

Abstract
A redox- and light-sensitive, T1-weighted magnetic resonance imaging (MRI) contrast agent which tethers a spiropyran(SP)/merocyanine(MC) motif to a Gd-DO3A moiety was synthesized and characterized. When in the dark, the probe is in its MC form which has an r1 relaxivity of 2.51 mM−1s−1 (60MHz, 37°C). After irradiation with visible light or mixing with NADH, the probe experiences an isomerization and the r1 relaxivity decreased 18% and 26%, respectively. Additionally, the signal intensity in MRI showed an observable decrease after the compound was mixed with NADH.
Introduction
Redox reactions are one of the two energy resources available to living organisms. Biochemical redox couples are responsive to changes in oxidizing-reducing conditions which can result from changes in blood flow, oxygenation, and other variables.1 Thus, the ability to non-invasively monitor redox activity in a living system would provide a foundation for detailed studies of metabolism in relation to disease processes and therapies. Among various biochemical redox couples, the reduced nicotinamide adenine dinucleotide NADH is the principal electron donor for the respiratory chain in mammalian cells.1 It plays a key role in the energy production of cells and also has the function of stimulating cellular production of the neurotransmitters dopamine, noradrenaline and serotonin, etc.1,2 Although the NADH activities in living organisms have been investigated by optical imaging for studies of metabolic responses of tissue, the harmful levels of ultraviolet radiation or intense laser pulses which were commonly used to induce the fluorescence needed for the measurements limit its clinical application.3–6
Magnetic resonance imaging (MRI) is one of the most important modern diagnosis tools available in medicine. Due to its high spatial resolution, this non-invasive and multidimensional imaging modality can explore the internal structural features of living systems.7 Although MRI typically is not capable of sensing biochemical activity, recent advances in contrast agents, which can increase or decrease signal intensity in MRI thus improving contrast between that tissue containing the agent and surrounding tissues, hold the promise that MR contrast agents can be designed to be indicators of biological processes.8–12 Meade et al have designed an analog of DOTA, i.e. galactopyranosyl substituted 1-hydroxyethyl-1,4,7,10-tetraazacyclododecane-4,7,10-triacetic acid. When this ligand binds Gd(III), all nine coordination sites of the metal ion are occupied and there is no water molecule in the first coordination sphere. When β-galactosidase cleaves the galactopyranose moiety enzymatically, a free coordination site appears that is quickly taken by a water molecule, hence the increase in relaxivity observed in the presence of the enzyme.8,9 The agent is promising in applications as an indicator for gene expression as the enzyme β-galactosidase has been used as a marker of gene delivery and expression in gene therapy and developmental biology.
To date the report of molecular imaging contrast agents sensitive to redox/electrical activity is rare. The relaxivities of DOTA-based complexes of gadolinium bearing a thiol moiety on a propyl or hexyl arm increased in saline when interacting with human serum albumin (HSA), probably by forming reversible covalent linkages with the reactive thiol at cysteine-34 of HSA.12 Optical techniques to measure electrical activity were first suggested in the 1960’s based on the discovery that changes in electrical activity may be accompanied by innate optical changes.13 Today, exogenous dyes for measurement of electrical activity are a large and well-studied group of indicators. Among them spiropyran attracted us because they experience a significant structural rearrangement in response to changes in light and electrical activity.14–16 Recently, we reported an activatable MR/optical imaging contrast agent which is a combination of a spiropyran group and a Gd-DO3A complex. The structural change of the spiropyran moiety in the contrast agent influences the accessibility of water to Gd(III), thus causing the two isomers to have different contrast enhancement properties.17 In this paper, we report the synthesis and characterization of compound 7, a T1-weighted MRI contrast agent that tethers a dinitrospiropyran molecule to a Gd-DO3A moiety. By adding a strong electron-withdrawing nitro group to the indole ring, the isomerization of compound 7 to the SP form should be more thermodynamically favorable. We expect that a more efficient conversion between MC and SP isomers would increase the effect on relaxivity.
Results and Discussion
Contrast agent 7 was synthesized as shown in Scheme 1. The dinitrospiropyran 3 was prepared by condensation of the salicylaldehyde derivative 2 with 5-nitroindolenine in refluxing THF.18,19 The electron-withdrawing nature of the nitro group was emphasized by the low reactivity of the eneamine group, which resulted in a moderate yield (49%) after a relatively long reflux time (24 h) as compared to our previously reported spiropyran Gd-DO3A. The chloride 3 was converted into the corresponding iodide 4 by halogen exchange, then refluxed with 1,4,7,10-Tris(tert-butoxycarbonylmethyl)-1,4,7,10-tetraazacyclododecane HBr salt20 (tri-ester cyclen) to give crowned dinitrospiropyran 5 in good yield. Compound 5 was hydrolyzed under acidic conditions to give tri-acid 6. Complexation was carried out in methanol using gadolinium(III) trifluoromethanesulfonate to yield complex 7.
SCHEME 1.

Synthesis of dinitrospiropyran-Gd-DO3A complex 7.
The photochromic behavior of spiropyran compounds has been well investigated.15,21 The compound is stable in its closed-ring (SP) isomeric form as a colorless or pale yellow solution, while UV irradiation produces a metastable open-ring isomer (MC), which has an optical absorption peak at 550–600 nm. The original colorless form of SP can be restored either via visible light irradiation or thermally, as shown in Scheme 2.
SCHEME 2.

Photoisomerization of spiropyran.
The photoisomerization of complex 7 in water is shown in Figure 1. Storage in the dark affords the red colored merocyanine isomer with a distinct absorption at 502 nm. UV light irradiation can not further increase the intensity of the absorption. Irradiation with visible light gives the pale yellow colored solution and the absorption spectrum shows that the peak at 502 nm dramatically decreases, indicating that the compound isomerizes to its closed SP form. After compound 7 was irradiated with visible light for 5 minutes, UV irradiation at 365 nm only partially restored the peak at 502 nm. However, the absorption at 502 nm was almost completely restored after the sample was placed in the dark for 30 minutes. Compared with the spiropyran-Gd-DO3A contrast agent we reported previously, irradiation with visible light results in almost complete loss of the 502 nm absorbance peak. This loss was only partial for the previous compound suggesting incomplete conversion to the SP isomer.17 Despite the strong electrostatic interaction between the high charge density, cyclen-complexed gadolinium cation and the phenoxide oxygen, which restricts the conversion of complex 7 from MC isomer to SP isomer (Scheme 3), the introduction of a strong electron-withdrawing nitro group onto the indole part of the complex destabilizes formation of the positive charge carried in the iminium form of the MC isomer, thus shifting the equilibrium of the complex back towards the SP isomer.19
FIGURE 1.

Absorption spectra of compound 7 (4.167 × 10−5 M) in water. 1: in the dark; 2: irradiated with visible light for 5 min; 3: in the dark for 30 min after irradiation with visible light for 5 min; 4: irradiated with UV at 365 nm for 30 min after irradiation with visible light for 5 min.
SCHEME 3.

Proposed isomerization of Gd-5′-NO2-SPDO3A (left) and Gd-5′-NO2-MCDO3A (right).
Fluorescence emission at 603 nm was detected when 7-MC was excited at 500 nm, as shown in Fig. 2. Compared with the complex we reported previously, the fluorescence intensity of complex 7 is much weaker.17 After irradiation with visible light for 5 minutes, the fluorescence of 7-MC was partially quenched. This result indicates that the fluorescence emission derives not only from the MC form, but also from SP form since the conversion from 7-MC isomer to 7-SP isomer is almost complete (Fig. 1). The result is different from those reported in literature, which show that both plain and metal ion complexed spiropyrans produce fluorescence emission only when they adopt the MC form.22–24 In complex 7, the strong electro-withdrawing nitro group on the indole ring and the possible interaction of complexed Gd(III) in SP form (Scheme 3) decrease the electron density on indole nitrogen atom, thus reducing its ability to quench fluorescence emission through PET and allows the SP form to retain some fluorescence emission.22
FIGURE 2.

Emission spectra of compound 7 (5.738 × 10−5 M) in water with excitation at 500 nm.
Most spiropyran compounds are sensitive to electrical activity and have the property of electrochromism. The redox property of contrast agent 7 was tested in water using a series of biological redox agents, such as NAD+/NADH, hydrogen peroxide, Fe(III) complexes, etc. NADH was found to be an effective reducing agent for the MC isomer of the agent. Reactions of compound 7 and NADH (molar ratio 1/1) in water were monitored by absorption change of compound 7 at 502 nm, as shown in Fig. 3. Upon introduction of NADH, compound 7 exhibited a steady decrease in absorbance at 502 nm through the 60 min. timepoint. When more or less concentrated NADH was employed, the reaction rate was raised or lowered, respectively. In addition, this experiment was repeated in phosphate buffer to assess the complex’s stability and illuminate whether its redox response to NADH is affected. The results in pH 7.0 Na2HPO4-NaH2PO4 buffer solution were similar to that of water indicating that phosphate does not affect the stability or redox sensitivity of the complex.
FIGURE 3.

Redox reaction of compound 7 (5.62 × 10−5 M) with NADH in water. Gd:NADH = 1:1. Lines from top: compound 7 in dark; mixture of compound 7 and NADH at 5 min, 10 min, 20 min, 30 min and 60 min.
The effect of light or NADH on the relaxivity of protons (r1) of compound 7 in aqueous solution was evaluated. The r1 value of compound 7 in dark is 2.51 ±.07 mM−1s−1, which is smaller than the r1 values of Gd-DOTA and spiropyran-Gd-DO3A we reported previously.17,25,26 The value decreased to 2.05 ±.07 mM−1s−1 after irradiation with visible light for 10 min or 1.86 ±.05 mM−1s−1 after mixing with NADH for 10 min. A control experiment showed that NADH has no influence on T1 relaxivity. Thus, visible light or NADH can decrease the r1 relaxivity value of compound 7 by 18% or 26%, respectively. The decrease in relaxivity observed for both light irradiation and mixture with NADH is statistically significant with p-values < 0.001. Addition of the nitro group to indole moiety produces a response to NADH that was not observed for our previously reported spiropyran-Gd-DO3A.17 Table 1 provides a comparison of these relaxivity values for our current and previous compound.
TABLE 1.
Comparison of r1 relaxivities for Dinitrospiropyran-Gd-DO3A and Spiropyran-Gd-DO3A
| r1 (mM-1s-1) | |||
|---|---|---|---|
| in dark | light | NADH | |
| Dinitrospiropyran-Gd-DO3A | 2.51 | 2.05 | 1.86 |
| Spiropyran-Gd-DO3A17 | 3.72 | 2.93 | x |
T1-weighted MR images of compound 7 were taken on a Biospec 7 T (300MHz) system (Bruker, Billerica, MA) in aqueous solution at room temperature. Aqueous solutions with a series of concentrations of compound 7 were prepared and stored in dark. Images were taken before and after irradiation with visible light for 10 min or mixing with NADH for 30 min. The signal intensity in MRI of compound 7 did not have observable change after visible light irradiation. However, the intensity did decrease after mixing with NADH for 30 min. as shown in Fig. 4.
FIGURE 4.
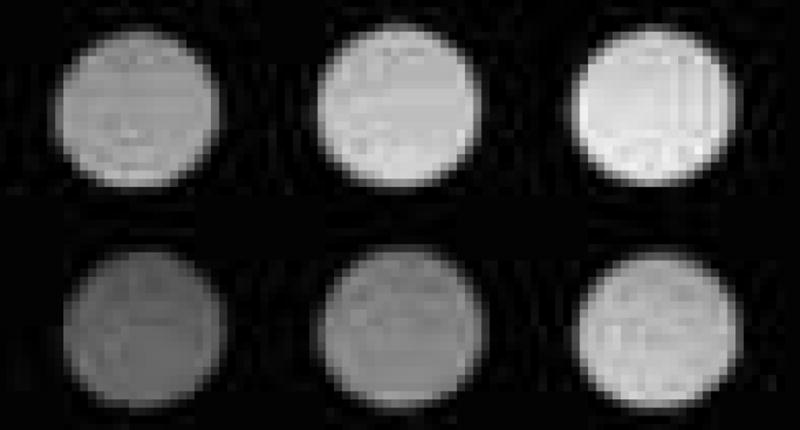
Compound 7 in water imaged by T1-weighted MRI show different brightness (pixel) before and after mixing with NADH: upper row (in dark): 152 ± 8; 187 ± 10; 208 ± 14; bottom row (mixed with NADH for 30 minutes): 92 ± 9; 126 ± 9; 175 ± 13. (Pixels were measured by image J. For both lines from left to right, the concentrations of compound 7 were 0.13, 0.26, 0.52 mM, individually. The concentration of NADH was 0.64 mM.).
The ability of a Gd contrast agent to create contrast in an MRI image depends on the interaction of water with the gadolinium in the first coordination sphere.8,9 The decrease in the r1 relaxivity of contrast agent 7 followed by visible light irradiation or reaction with NADH may be explained by the structural distinction between 7-MC and 7-SP isomeric forms.17 In the 7-SP form, the complexed Gd(III) may be attracted to the indoline part of the molecule where the metal ion has electrostatic interaction with nonbonding electrons in oxygen and nitrogen atoms, and perhaps the π electrons on the aromatic ring.17 Thus the indoline part “covers” Gd(III) and the “indoline cap” prevents the water molecules from accessing the metal ion and inhibits MRI contrast enhancement. In the 7-MC form, cleavage of the Csp3–O bond and rehybridization of the spiro carbon atom from sp3 to sp2 gives coplanar rings; thus, removal of “indoline cap” allows the water molecules to access the metal ion more readily and increases relaxivity, as shown in Scheme 3. In order to investigate our hypothesis, we performed 17ONMR experiments to determine the hydration number, q, for the contrast agent 7 before and after reaction with NADH. Complex 7 in dark conditions gave a hydration number of 1.16; upon reaction with equimolar NADH, the q value decreased to 0.44. The control experiment determined that NADH has no influence on hydration number. This result supports our speculation that a conformation change brought about by reaction with NADH is able to block water access to the first coordination sphere of gadolinium, as evidenced by the decrease in hydration number and corresponding decrease in r1 relaxivity.
Conclusion
This work describes a T1-weighted, activatable MRI contrast agent triggered by either light or NADH. When in the dark, the contrast agent is in its MC form and has higher r1 relaxivity. After irradiation with visible light or mixing with NADH, the contrast agent experiences a color change due to isomerization to SP form (or other conformation) and r1 relaxivity decreases by 18% or 26%, respectively. The light induced isomerization is reversible, but the NADH induced process is not. The contrast agent’s signal intensity in MRI has an observable decrease after mixture with NADH. This novel MRI contrast agent may have unique potential to respond to NADH related biochemical activities and may lead to non-invasive investigation of metabolic activities and cell signaling in vivo.27,28 Currently, almost all investigation related to redox activities is performed by optical imaging. Since optical imaging is limited to observing events near the tissue surface and is generally not applicable to living mammalian systems, MRI contrast agents sensitive to redox would open MRI to a host of new applications in functional imaging.
Experimental
General
Reagents were obtained from commercial suppliers and used directly, unless otherwise noted. The absorption spectra were taken with Cary 100-Bio UV-Vis spectrophotometer. Fluorescent spectra were measured on a Jobin Yvon Horiba FluoroMax-P spectrophotometer. ESI+ MS was obtained on a Thermo LCQ ion trap (San Jose, CA) operated with an electrospray source in positive ion mode under standard conditions. HR-MS was obtained on a Thermo LTQ-ORBITRAP (Waltham, MA). NMR spectra were measured with a Bruker FT-500 spectrometer (499.7 MHz for 1H, 125.7 MHz for 13C, 67.8 MHz for 17O) at 298.0 K. Chemical shifts (δ) are expressed in ppm downfield from TMS and coupling constants (J) values are given in Hz. The 17ONMR hydration experiments were run with 2% 17O enrichment in nanopure water without frequency locking of the spectrometer. T1 relaxation time was measured on a 1.5 T Bruker Minispec relaxometer at 37°C. MRI images were taken at room temperature on a Biospec 7 T system (Bruker, Billerica, MA) equipped with the standard gradient set, 95 mT/m maximum gradient, and 72 mm inner diameter (i.d.) volume coil.
Synthesis of 1′,3′,3′-Trimethyl-5′,6-dinitro-8-chloromethyl-spiro(2H-1-benzopyran-2,2′-indoline) 3: A solution of 3-chloromethyl-5-nitrosalicylaldehyde 2 (1.0 eq, 0.05 g, 0.23 mmol) and 5-nitro-1,3,3-trimethyl-2-methyleneindoline 1 (1.0 eq, 0.05 g, 0.23 mmol) in THF (180 mL) was refluxed for 24 hours. After cooling to room temperature, the solvent was removed. The crude product was purified by column chromatography on silica gel with dichloromethane as eluent to give 0.047 grams (49% yield) of yellow solid. Mp: 213-215°C. ESI+ MS (50% MeOH and 0.1% Formic acid): m/z 416 (M, 51%), 380 (100%). 1H NMR (500 MHz, CDCl3) δ 8.22 (s, 1H, ArH), 8.18 (s, 1H, ArH), 8.06 (s, 1H, ArH), 8.00 (s, 1H, ArH), 7.05 (d, J = 9.7 Hz, 1H, ArH), 6.57 (d, J = 8.2 Hz, 1H, =CH), 5.93 (d, J = 9.9 Hz, 1H, =CH), 4.43 (d, J = 11.0, 1H, CH2), 4.35 (d, J = 9.5 Hz, 1H, CH2), 2.86 (s, 3H, NCH3), 1.41 (s, 3H, CH3), 1.28 (s, 3H, CH3). 13C NMR (126 MHz, CDCl3) δ 156.3, 152.8, 141.5, 141.3, 137.0, 129.3, 126.8, 126.6, 125.0, 123.3, 120.3, 118.8, 118.5, 106.6, 106.2, 51.8, 39.7, 29.1, 26.2, 20.1. HRMS (ESI, M+) calc. for C20H18O5N3Cl: 416.1008. Found: 416.0986.
Synthesis of 1′,3′,3′-Trimethyl-5′,6-dinitro-8-iodomethyl-spiro(2H-1-benzopyran-2,2′-indoline) 4: The compound 3 (1.0 eq, 1.2 g, 2.88 mmol) was mixed with potassium iodide (4.17 eq, 1.99 g, 12 mmol) and stirred for 24 hours in 80 mL of acetone. The solvent was evaporated in vacuo and the residue was dissolved in dichloromethane. After filtration, the filtrate was evaporated to dryness to give 1.42 g (96% yield) of orange solid. Mp: 158–161°C. ESI+ MS (50% MeOH and 0.1% Formic acid): m/z 380 (100%). 1H NMR (500 MHz, CDCl3) δ 8.20 (s, 1H, ArH), 8.10 (s, 1H, ArH), 7.99 (s, 2H, ArH), 7.05 (d, J = 10.2 Hz, 1H, ArH), 6.60 (d, J = 8.4 Hz, 1H, =CH), 5.95 (d, J = 10.2 Hz, 1H, =CH), 4.24 (m, 1H, CH2), 4.14 (m, 1H, CH2), 2.91 (s, 3H, NCH3), 1.44 (s, 3H, CH3), 1.29 (s, 3H, CH3). 13C NMR (126 MHz, CDCl3) δ 156.0, 152.9, 141.4, 141.2, 137.1, 129.4, 126.8, 126.6, 126.1, 122.8, 120.0, 118.9, 118.5, 106.7, 106.4, 51.7, 29.2, 26.2, 20.2, 20.1. HRMS (ESI, M+) calc. for C20H18O5N3I: 508.0364. Found: 508.03415.
Synthesis of 1′,3′,3′-Trimethyl-5′,6-dinitro-8-{1-[4,7,10-tris(tert-butoxycarboxymethyl)-1,4,7,10-tetraazacyclododecyl]methyl}spiro(2H-1-benzopyran-2,2′-indoline) 5 A solution of triester cyclen (1.2 eq, 1.73 g, 2.908 mmol) and sodium carbonate (3.0 eq, 0.77 g, 7.27 mmol) in 25 ml of dry THF was stirred in a 100-ml of flask equipped with a calcium chloride drying tube for 30 minutes. 8-(Iodomethyl)spirobenzopyran 4 (1.0 eq, 1.23 g, 2.42 mmol) in 15 ml of dry THF was added to the flask dropwise. The mixture was refluxed for 5 hours. The solvent was evaporated in vacuo to dryness. The residue was dissolved in dichloromethane and the resulting solution was washed with 1N hydrochloric acid, 5% aqueous NaHCO3 solution and water. The organic layer was separated and dried over magnesium sulfate. The crude product was purified by column chromatography on silica gel with dichloromethane/methanol (98/2) as eluent to give 1.372 grams (63% yield) of yellow solid. Mp: 184–185°C. ESI+ MS (50% MeOH and 0.1% Formic acid): m/z 896 (M+2H, 100%), 897 (M+3H, 44%), 380 (22%). 1H NMR (500 MHz, CDCl3) δ 8.30 (s, 1H, ArH), 8.23 (d, J = 8.2 Hz, 1H, ArH), 8.00 (s, 1H, ArH), 7.95 (s, 1H, ArH), 7.06 (d, J = 9.8 Hz, 1H, ArH), 6.71 (d, J = 7.9 Hz, 1H, =CH), 5.89 (d, J = 10.0 Hz, 1H, =CH), 3.68 (d, J = 13.0 Hz, 1H, ArCH2), 3.49 (d, J = 12.2 Hz, 1H, ArCH2), 3.06-2.81 (m, 14H, NCH2 & NCH3), 2.44-2.09 (m, 11H, NCH2), 1.49-1.43 (m, 27H, C(CH3)3), 1.30 (s, 3H, CH3), 1.22 (s, 3H, CH3). 13C NMR (126 MHz, CDCl3) δ 174.1, 173.7, 173.0, 157.6, 152.8, 141.2, 141.1, 136.7, 129.9, 129.7, 128.7, 126.9, 123.9, 122.8, 118.9, 107.0, 106.6, 106.3, 83.4, 83.3, 82.8, 57.4, 56.6, 56.1, 54.9, 53.7, 52.3, 50.8, 50.3, 29.8, 28.8, 28.7, 28.5, 28.4, 28.2, 28.1, 28.0, 27.8, 27.6, 27.4, 20.3, 20.1. HRMS (ESI, M+) calc. for C46H67O11N7: 894.4971. Found: 894.4988.
Synthesis of 1′,3′,3′-Trimethyl-5′,6-dinitro-8-{1-[4,7,10-tris(carboxymethyl)-1,4,7,10-tetraazacyclododecyl]methyl}spiro(2H-1-benzopyran-2,2′-indoline) 6 A solution of triester cyclen spiropyran 5 (1.24 g, 1.39 mmol) in 12 mL of chloroform and 15 mL of trifluoroacetic acid was refluxed for 24 hours. The solvent was evaporated in vacuo to dryness. The residue was taken up in methanol (3 × 15 mL) and each time the resulting solution was evaporated to dryness to give 1.0 grams (99% yield) of red solid. Mp: 138–140°C. ESI+ MS (50% MeOH and 0.1% Formic acid): m/z 727 (M+H, 100%), 728 (M+2H, 89%), 380 (21%). 1H NMR (500 MHz, DMSO-d6) δ 8.53 (s, 1H, ArH), 8.40 (s, 1H, ArH), 8.21 (s, 1H, ArH), 8.05 (s, 1H, ArH), 7.37 (s, 1H, ArH), 6.94 (s, 1H, =CH), 6.14 (s, 1H, =CH), 4.26-4.12 (m, 4H, NCH2), 3.94-3.86 (m, 2H, NCH2), 3.65-3.51 (m, 7H, NCH2), 3.20 (s, 7H, NCH3 & NCH2), 2.93 (s, 7H, NCH2), 1.22 (s, 3H, CH3), 1.16 (s, 3H, CH3). 13C NMR (126 MHz, DMSO-d6) δ 173.5, 172.8, 169.3, 159.1, 158.8, 153.0, 141.6, 140.9, 136.9, 131.0, 127.0, 125.1, 121.6, 120.5, 119.2, 108.5, 107.0, 106.8, 54.9, 53.4, 52.9, 51.9, 51.2, 49.7, 29.9, 29.4, 28.8, 26.4, 25.8, 19.9, 19.8. HRMS (ESI, M+) calc. for C34H43O11N7: 726.3093. Found: 726.3095.
Compound 7: The triacid 6 (1.0 eq, 0.20 g, 0.28 mmol) and Gd(OTf)3 (1.0 eq, 0.17 g, 0.28 mmol) in 10 mL of methanol was stirred for 24 hours at 60°C. The solvent was evaporated. Methanol was added to the residue and then the solvent was evaporated (twice). The crude product was purified by column chromatography on silica gel with dichloromethane/methanol/water (1/4/1) as eluent to give 0.19 grams (79% yield) of red solid. ESI+ MS (50% MeOH and 0.1% Formic acid): m/z 880 (M, 82%), 882 (M+2H, 100%), 883 (M+3H, 78%). HRMS (ESI, M+) calc. for C34H40O11N7Gd: 878.7229. Found: 878.7091 with expected isotope distribution pattern for Gd.
Longitudinal (r1) relaxivity
A series of aqueous solutions (0.4 mL each) were prepared in which the concentrations of compound 7 range from 0 to 0.68 mM, respectively. The solutions were stored in the dark. T1 relaxation time of these solutions was measured before and after irradiation with visible light for 10 min. The longitudinal (r1) relaxivity was determined as the slope of the line for plots of 1/T1, against increasing gadolinium concentration with a correlation coefficient greater than 0.95. The same method was used for measuring the effect of NADH on the r1 relaxivity of compound 7. An aqueous NADH solution was added to the Gd solutions (NADH:Gd 1:1) and the mixture was incubated for 10 min at 37°C before the T1 relaxation time measurement. The statistical significance of the decrease in r1 relaxivity values was ascertained with a two-tailed paired t-test.
Magnetic Resonance Imaging
The effect of NADH or light on the magnetic resonance properties of compound 7 was evaluated by NMR relaxivity. The solutions of compound 7 were prepared as described before. Images were taken before and after aqueous solutions of compound 7 were irradiated with visible light for 10 min or mixed with NADH for 30 min. T1 was measured using a sequence of spin echo images with independently varying recovery times (10 data points, TR, 400–5000 ms). Imaging parameters for T1-weighted (T1W) images were FOV = 4.5 cm, slice thickness 1.2 mm, TR = 1000 ms, TE = 15 ms.
Determination of q
A series of concentrations of dinitrospiropyran-Gd-DO3A and GdCl3 were prepared in nanopure water with 2% 17O enrichment. The molar concentration of Gd varied from 0 to 40mM. The samples were analyzed at 298 ± 0.3 K in 5mm sample tubes without frequency locking of the spectrometer. Gadolinium-induced 17O ppm shifts were recorded versus the 17O shift of water with a precision of 0.03 ppm. These ppm shifts were plotted versus concentration to obtain the slope, and the hydration number was calculated according to literature methods.29
Supplementary Material
Acknowledgments
The authors wish to acknowledge the National Institute of Health (RO3 EY13941-01) and NMR award from the NMR Facility of the University of California, Davis for support of this work.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Stryer L. Biochemistry. W. H. Freeman and Company; New York: 1995. [Google Scholar]
- 2.Klaidman LK, Leung AC, Adams JD. Anal Biochem. 1995;228:312. doi: 10.1006/abio.1995.1356. [DOI] [PubMed] [Google Scholar]
- 3.Vishwasrao HD, Heikal AA, Kasischke KA, Webb WW. J Biol Chem. 2005;280:25119. doi: 10.1074/jbc.M502475200. [DOI] [PubMed] [Google Scholar]
- 4.Sud D, Zhong W, Beer DG, Mycek MA. Opt Express. 2006;14:4412. doi: 10.1364/oe.14.004412. [DOI] [PubMed] [Google Scholar]
- 5.Mottin S, Laporte P, Cespuglio R. J Neurochem. 2003;84:633. doi: 10.1046/j.1471-4159.2003.01508.x. [DOI] [PubMed] [Google Scholar]
- 6.Patterson GH, Knobel SM, Arkhammar P, Thastrup O, Piston DW. Proc Natl Acad Sci USA. 2000;97:5203. doi: 10.1073/pnas.090098797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Westbrook C, Roth CK, Talbot J. MRI in Practice. 3. Blackwell Publishing Inc: Malden, MA; 2005. [Google Scholar]
- 8.Moats RA, Fraser SE, Meade TJ. Angew Chem Int Ed. 1997:726. [Google Scholar]
- 9.Louie AY, Hüber M, Ahrens ET, Rothbächer U, Moats RA, Jacobs R, Fraser SE, Meade TJ. Nat Biotechnol. 2000;18:321. doi: 10.1038/73780. [DOI] [PubMed] [Google Scholar]
- 10.Blankenberg FG. J Cell Biochem. 2003;90:443. doi: 10.1002/jcb.10635. [DOI] [PubMed] [Google Scholar]
- 11.Aime S, Cabella C, Colombatto S, Crich SG, Gianolio E, Maggioni F. J Magn Reson Imag. 2002;16:394. doi: 10.1002/jmri.10180. [DOI] [PubMed] [Google Scholar]
- 12.Raghunand N, Jagadish B, Trouard TP, Galons JP, Gillies RJ, Mash EA. Magn Reson Med. 2006;55:1272. doi: 10.1002/mrm.20904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu JY, Cohen LB. Fast Multisite Optical Measurement of Membrane Potential: Fluorescent and Luminescent Probes for Biological Activity. Academic Press; San Diego, CA: 1993. pp. 389–403. [Google Scholar]
- 14.Preigh MJ, Stauffer MT, Lin FT, Weber SG. J Chem Soc Faraday Trans. 1996;92:3991. [Google Scholar]
- 15.Zhi JF, Baba R, Hashimoto K, Fujishima A. J Photochem Photobio. 1995;92:91. [Google Scholar]
- 16.Berkovic G, Krongauz V, Weiss V. Chem Rev. 2000;100:1741. doi: 10.1021/cr9800715. [DOI] [PubMed] [Google Scholar]
- 17.Tu CQ, Louie AY. Chem Commun. 2007:1331. doi: 10.1039/b616991k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Taylor LD, Davis RB. J Org Chem. 1963:1713. [Google Scholar]
- 19.Roxburgh CJ, Sammes PG. Dyes and Pigments. 1995;28:317. [Google Scholar]
- 20.Dadabhoy A, Faulkner S, Sammes PG. J Chem Soc, Perkin Trans. 2;2002:348. [Google Scholar]
- 21.Tyer NW, Becker RS., Jr J Am Chem Soc. 1970;92:5605. [Google Scholar]
- 22.Ahmed SA, Tanaka M, Ando H, Tawa K, Kimura K. Tetrahedron. 2004;60:6029. [Google Scholar]
- 23.Raymo FM, Giordani S. J Am Chem Soc. 2001;123:4651. doi: 10.1021/ja005699n. [DOI] [PubMed] [Google Scholar]
- 24.Zhu MQ, Zhu LY, Han JJ, Wu WW, Hurstand JK, Li ADQ. J Am Chem Soc. 2006;128:4303. doi: 10.1021/ja0567642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shukla RB, Kumar K, Weber R, Zhang X, Tweedle M. Acta Radiologica. 1997;38:121. [PubMed] [Google Scholar]
- 26.Aime S, Botta M, Panero M, Grandi M, Uggeri F. Magn Reson Chem. 1991;29:923. [Google Scholar]
- 27.Belenky P, Bogan KL, Brenner C. Trends Biochem Sci. 2007;32:12. doi: 10.1016/j.tibs.2006.11.006. [DOI] [PubMed] [Google Scholar]
- 28.Pollak N, Dölle C, Ziegler M. Biochem J. 2007;402:205. doi: 10.1042/BJ20061638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Alpoim MC, Urbano AM, Geraldes CFGC, Peters JA. J Chem Soc, Dalton Trans. 1992:463. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.