

Abstract
Intramembrane proteases hydrolyze peptide bonds within cell membranes. Recent crystal structures revealed that rhomboid intramembrane proteases contain a hydrated active site that opens to the outside of the cell, but is protected laterally from membrane lipids by protein segments. Using E. coli rhomboid (GlpG) structures as a guide, we previously took a mutational approach to identify the GlpG gating mechanism that allows substrates to enter the active site laterally from the membrane. Mutations that weaken contacts keeping the gate closed increase enzyme activity, and implicate transmembrane segment 5 as the substrate gate. Since these analyses were performed in vitro with pure proteins in detergent micelles, we have now examined GlpG in its natural environment, within the membrane of live E coli cells. In striking congruity with in vitro analysis, gate-opening mutants in transmembrane segment 5 display up to a 10-fold increase in protease activity in living cells. Conversely, mutations in other parts of the protease, including the membrane-inserted L1 loop previously thought to be the gate, decrease enzyme activity. These observations provide evidence for the existence of both closed and open forms of GlpG in cells, and show that inter-conversion between them via substrate gating is rate-limiting physiologically.
Keywords: presenilin, regulated intramembrane proteolysis, site-2 protease, substrate gating
INTRODUCTION
Cleavage of proteins within their membrane-spanning segments has emerged as the regulation point of many cellular processes (Brown et al., 2000; Urban, 2006; Wolfe and Kopan, 2004). While the spectrum of biological roles that intramembrane proteolysis regulates have become increasingly clear, elucidating the underlying biochemical mechanisms that govern this improbable hydrolytic reaction have seen limited progress until recently.
Three mechanistic classes of enzyme are currently known to catalyze intramembrane proteolysis. Site-2 protease was first to be discovered, and is the founding member of a family of metalloproteases conserved from bacteria to humans (Akiyama et al., 2004; Rawson et al., 1997). Signal peptide peptidase and gamma-secretase are aspartyl proteases (Fluhrer and Haass, 2007; Weihofen et al., 2002; Wolfe et al., 1999), while rhomboid enzymes are a family of serine proteases (Urban et al., 2001; Urban et al., 2002). Progress over the past few years have culminated in defined, pure enzyme reconstitution systems with which to study the detailed mechanism of all of three superfamilies of intramembrane proteases (Akiyama et al., 2004; Lemberg et al., 2005; Narayanan et al., 2007; Urban and Wolfe, 2005). These studies showed that intramembrane proteases function as single subunit enzymes, without the need for protein partners, and activity can be reconstituted from bacterially expressed and purified components. The notable exception to this rule is gamma-secretase, which functions as a complex comprised of at least four subunits (Edbauer et al., 2003; Kimberly et al., 2003; Sato et al., 2007).
Armed with this advance, crystallization was initiated using the pure and active bacterial preparations. The rhomboid enzyme GlpG from Escherichia coli and its homolog from Haemophilus influenzae were the first intramembrane proteases whose high-resolution structures were solved (Ben-Shem et al., 2007; Lemieux et al., 2007; Wang et al., 2006; Wu et al., 2006). Remarkably, the resulting structures were revealed in four different space groups and in a variety of detergents, but were highly similar to each other along the length of the enzyme core. This suggested a degree of confidence with respect to the general architecture of the enzyme structure as it exists in its native membrane environment. However, the structures revealed that the active site is contained within a central hydrophilic cavity that opens to the outside, but delimited from membrane lipids by six transmembrane segments and the membrane-submerged L1 loop. The challenge thus moved from structure to function; this architecture immediately posed the question of how gating by rhomboid proteases occurs to facilitate substrate entry into the internal active site.
The first clues about substrate gating came from the crystal structures themselves. The external loop L1 was unexpectedly found to be membrane-submerged, and in this conformation it plugged a gap between transmembrane segments 1 and 3 that could otherwise accommodate a polypeptide chain. This observation led to the hypothesis that displacement of the L1 loop is the gating mechanism (Fig. 1A) (Wang et al., 2006). Conversely, comparison of rhomboid in a second crystal form revealed that the L1 loop remains remarkably similar, but transmembrane helix 5 (and the overlying L5 loop) on the opposite face of the enzyme tilts outwards at its top half by about 35 degrees (Fig. 1B) (Wu et al., 2006). This movement could accommodate insertion of a substrate transmembrane region to allow cleavage within its transmembrane segment. Such tilting by transmembrane helix 5 also displaces loop 5, termed the Cap, which is continuous with helix 5 and shields the active site from above (Wang and Ha, 2007). However, structural inferences face possible artefactual causes because of contacts imposed by crystal packing, as noted previously (Ha, 2007; Wu et al., 2006).
Figure 1. Structure and gating models of the GlpG rhomboid protease from Escherichia coli.
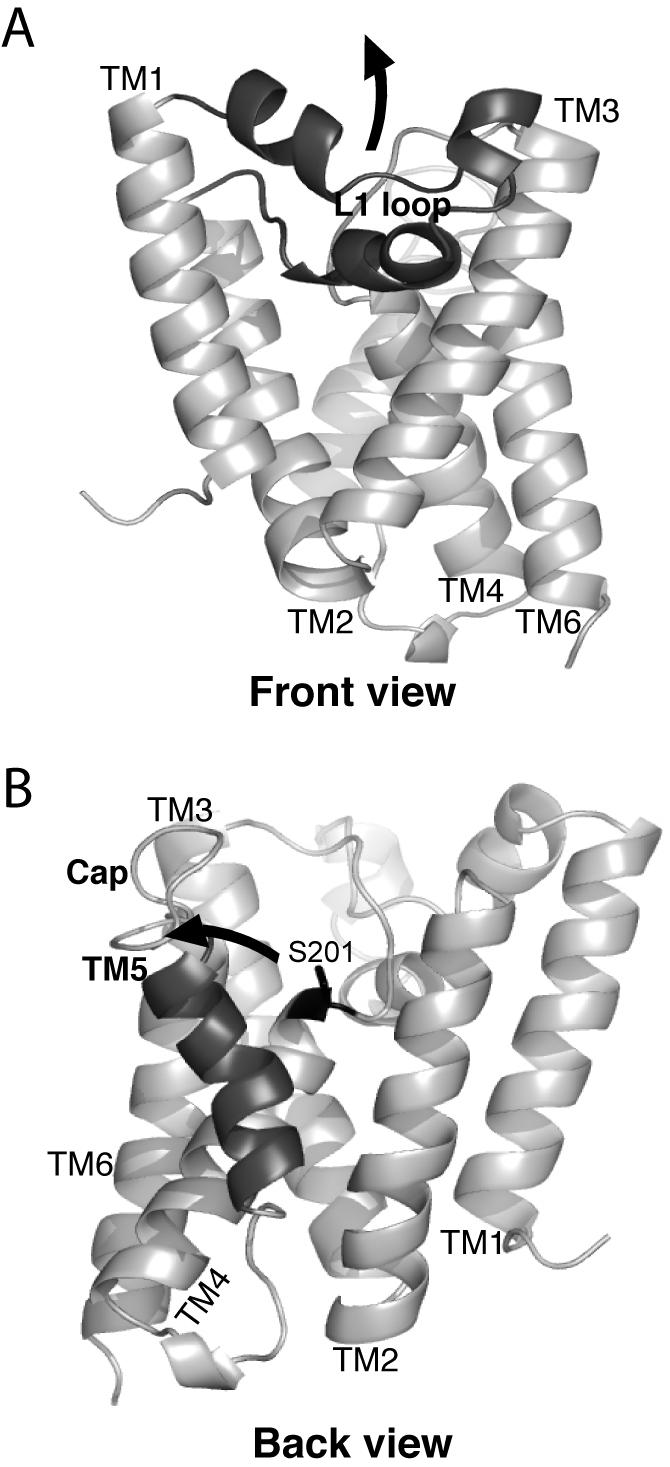
A. The ‘front view’ of the core domain of GlpG is shown laterally from the plane of the membrane, with the extracellular side up and the cytosolic side down. The membrane-submerged L1 loop is highlighted in black, and the hypothetical conformation change to allow substrate access between transmembrane helices 1 and 3 is depicted by an upward arrow. B. The ‘back view’ of the GlpG protease is shown laterally as above, with the transmembrane helix 5 highlighted in black. The open conformation is shown (coordinates from PDB N2RF), with an arrow depicting the proposed transmembrane segment 5 tilting and Cap movement that gates substrate access to the active site serine (in black ball-and-stick).
Ultimately these different models could be resolved with a structure of an enzyme-substrate complex, but given the difficulty in achieving this complex, other approaches are useful for addressing this problem. Additionally, functional analyses are necessary to delineate the sequence of events in substrate access and subsequent catalysis. We therefore pursued a structure-function analysis of rhomboid to begin addressing these gaols, by generating and analyzing defined mutants in the protease. Our rationale was that gate opening would be a rate-limiting event, and thus mutating some of the many contacts that keep the gate closed might help it to open and thus result in mutant enzymes with enhanced proteolytic activity (Baker et al., 2007). Isolation of such activating mutants would reveal which region of the protease is responsible for gating. Using an extensive mutagenesis strategy with over 40 rhomboid mutants and a defined substrate that is cleaved at intramembrane sites, we discovered that four different mutations in transmembrane helix 5 increase substrate cleavage by up to 10-fold in vitro, while 18 different mutations in the L1 loop all decreased activity (Baker et al., 2007). These in vitro perturbations strongly suggested that substrate gating is accomplished by movement of transmembrane segment 5 and the overlying Cap, and that this conformation change is a major rate-limiting step in intramembrane proteolysis.
One limitation of these studies was the fact that rhomboid enzymes were analyzed in an artificial environment, as pure proteins in detergent micelles in vitro. This made it difficult to understand whether the effects of the mutations, although clear in terms of their implications on the biophysical mechanism, were directly relevant to the function of these enzymes in their natural environment, within the membranes of living cells. We sought to address this directly by examining the proteolytic activity of GlpG mutants on a Drosophila Spitz-based substrate that is cleaved by GlpG when co-expressed and targeted to the inner membrane of living E coli cells (Urban and Freeman, 2003; Urban and Wolfe, 2005). The resulting experiments confirm previous conclusions based on in vitro work, and further indicate that gating is rate-limiting during intramembrane proteolysis in cells.
RESULTS
Analysis of GlpG Activity In vivo
In order to analyze GlpG protease activity in E. coli cells, we targeted a Drosophila Spitz substrate to the inner membrane of E. coli (Urban and Wolfe, 2005) because the natural substrate for GlpG has not yet been identified (Clemmer et al., 2006; Maegawa et al., 2005). This construct contained the PelB leader peptide to facilitate signal-dependent insertion into the membrane, followed by GFP, Spitz lacking its N terminal EGF domain (because disulfide bridges are often not faithfully reproduced in bacteria), and a single Flag tag for detection (Urban and Wolfe, 2005). GlpG and the GFP-Spitz-Flag substrate were co-expressed from two different low-copy number plasmids, which were maintained by double antibiotic selection. Cultures were grown to early-log phase, induced to co-express GlpG and the Spitz substrate, and cells were harvested and lysed in denaturing Laemmli buffer hourly from 0 to 6 hours post-induction. The serine-to-alanine GlpG mutant S201A served as a negative control for assessing substrate processing. Co-expression of wildtype GlpG, but not the S201A GlpG mutant, resulted in GFP-Spitz-Flag substrate cleavage in a time-dependent manner, as assessed by anti-Flag western analysis of bacterial cells (Fig. 2A). This observation confirmed that this system was applicable for monitoring GlpG protease activity specifically in vivo in E. coli cells.
Figure 2. Transmembrane 5 segment mutations enhance GlpG protease activity in E. coli cells.
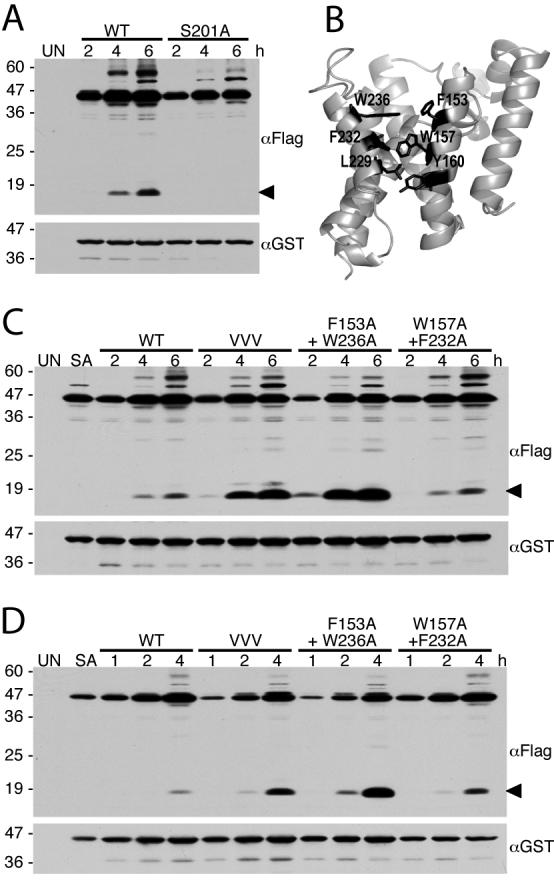
A. Western analysis of Spitz substrate processing (anti-Flag) and GlpG protease expression (anti-GST) in E. coli cells following 2, 4 and 6 hours post-induction. Uninduced E. coli cells (UN) were used as a negative control. The black arrow on the right denotes the cleavage product. Higher molecular weight substrate bands likely represent aggregates. Protein mass standards (in kDa) are depicted to the left of the images. B. Back view of GlpG (open conformation, PDB N2RF) with transmembrane residues on helix 2 and 5 highlighted in black. C and D. Western analysis of cultures expressing wildtype or GlpG helix 5 mutant enzymes with the Spitz substrate revealed a dramatic increase of protease activity of the mutant GlpG enzymes. VVV is the triple transmembrane helix 5 mutant L229V+F232V+W236V. Note that wildtype and mutant-expressing cultures were grown and analyzed in parallel, with data in C and D being from two independent experiments (samples in D were analyzed at shorter times post-induction). UN and SA denote uninduced wildtype GlpG and S201A GlpG mutant cultures, respectively, that served as general negative controls.
Transmembrane Segment 5 Mutants Enhance Proteolytic Activity In vivo
In order to identify the region of rhomboid proteases that functions as a substrate gate, we previously mutated interactions in different parts of GlpG and analyzed the effect on intramembrane proteolytic activity (Baker et al., 2007). Transmembrane segment 5 interacts with its neighbouring transmembrane segment 2 via a series of large hydrophobic residue sidechains that interdigitate in the closed form of the enzyme (Fig. 2B). Mutation of the helical face of transmembrane segment 5 that is involved in this interaction to valine increased activity 4-fold in vitro, while mutating pairs of residues that interact across helices 2 and 5 resulted in a 10-fold increase in activity for the upper F153A+W236A pair, and 7-fold for the middle W157A+F232A pair (Baker et al., 2007). However, whether similar activating effects would occur in the membranes of living cells remained unclear.
We thus examined the activity of these activating mutants in vivo relative to the wildtype enzyme, and discovered that the triple valine helix 5 mutant indeed displayed increased activity, while the F153A+W236A double mutant dramatically increased cleavage of the Spitz substrate in E. coli cells (Fig. 2C). This mutant had the most profound increase in activity in vivo that was pronounced even under shorter induction times (Fig. 2D). We also quantified the relative increase caused by these mutants by quantitative western analysis using infrared fluorescence (Fig. 3). The triple valine mutant increased activity ∼3.3-fold, while the F153A+W236A mutant increased substrate cleavage by ∼11-fold in cells. The W157A+F232A double mutant had a weaker and more variable effect, producing up to a ∼2.5-fold increase in enzyme activity in vivo, whereas it displays a ∼7-fold increase in activity in vitro. Collectively, mutations within transmembrane segment 5 have a dramatic activating effect on the protease activity of GlpG in vivo in living E. coli cells, similar to their effect in vitro in the reconstitution assay (Baker et al., 2007).
Figure 3. Quantification of transmembrane segment 5 mutant GlpG activity.
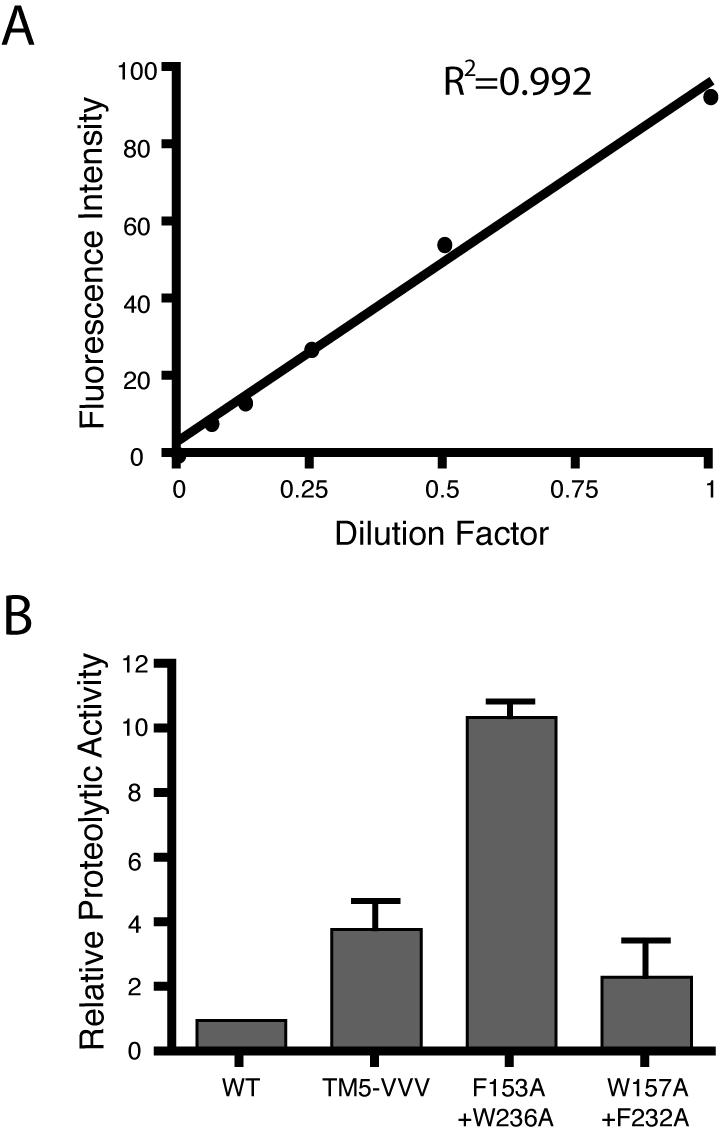
A. Cleaved Spitz was quantified using infrared fluorescence western analysis. Standard curve of 2-fold dilutions of the F153A+W236A signal from 1 (undiluted) to a 1/16 dilution revealed linearity in detection with an R2 value of 0.992. Note that this range represented the highest signal detected in the samples (undiluted) to 16-fold below the signal, which was below the lowest analyzed sample signal. B. The relative activity of L229V+F232V+W236V (VVV), F153A+W236A, and W157A+F232A were quantified relative to wildtype activity, which was arbitrarily set to 1. The error bars show standard deviation.
Substrate-Gating GlpG Mutants also Display Increased Activity in Mammalian Cells
In addition to analysis in E. coli cells, rhomboid activity can also be monitored in transfected mammalian cells. Although this is not the native environment for GlpG, the activity of many rhomboid proteases across evolution have been studied in this heterologous assay. In order to examine the effect of substrate gating mutants in a second biological context, we examined GlpG activity against Spitz in transfected COS cells. We previously found that wildtype GlpG displays very weak activity in this assay (Urban et al., 2002); a cleaved Spitz band can barely be detected in transfected cells early in the transfection cycle, and unlike for other rhomboid enzymes, no cleaved Spitz could be detected as a secreted product in the cell culture media.
We co-transfected hemagglutinin-tagged wildtype, L229V+F232V+W236V, F153A+W236A, and W157A+F232A mutant GlpG enzymes in parallel with Drosophila rhomboid-1 and monitored Spitz cleavage in COS cells. Wildtype GlpG was expressed at a very low level compared to Drosophila rhomboid-1, and Spitz cleavage could barely be detected in cell lysates with wildtype GlpG, and failed to be detected as a secreted product in cell culture media (Fig. 4). However, all three different substrate-gating GlpG mutants displayed increased activity in transfected COS cells, resulting in much higher levels of Spitz cleavage in cell lysates relative to wildtype GlpG, and even detection of cleaved Spitz as a secreted product in media. As in E. coli and in vitro, the F153A+W236A mutant displayed the greatest increase in activity. These experiments further confirm that substrate-gating mutants of GlpG display dramatically increased activity in an independent cellular assay, and in a different (eukaryotic) membrane environment.
Figure 4. Substrate-gating mutants of GlpG display increased activity in mammalian cells.
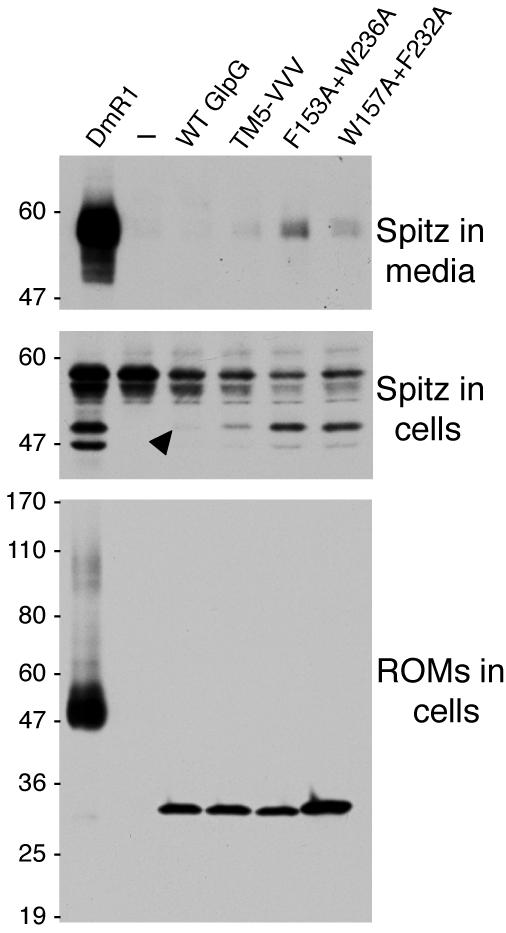
Western analysis of GlpG expression and activity against Spitz in transfected COS cells. The top panel shows accumulation of cleaved GFP-Spitz as a secreted product in cell culture media, as detected by anti-GFP. Note that while wildtype GlpG did not produce a cleaved GFP-Spitz product above background, the cleaved product can be detected in the media fraction with all three GlpG mutants. The positive control, Drosophila rhomboid-1 (DmR1) displayed strong activity against Spitz. The middle panel represents GFP-Spitz in transfected cells, with the upper band being uncleaved GFP-Spitz, and the lower band being cleaved GFP-Spitz prior to being secreted. The arrow indicates the barely detectable GFP-Spitz cleavage band produced by wildtype GlpG. Note that all three GlpG mutants increase the amount of cleaved Spitz in the cell lysates. The lower panel depicts expression levels of each rhomboid, all of which were tagged with a triple hemagglutinin tag and detected by anti-HA western. Note that only 1/4 of the DmR1 cell lysate volume was loaded compared to the GlpG lysates since its expression was already so much greater than that of GlpG proteins. The W157A+F232A GlpG mutant was expressed at a higher level than other GlpG proteins. The location of protein mass standards (in kDa) are depicted on the left.
Mutations within the L1 Loop Impair Proteolytic Activity In vivo
We next examined the effect of mutations within the L1 loop, which had originally been proposed to be the substrate gate (Wang et al., 2006). In order for the L1 loop to function as the substrate gate, it would have to be displaced from its membrane-submerged conformation. To facilitate this displacement, we mutated the hydrophobic underside of the loop that would be expected to contact lipid tails (Fig. 5A) to serine, a non-charged but hydrophilic sidechain. The resulting mutants Y138S, F139S, and L143S all decreased GlpG activity in vivo in E. coli cells relative to the wildtype enzyme (Fig. 5B). Introducing charged residues like Y138D, which should destabilize the membrane-inserted conformation of the L1 loop further, strongly hindered activity, as did combining the serine mutations in double or triple mutants (Fig. 5B).
Figure 5. Mutations within the L1 loop decrease GlpG protease activity in E. coli cells.

A.Lateral view of GlpG, with the membrane-inserted L1 loop highlighted in black. Sidechains of residues Y138, F139, and L143 that would be expected to contact membrane lipid and thus stabilize the membrane inserted conformation of the loop are shown. B. Western analysis of cultures expressing wildtype and L1 loop mutant GlpG enzymes with the Spitz substrate. Anti-flag was used to reveal the Spitz substrate while GlpG protease expression levels were monitored using anti-GST. The black arrow denotes the cleavage product. Note that all L1 loop mutants decrease GlpG protease activity. C. Lateral view of the GlpG structure (PDB N2RF) highlighting the W136 and R137 residue pair (in black) within the L1 loop. D. Western analysis of cultures co-expressing wildtype versus W136A or R137A mutant GlpG with the Spitz substrate. Note that the R137A mutation strongly decreased GlpG protease activity. In all B and D panels, UN and SA denote uninduced wildtype and S201A-expressing cultures, and location of protein mass standards (in kDa) are depicted on the left.
The L1 loop also contains a conserved WR motif that had long been mysterious in its function (Urban et al., 2001). Structural analysis revealed that the R137 lies on the membrane-submerged half of the loop, and its sidechain reaches up to make five hydrogen bonds, thus stabilizing the sideways hairpin-like, membrane-submerged structure of the loop (Fig. 5C) (Wang et al., 2006). Accordingly, mutation of R137 to alanine nearly abolished proteolytic activity in vivo, while W136A only decreased activity ∼2-fold (Fig. 5D). The effect of the W136A mutant was more pronounced in vivo than in vitro, where it has no or little effect (Baker et al., 2007). Collectively, these observations indicate that the membrane-submerged conformation of the L1 loop is important for enzyme stability, and inconsistent with it serving a mobile gating function.
Active Site Mutations Abrogate Proteolytic Activity In vivo
We also used the in vivo assay to examine the effect of mutants in other regions of the enzyme. The inner cavity of rhomboid enzymes is lined with conserved hydrophilic residues (Fig 6A), the function of most of which remain unclear (Baker et al., 2007; Lemberg et al., 2005; Wang et al., 2006). We mutated a number of these residues and examined the effect on proteolytic activity of the E. coli GlpG enzyme in E. coli cells, its natural environment. Mutating G199, which lies just above the active serine, to alanine nearly abolished activity, while mutating tyrosine 205, which stabilizes the active site histidine by base stacking, greatly reduced activity (Fig. 6B). Rhomboid enzymes contain a conserved HxxxxHxxxN motif that spans the end of loop 1 and beginning of transmembrane helix 2. The function of these residues is unknown but may involve stabilizing the oxyanion reaction intermediate. We found that mutating any of these residues individually to alanine greatly reduced GlpG activity in E. coli cells (Fig. 6C). These effects are similar to those observed in vitro (Baker et al., 2007), as well as when these conserved residues were mutated in Drosophila rhomboid-1 (Urban et al., 2001), the prototype rhomboid enzyme, and assayed in transfected mammalian cells. While these observations reinforce the functional importance of these residues for proteolytic activity, they do not provide further clues regarding their detailed roles.
Figure 6. Mutation of non-catalytic residues lining the GlpG active site hinder protease activity.
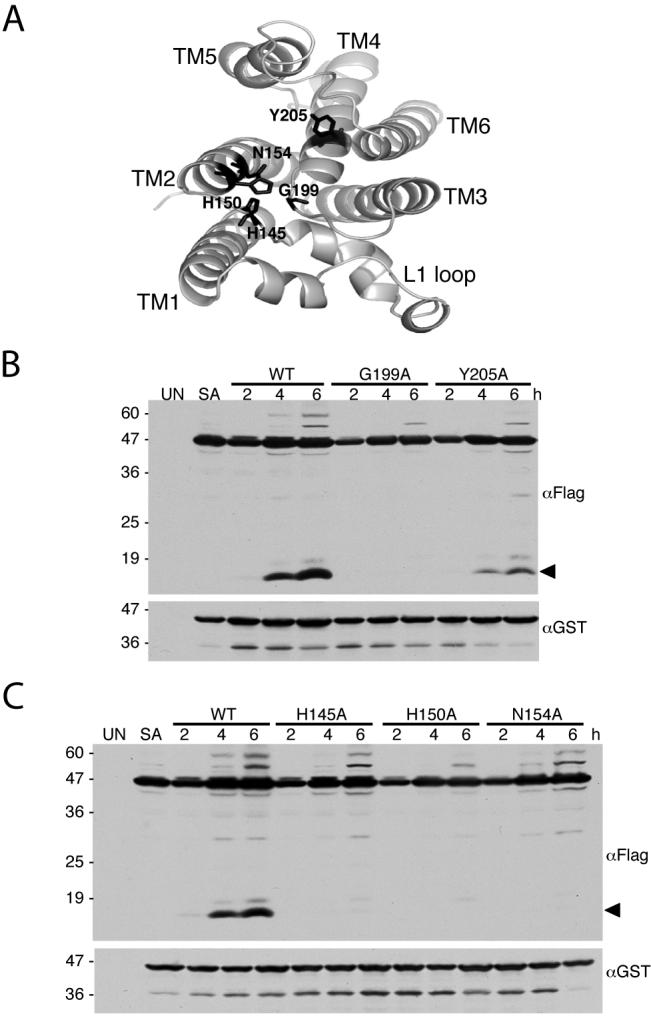
A. Top view of GlpG (PDB N2RF), with the L1 loop down. Side-chains of conserved non-catalytic active sites residues under investigation are highlighted in black. B and C. Western analysis of cultures expressing either wildtype of mutant GlpG proteases with the Spitz substrate. All mutant enzymes show a decrease in substrate processing (cleavage product denoted by black arrow). UN and SA denote uninduced wildtype and S201A-expressing cultures, respectively. Location of protein mass standards (in kDa) are depicted on the left.
Analysis of Single and Combined Activating Mutants In vivo
Transmembrane helix 5 is connected to loop 5 (termed the Cap), which covers the active site cavity from the top in the closed conformation of the enzyme, with two methionines inserting directly near the active serine and phenylalanine 245 just above helix 5 bending back to contribute to closing further the region above helices 5 and 2 (Fig. 7A) (Wang and Ha, 2007). The Cap is moved by tilting away of transmembrane helix 5 to allow substrate access. But it is not clear how much contribution each residue of this region makes to opening the gate. We addressed this issue by mutating F245 of the Cap, F153 of helix 2, and W236 of helix 5 singly and assessing the effect on proteolytic activity of the mutant enzymes in cells using quantitative infrared fluorescence western analysis.
Figure 7. Single and combined Helix 2, Helix 5 and Cap mutations enhance GlpG protease activity in E. coli cells.

A. Lateral back view of the closed conformation of GlpG (PDB 2IC8) showing the sidechain (in black) interactions of residues W236 on helix 5, F153 on helix 2, and F245 on the L5 Cap. B. Western analysis of bacteria expressing wildtype, single and double mutants of GlpG and the Spitz substrate. Note that, of the single mutants, the most profound increase in protease activity was observed with the transmembrane helix 5 mutant W236A. C. Combining the F153A mutant with the W236A mutant increases GlpG protease activity relative to the single W236A mutant in E. coli cells. D. Combining the Cap mutant F245A with the double transmembrane helix mutant F153A+W236A did not increase the GlpG protease activity of the triple mutant relative to that of the F153A+W236A double mutant in bacterial cells. E. Removing the sidechains of Cap residues 243-250 by mutating all to glycine (L5-polyG) abolished GlpG protease activity in E. coli cells. In all panels, the cleavage product is denoted by a black arrow, UN and SA are uninduced wildtype and S201A-expressing cultures, respectively, and the location of protein mass standards (in kDa) are depicted on the left.
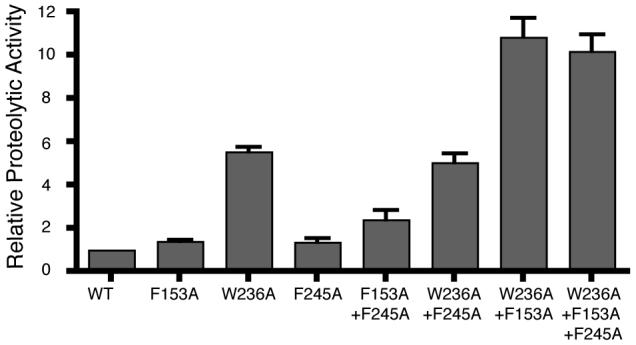
Mutating F245 to alanine resulted in a small increase in proteolytic activity that varied between no stimulation to less than a 2-fold stimulation (Fig. 7B and 8). Mutating F153 on helix 2 alone to alanine also had a mild stimulatory effect of up to 2-fold. In contrast, mutating W236 on helix 5 alone to alanine dramatically increased GlpG activity by ∼6-fold. This is the strongest stimulation observed with any single residue mutation. (Fig. 7B and 8).
Figure 8. Quantification of single and combined Helix 2, Helix 5 and Cap mutations on GlpG activity.
Levels of cleaved Spitz generated by wildtype and mutant GlpG proteases at both 4 and 6 hour timepoints (see Fig. 7) was quantified using infrared fluorescence western analysis. Levels are relative to wildtype GlpG, which was arbitrarily set to 1. A standard curve revealed linearity in the analysis range with an R2 value of 0.992. Error bars show standard deviation.
To address this issue further, we examined the effect of pairwise combinations of W236A, F245A and F153A on proteolytic activity in vivo. Combining W236A and F153A resulted in an enzyme with ∼ 11-fold higher activity than wildtype, and ∼2-fold higher activity than W236A alone (Fig. 7C and 8). Conversely, combining F245A with W236A actually decreased activity slightly compared to W236A alone (Fig 7B and 8), while combining the F245A mutation with F153A resulted in an additive increase of enzyme activity up to ∼2.4-fold that of wildtype GlpG (Fig. 7B and 8). Finally, combining F245A with the double F153A+W236A mutant in a triple mutant resulted in no further stimulation of activity in cells compared to the F153A+W236A double mutant (Fig. 7D and 8). These single and combined mutant analyses suggest that residue W236 on transmembrane helix 5 has the strongest effect on increasing proteolytic activity in vivo. Moreover, only the W236A and F153A mutations had a synergistic effect on activity.
One intriguing observation is the apparent importance of the loop 5 Cap. The Cap covers the hydrophilic active site cavity from above in the closed conformation of the enzyme. Although the importance of the open, Cap-displaced conformation is obvious, the closed conformation may also be important, since mutating the Cap residues to polyglycine (thus maintaining loop length but removing sidechains) abolished any detectable GlpG protease activity in cells (Fig. 7E). It should be noted that we cannot exclude the possibility that this mutant affects the folding of the enzyme rather than its function specifically, although the mutant protein was expressed well in cells (Fig. 7E). This is also consistent with no detectable activity of this mutant in vitro (Baker et al., 2007), and now implies that the closed form of the enzyme is also important for function in cells.
DISCUSSION
Determining the effects of defined mutants on enzyme activity in vitro has served as a useful approach towards interpreting the mechanistic function of rhomboid proteases in the context of the wealth of recent structural information (Baker et al., 2007; Ben-Shem et al., 2007; Lemberg et al., 2005; Lemieux et al., 2007; Maegawa et al., 2007; Wang et al., 2007; Wang et al., 2006; Wu et al., 2006). These analyses have highlighted both residues and interactions that are required for catalysis, as well as a new class of mutants that dramatically increase enzymatic activity (Baker et al., 2007). These in vitro perturbations aided in identifying the gating mechanism of rhomboid enzymes, but the possible effects of the activating mutants in vivo in their natural environment have not been explored. We examined the enzymatic activity of engineered variants of the GlpG rhomboid protease within the inner membrane of living E. coli cells.
Interestingly, both compromising and activating mutants displayed similar effects in vivo in bacterial membranes as they had in vitro in pure form within detergent micelles (Baker et al., 2007), with two possible exceptions. First, transmembrane helix 5 mutants located deeper within the membrane had milder effects in vivo than in vitro. Specifically, while the paired helix 2-helix 5 mutant W157A-F232A resulted in a strong ∼7-fold increase in enzyme activity in vitro (Baker et al., 2007), its stimulation in vivo was only approximately 2-fold. This discrepancy could be due to the lateral pressure applied on proteins by the membrane (van den Brink-van der Laan et al., 2004), which would push helix 5 into its closed conformation. Mutants within the middle portions of helix 5 might be less effective in counteracting this pressure, which is not present in detergent micelles.
Secondly, while mutating the tryptophan of the conserved WR motif in the L1 loop resulted in no decrease of activity in vitro (Baker et al., 2007), in cells this W136A mutant displayed a decrease in activity. This might again reflect the difference between a membrane and detergent environment: the neighbouring W136 possibly shields the sidechain of R137 from membrane lipid in the lipid-submerged conformation (Wang et al., 2006). As such, its mutation would have more drastic effect in a membrane context where this shielding from lipid might be more important, in contrast to the situation in detergent micelles. Mutating R137 itself to alanine resulted in virtually no enzyme activity both in vitro and in vivo (Baker et al., 2007; Wang et al., 2007). This effect is also consistent with mutations analyzed in Drosophila rhomboid-1 and human RHBDL2 in transfected mammalian cells (Urban et al., 2001), and YqgP in detergent (Lemberg et al., 2005), where the arginine was shown to have a critical role in catalysis for all three enzymes.
Importantly, observing activity-enhancing effects of helix 5 mutants in vivo provides the first evidence for the existence of both the closed and open conformations of GlpG in biological membranes of living cells. Moreover, the stimulating nature of helix 5 mutants implies that switching between these two states through gate opening is likely to be rate-limiting during intramembrane proteolysis in cellular membranes. In addition to this insight, being able to examine rhomboid activity in vivo via gating mutants described here should also provide new tools towards further assessing the detailed mechanism of rhomboid proteases in a physiological context.
While it is not clear whether other intramembrane proteases use similar gating mechanisms, the recently solved structure of an Archaeal site-2 protease homolog also revealed two different conformations that could correspond to gate-closed and gate-open forms (Feng et al., 2007). The resulting gating model involves more extensive conformation changes of transmembrane helices 1 and 6 to produce an open crevice that could allow substrate to pass between them. But the basic principles may actually be similar (Urban and Shi, 2008); gating was also proposed to involve unzippering of interdigitating hydrophobic residues on helices 1 and 6. Moreover, these regions of the protease also do not contain well-conserved sequence elements among the other site-2 proteases (Feng et al., 2007), which is similar to the lack of sequence conservation in transmembrane helix 5 and Cap of rhomboid proteases (Koonin et al., 2003; Lemberg and Freeman, 2007). Despite these tempting observations, it must be stressed that these possible similarities in site-2 protease gating are speculative, and await further structural and enzymatic analysis. In particular, interrogating site-2 protease gating models using the activity-enhancing mutant strategy developed for rhomboid proteases both in vitro and now in vivo may be a useful approach towards delineating the molecular mechanism of substrate gating in an unrelated intramembrane protease.
MATERIALS AND METHODS
Plasmid constructs
All GlpG rhomboid genes were cloned as full-length open reading frames into the pGEX-6P-1 vector (GE Healthcare, Uppsala, Sweden) to provide an N-terminal GST tag, expression from the ptac promoter, and ampicillin resistance, as described previously (Urban and Wolfe, 2005). The GFP-Spitz substrate gene was cloned into the pET27b(+) vector (Novagen, Madison, USA) to provide an N-terminal PelB leader peptide, expression from the T7 promoter, and kanamycin resistance. The PelB-GFP-Spitz-Flag substrate contained Spitz from residue D119 to its natural end, as described previously (Urban and Wolfe, 2005). Mutations were introduced into GlpG using Quik-Change and Multi-Site Quik-Change Mutagenesis methods (Stratagene, Cedar Creek, USA), and the constructs were verified by sequencing the entire GlpG open reading frame.
Bacterial culture
Expression plasmids were transformed into C43(DE3) competent cells, and liquid cultures were grown shaking at 37 degrees Celsius, 250 rpm under double selection in Lauria Bertani (LB) broth supplemented with 100 μg/ml amplcillin and 50 μg/ml kanamycin. The cultures were grown to an OD600 of approximately 0.7, and co-expression of substrate and GlpG was induced with 250 μM isopropyl-1-thio-β-D-galactopyranoside. Cultures were shifted to 27 degrees Celsius upon induction, and cells were harvested and lysed in denaturing and reducing Laemmli buffer either immediately prior to induction, or 1 hour, 2 hours, 4 hours and 6 hours following induction.
Mammalian cell transfection
Mammalian cells were grown and transfected as described in detail previously (Baker et al., 2006). Briefly, COS cells grown in 6-well plates were transfected using FuGene6 (Roche, Basel, Switzerland) with pcDNA3.1 (Invitrogen, Carlsbad, USA) plasmids driving expression from the pCMV promoter of GFP-Spitz, Star (transport factor for Spitz) and triple HA-tagged rhomboid-1, or triple HA-tagged wildtype or mutant GlpG. 18 hours post-transfection, cells were washed and incubated in serum-free media containing metalloprotease inhibitors (to decrease cellular cleavage of GFP-Spitz). 24 hours later, cell and media samples were harvested in denaturing and reducing Laemmli buffer and subjected to western analysis.
Western Analysis
Cell lysates were loaded on 4-20% polyacrylamide gradient tris-glycine SDS gels (Invitrogen), electro-transferred to nitrocellulose, and probed with anti-GST, anti-Flag, anti-GFP or anti-HA antibodies. Benchmark (Invitrogen) prestained protein markers were used as mass standards. The resulting immunocomplexes were revealed by enhanced chemiluminescence (GE Healthcare) and imaged with film. For quantitative western analysis, membranes were probed with secondary antibodies conjugated to infrared IRDye800, developed with a LiCor Odyssey scanner, and quantified using Odyssey software (LiCor, Lincoln, USA). A standard curve revealed linearity in the analysis range with an R2=0.99.
Structural analysis
All structure images were analyzed and prepared with MacPymol, using published N2RF coordinates for the GlpG open form (Wu et al., 2006), and 2IC8 for the GlpG closed form (Wang et al., 2006).
ACKNOWLEDGEMENTS
We are grateful to Keith Young for assistance with culture growth and lysate preparation, Chelsea Newhouse for administrative assistance, and all members of the Urban lab for valuable discussions and encouragement. Research in the Urban lab is supported by NIH grant R01AI066025 (to SU), a career award from the Burroughs-Wellcome Fund (to SU), an award from the David and Lucile Packard Foundation (to SU), and by Johns Hopkins University School of Medicine.
REFERENCES
- Akiyama Y, Kanehara K, Ito K. RseP (YaeL), an Escherichia coli RIP protease, cleaves transmembrane sequences. Embo J. 2004;23:4434–4442. doi: 10.1038/sj.emboj.7600449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker RP, Wijetilaka R, Urban S. Two Plasmodium Rhomboid Proteases Preferentially Cleave Different Adhesins Implicated in All Invasive Stages of Malaria. PLoS Pathog. 2006;2:922–932. doi: 10.1371/journal.ppat.0020113. e113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker RP, Young K, Feng L, Shi Y, Urban S. Enzymatic analysis of a rhomboid intramembrane protease implicates transmembrane helix 5 as the lateral substrate gate. Proc Natl Acad Sci U S A. 2007;104:8257–8262. doi: 10.1073/pnas.0700814104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Shem A, Fass D, Bibi E. Structural basis for intramembrane proteolysis by rhomboid serine proteases. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:462–466. doi: 10.1073/pnas.0609773104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown MS, Ye J, Rawson RB, Goldstein JL. Regulated intramembrane proteolysis: a control mechanism conserved from bacteria to humans. Cell. 2000;100:391–398. doi: 10.1016/s0092-8674(00)80675-3. [DOI] [PubMed] [Google Scholar]
- Clemmer KM, Sturgill GM, Veenstra A, Rather PN. Functional characterization of Escherichia coli GlpG and additional rhomboid proteins using an aarA mutant of Providencia stuartii. J Bacteriol. 2006;188:3415–3419. doi: 10.1128/JB.188.9.3415-3419.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edbauer D, Winkler E, Regula JT, Pesold B, Steiner H, Haass C. Reconstitution of gamma-secretase activity. Nat Cell Biol. 2003;5:486–488. doi: 10.1038/ncb960. [DOI] [PubMed] [Google Scholar]
- Feng L, Yan H, Wu Z, Yan N, Wang Z, Jeffrey PD, Shi Y. Structure of a site-2 protease family intramembrane metalloprotease. Science. 2007;318:1608–1612. doi: 10.1126/science.1150755. [DOI] [PubMed] [Google Scholar]
- Fluhrer R, Haass C. Signal peptide peptidases and gamma-secretase: cousins of the same protease family? Neurodegener Dis. 2007;4:112–116. doi: 10.1159/000101835. [DOI] [PubMed] [Google Scholar]
- Ha Y. Structural principles of intramembrane proteases. Curr Opin Struct Biol. 2007;17:405–411. doi: 10.1016/j.sbi.2007.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimberly WT, LaVoie MJ, Ostaszewski BL, Ye W, Wolfe MS, Selkoe DJ. Gamma-secretase is a membrane protein complex comprised of presenilin, nicastrin, Aph-1, and Pen-2. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:6382–6387. doi: 10.1073/pnas.1037392100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koonin EV, Makarova KS, Rogozin IB, Davidovic L, Letellier MC, Pellegrini L. The rhomboids: a nearly ubiquitous family of intramembrane serine proteases that probably evolved by multiple ancient horizontal gene transfers. Genome Biol. 2003;4:R19. doi: 10.1186/gb-2003-4-3-r19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemberg MK, Freeman M. Functional and evolutionary implications of enhanced genomic analysis of rhomboid intramembrane proteases. Genome Res. 2007;17:1634–1646. doi: 10.1101/gr.6425307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemberg MK, Menendez J, Misik A, Garcia M, Koth CM, Freeman M. Mechanism of intramembrane proteolysis investigated with purified rhomboid proteases. Embo J. 2005;24:464–472. doi: 10.1038/sj.emboj.7600537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemieux MJ, Fischer SJ, Cherney MM, Bateman KS, James MN. The crystal structure of the rhomboid peptidase from Haemophilus influenzae provides insight into intramembrane proteolysis. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:750–754. doi: 10.1073/pnas.0609981104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maegawa S, Ito K, Akiyama Y. Proteolytic action of GlpG, a rhomboid protease in the Escherichia coli cytoplasmic membrane. Biochemistry. 2005;44:13543–13552. doi: 10.1021/bi051363k. [DOI] [PubMed] [Google Scholar]
- Maegawa S, Koide K, Ito K, Akiyama Y. The intramembrane active site of GlpG, an E. coli rhomboid protease, is accessible to water and hydrolyses an extramembrane peptide bond of substrates. Mol Microbiol. 2007;64:435–447. doi: 10.1111/j.1365-2958.2007.05679.x. [DOI] [PubMed] [Google Scholar]
- Narayanan S, Sato T, Wolfe MS. A C-terminal region of signal peptide peptidase defines a functional domain for intramembrane aspartic protease catalysis. J Biol Chem. 2007;282:20172–20179. doi: 10.1074/jbc.M701536200. [DOI] [PubMed] [Google Scholar]
- Rawson RB, Zelenski NG, Nijhawan D, Ye J, Sakai J, Hasan MT, Chang TY, Brown MS, Goldstein JL. Complementation cloning of S2P, a gene encoding a putative metalloprotease required for intramembrane cleavage of SREBPs. Mol Cell. 1997;1:47–57. doi: 10.1016/s1097-2765(00)80006-4. [DOI] [PubMed] [Google Scholar]
- Sato T, Diehl TS, Narayanan S, Funamoto S, Ihara Y, De Strooper B, Steiner H, Haass C, Wolfe MS. Active gamma-secretase complexes contain only one of each component. J Biol Chem. 2007;282:33985–33993. doi: 10.1074/jbc.M705248200. [DOI] [PubMed] [Google Scholar]
- Urban S. Rhomboid proteins: conserved membrane proteases with divergent biological functions. Genes Dev. 2006;20:3054–3068. doi: 10.1101/gad.1488606. [DOI] [PubMed] [Google Scholar]
- Urban S, Freeman M. Substrate specificity of rhomboid intramembrane proteases is governed by helix-breaking residues in the substrate transmembrane domain. Mol Cell. 2003;11:1425–1434. doi: 10.1016/s1097-2765(03)00181-3. [DOI] [PubMed] [Google Scholar]
- Urban S, Lee JR, Freeman M. Drosophila rhomboid-1 defines a family of putative intramembrane serine proteases. Cell. 2001;107:173–182. doi: 10.1016/s0092-8674(01)00525-6. [DOI] [PubMed] [Google Scholar]
- Urban S, Schlieper D, Freeman M. Conservation of intramembrane proteolytic activity and substrate specificity in eukaryotic and prokaryotic Rhomboids. Current Biology. 2002;12:1507–1512. doi: 10.1016/s0960-9822(02)01092-8. [DOI] [PubMed] [Google Scholar]
- Urban S, Shi Y. Core principles of intramembrane proteolysis: comparison of rhomboid and site-2 family proteases. Current Opinion in Structural Biology. 2008 doi: 10.1016/j.sbi.2008.03.005. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urban S, Wolfe MS. Reconstitution of intramembrane proteolysis in vitro reveals that pure rhomboid is sufficient for catalysis and specificity. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:1883–1888. doi: 10.1073/pnas.0408306102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Brink-van der Laan E, Killian JA, de Kruijff B. Nonbilayer lipids affect peripheral and integral membrane proteins via changes in the lateral pressure profile. Biochimica et biophysica acta. 2004;1666:275–288. doi: 10.1016/j.bbamem.2004.06.010. [DOI] [PubMed] [Google Scholar]
- Wang Y, Ha Y. Open-cap conformation of intramembrane protease GlpG. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:2098–2102. doi: 10.1073/pnas.0611080104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Maegawa S, Akiyama Y, Ha Y. The role of L1 loop in the mechanism of rhomboid intramembrane protease GlpG. J Mol Biol. 2007;374:1104–1113. doi: 10.1016/j.jmb.2007.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Zhang Y, Ha Y. Crystal structure of a rhomboid family intramembrane protease. Nature. 2006;444:179–180. doi: 10.1038/nature05255. [DOI] [PubMed] [Google Scholar]
- Weihofen A, Binns K, Lemberg MK, Ashman K, Martoglio B. Identification of signal peptide paptidase, a presenilin-type aspartic protease. Science. 2002;296:2215–2218. doi: 10.1126/science.1070925. [DOI] [PubMed] [Google Scholar]
- Wolfe MS, Kopan R. Intramembrane proteolysis: theme and variations. Science. 2004;305:1119–1123. doi: 10.1126/science.1096187. [DOI] [PubMed] [Google Scholar]
- Wolfe MS, Xia W, Ostaszewski BL, Diehl TS, Kimberly WT, Selkoe DJ. Two transmembrane aspartates in presenilin-1 required for presenilin endoproteolysis and gamma-secretase activity. Nature. 1999;398:513–517. doi: 10.1038/19077. [DOI] [PubMed] [Google Scholar]
- Wu Z, Yan N, Feng L, Oberstein A, Yan H, Baker RP, Gu L, Jeffrey PD, Urban S, Shi Y. Structural analysis of a rhomboid family intramembrane protease reveals a gating mechanism for substrate entry. Nat Struct Mol Biol. 2006;13:1084–1091. doi: 10.1038/nsmb1179. [DOI] [PubMed] [Google Scholar]