 hexanes/EtOAc) to afford 0.0437 g (53% yield) of 9 as a bright red solid. Rf 0.70 (1:1 hexanes/EtOAc); 1H NMR (400 MHz, CDCl3) δ 7.48 (d, J = 8.5 Hz, 2H), 7.07 (d, J = 8.5 Hz, 3H), 7.03 (s, 1H), 6.99 (s, 1H), 6.92 (d, J = 5.0 Hz, 2H), 6.79 (m, 2H), 6.41 (m, 2H), 6.37 (m, 1H), 6.29 (s, 1H), 3.84 (s, 3H), 3.77 (s, 3H), 3.70 (m, 3H), 3.62 (s, 6H), 3.59 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 160.15, 159.44, 159.27, 158.44, 158.33, 141.19, 140.64, 139.63, 138.90, 137.95, 137.40, 132.47, 131.38, 131.10, 129.46, 127.90, 124.58, 124.43, 113.98, 113.96, 113.44, 112.44, 109.82, 108.49, 102.04, 99.70, 99.58, 99.03, 55.83, 55.66, 55.64, 55.55, 55.42, 55.32; IR (film) νmax 2998, 2936, 2836, 1591, 1559, 1540, 1509, 1462, 1421, 1282, 1247, 1203, 1174, 1153, 1105, 1065 cm−1; HRMS (ESI) m/z 536.2198 [(M+); calculated for [C34H32O6]+: 536.2199]; MP 158–159 °C.
- 17.15: To a solution of 9 (0.0171 g, 0.0319 mmol) in CH2Cl2 (1 mL) was added FeCl3•6H2O (0.009 g, 0.0319 mmol). The reaction mixture was stirred at rt for 18 h, diluted with CH2Cl2 (5 mL), washed with water (3 mL), brine (5 mL), dried over MgSO4, filtered and concentrated in vacuo. The resulting residue was purified by flash chromatography (9:1 hexanes/EtOAc) to give 0.0065 g (38% yield) of 15 as a maroon solid. Rf 0.50 (1:1 hexanes/EtOAc); 1H NMR (400 MHz, CDCl3) δ 7.52 (d, J = 8.8 Hz, 4H), 6.91 (d, J = 8.8 Hz, 4H), 6.37 (d, J = 2.0 Hz, 2H), 5.95 (d, J = 2.0 Hz, 2H), 3.85 (s, 6H), 3.64 (s, 6H), 3.52 (s, 6H); 13C NMR (125 MHz, CDCl3) δ 161.71, 159.61, 156.22, 146.79, 142.30, 140.10, 139.14, 130.78, 128.11, 118.08, 112.79, 101.68, 98.47, 97.19, 69.72, 55.57, 55.47, 55.45, 53.98; IR (film) νmax 2927, 1596, 1508, 1462, 1390, 1360, 1246, 1200, 1151, 1085 cm−1; HRMS (ESI) m/z 534.2035 [(M+); calculated for [C34H30O6]+: 534.2042]; MP 242–243 °C.
- 18.Kovacic P, Jones MB. Chem Rev. 1987;87:357–379. [Google Scholar]