

Abstract
Histone acetylation and nucleosome remodeling regulate DNA damage repair, replication and transcription. Rtt109, a recently discovered histone acetyltransferase (HAT) from Saccharomyces cerevisiae, functions with the histone chaperone Asf1 to acetylate lysine K56 on histone H3 (H3K56), a modification associated with newly synthesized histones. In vitro analysis of Rtt109 revealed that Vps75, a Nap1 family histone chaperone, could also stimulate Rtt109-dependent acetylation of H3K56. However, the molecular function of the Rtt109-Vps75 complex remains elusive. Here we have probed the molecular functions of Vps75 and the Rtt109-Vps75 complex through biochemical, structural and genetic means. We find that Vps75 stimulates the kcat of histone acetylation by ∼100-fold relative to Rtt109 alone and enhances acetylation of K9 in the H3 histone tail. Consistent with the In vitro evidence, cells lacking Vps75 showed a substantial reduction (60%) in H3K9 acetylation during S phase. X-ray structural, biochemical and genetic analyses of Vps75 indicate a unique, structurally dynamic Nap1-like fold that suggests a potential mechanism of Vps75-dependent activation of Rtt109. Together, these data provide evidence for a multifunctional HAT-chaperone complex that acetylates histone H3 and deposits H3-H4 onto DNA, linking histone modification and nucleosome assembly.
Acetylation of chromatin is crucial for regulating replication, transcription and DNA repair in all eukaryotic organisms (reviewed in refs. 1-4). Typically, this post-translational modification occurs on the ε-amino group of lysines within N-terminal ‘tails’ of histones. However, recent work has shown that modifications to the histone-fold domain are prevalent and function to regulate genomic transactions5-7. One modification to the histone-fold domain, acetylation of lysine 56 of histone H3 (H3K56ac), is a global modification of nascent histone H3 during replication, and a targeted modification at gene promoters6,8,9. H3K56 contacts the DNA backbone (via a water-mediated interaction), and neutralization of this charge by acetylation alters the wrapping of DNA around the histone octamer5. H3K56ac is abundant in budding and fission yeasts and in Tetrahymena thermophilus5,10,11. Although minor amounts have been detected in Drosophila melanogaster and humans, the proteins responsible for the regulation of this modification have been identified only in fungi6,10. H3K56ac occurs on newly synthesized molecules before deposition and appears on chromatin during histone-exchange events, both during and outside S phase5,8,9.
In S. cerevisiae and Schizosaccharomyces pombe, Rtt109 (also known as KAT11) was identified as the HAT responsible for H3K56 acetylation11-16. Interestingly, either of two histone chaperones, Asf1 or the Nap1 family protein Vps75, are necessary for high catalytic activity of Rtt109-dependent H3K56 acetylation In vitro13,17. Because many histone chaperones both transport histones to the nucleus and function in the assembly and disassembly of nucleosomes, these data suggest that the Rtt109-chaperone complexes may link histone acetylation and nucleosome assembly18-23. The interaction between Rtt109 and Vps75 seems to be a high-affinity one, as the proteins can be copurified from yeast and bacterial cell extracts; in contrast, Rtt109 and Asf1 do not copurify efficiently13,24-26. These data suggest Rtt109-Vps75 function as a complex in the cell.
Functional interactions between HATs and Nap1 family histone chaperones have been documented27,28. In humans, NAP1 and a complex termed inhibitor of histone acetyltransferases (INHAT) seem to function with the HATs p300 (also known as KAT3B) and CREB binding protein (CBP, also known as KAT3A) in transcriptional activation; however, the mechanism underlying the coordinated function of HATs and histone chaperones remains unresolved16,28-30. One proposal suggested that NAP1 removes H2A-H2B dimers from chromatin, allowing access by the transcriptional machinery31,32. Notably, In vitro both NAP1 and INHAT seem to inhibit the acetylation of histones by p300, CBP and p/CAF (also known as KAT2B), making the interplay between these HATS and NAP1 or INHAT unclear16,28,29. In contrast to NAP1 and INHAT, Vps75 stimulates acetylation of H3K56 by Rtt109, suggesting that Vps75 is a functionally distinct member of the Nap1 family of histone chaperones13,17.
In S. cerevisiae, deletion of VPS75 results in modest effects on H3K56ac levels during S phase and does not alter sensitivity to genotoxic agents13,17,24. However, deletion of ASF1 abolishes detectable H3K56ac, suggesting that the Rtt109-Vps75 complex functions separately from Asf1-dependent H3K56ac13,14. Therefore, the precise cellular function of the Rtt109-Vps75 complex remains unclear. Here, we have probed the molecular functions of Vps75 and the Rtt109-Vps75 complex through biochemical, structural and genetic means. We find that Vps75 stimulates the kcat (∼100-fold) of Rtt109 and enhances acetylation of the H3 histone tail, a previously unknown substrate of Rtt109-Vps75. Supporting the In vitro functions of the Rtt109-Vps75 complex, loss of Vps75 (vps75Δ in S. cerevisiae) resulted in a substantial drop (60%) in H3K9ac during S phase. X-ray structural analysis of two Vps75 crystal forms revealed a Nap1-like fold with significant differences between the structural states, suggesting conformational flexibility of Vps75 and a potential mechanism of Rtt109 activation by Vps75. Together, the presented data suggest that the Rtt109-Vps75 complex functions as a multifunctional HAT-chaperone complex that links histone modification and nucleosome assembly.
RESULTS
Vps75 dramatically stimulates catalysis by Rtt109
Previous work demonstrated that Vps75 enhances the acetyltransferase activity of Rtt109; however, the mechanism of activation was unknown13,17. Using detailed steady-state kinetic analyses, we examined the nature of Vps75-dependent activation of Rtt109 with three different types of histone substrates: an H3 tail peptide (residues 1-20), H3 and reconstituted H3-H4 tetramers (hereafter referred to as H3-H4). Initial rates of acetylation for Rtt109 and Rtt109-Vps75 were measured at increasing concentrations of histone substrate and saturating levels of acetyl coenzyme A (acetyl-CoA). Plots of rate versus concentration of substrate were fitted to the Michaelis-Menten equation, yielding the kinetic constants kcat, KM and kcat/KM (Table 1). In the absence of Vps75, Rtt109 shows kcat values 50-fold to 150-fold lower than those of the Rtt109-Vps75 complex, regardless of the substrate examined (Table 1). In contrast, the difference in KM values for the histone substrates between Rtt109 and Rtt109-Vps75 was modest (1.1-fold to 2.1-fold higher for Rtt109) when compared within each substrate type. At saturating H3 peptide concentrations and varied acetyl-CoA levels, similar steady-state results were obtained (Table 1). Rtt109 was 40-fold less efficient in acetylating the peptide than Rtt109-Vps75, largely owing to a decrease in the kcat value. To corroborate the kinetic results, we carried out two different binding assays. GST-tagged Rtt109 was used in ‘pull-down’-type binding assays for H3-H4. As evidenced by these data (Supplementary Fig. 1a and Supplementary Methods online), Rtt109 is equally capable of binding H3-H4 in the presence or absence of Vps75. Using fluorescently tagged H3, fluorescence polarization assays indicated that Rtt109 yielded a Kd value for H3 of 17 ± 8 nM (Supplementary Fig. 1b and Supplementary Methods). Because the affinity (23 ± 10 nM) of Vps75 for Rtt109 is comparable to that of Rtt109 for histones, fluorescence polarization data for H3 binding to Rtt109-Vps75 was not technically attainable. Collectively, these data indicate that Vps75 stimulates Rtt109 HAT activity by increasing catalysis (kcat), without a substantial change in the KM or Kd for each substrate.
Table 1. Kinetics of acetylation by Rtt109 and Rtt109-Vps75.
| Enzyme | Substrate | kcat (s-1)a | KM (μM)a | kcat/KM (M-1 s-1)a |
|---|---|---|---|---|
| Rtt109-Vps75 | H3b | 0.21 ± 0.04 | 5.8 ± 0.8 | 3.5 ± 0.9 × 104 |
| H3-H4 | 0.11 ± 0.05 | 1.4 ± 0.4 | 8.4 ± 2 × 104 | |
| H3 tail peptide | 0.13 ± 0.04 | 75 ± 15 | 1.8 ± 0.5 × 103 | |
| Acetyl-CoA | 0.19 ± 0.01 | 1.0 ± 0.2 | 1.9 ± 0.4 × 105 | |
| Rtt109 | H3 | 0.0033 ± 0.0003 | 8.1 ± 0.1 | 4.3 ± 0.2 × 102 |
| H3-H4 | 0.0044 ± 0.0009 | 2.9 ± 0.6 | 1.5 ± 0.6 × 103 | |
| H3 tail peptide | 0.0014 ± 0.0004 | 83 ± 29 | 1.7 ± 1 × 101 | |
| Acetyl-CoA | 0.0017 ± 0.0001 | 0.3 ± 0.09 | 5.2 ± 2 × 103 |
All values are averages of three or four experiments ± s.d.
Data published previously13.
Unexpectedly, both Rtt109 and Rtt109-Vps75 showed considerable activity on the H3 tail peptide, although the KM values for the peptide were ∼10-fold higher (83 μM and 75 μM) than those for full-length H3 (8.1 μM and 5.8 μM) (Table 1). These observations suggested that Rtt109-Vps75 has broader HAT specificity than previously reported and that the tail of histone H3 is a viable substrate.
Rtt109-Vps75 shows unique substrate specificity
The observation of efficient acetylation of an H3 tail peptide prompted us to explore the specificity of Rtt109-Vps75 (Table 1). Using MS/MS analysis to determine the acetylated sites on the H3 tail peptide, acetylation of H3K9 and H3K14 were clearly observed in the MS spectra (data not shown). Next, we determined whether Rtt109-Vps75 could acetylate the H3 tail within the context of full-length H3 histone. Using catalytic amounts of enzyme, we allowed Rtt109-Vps75 to acetylate H3 and analyzed selected time points by LC-MS/MS after trypsin digestion (Supplementary Fig. 2a and Supplementary Methods online). Acetylation of H3 on K56, K9, K14 and K23 were readily observed as the first set of sites modified, with H3K9ac appearing before H3K14ac (Fig. 1a and Supplementary Fig. 2a). After longer incubation times, we detected H3K79ac and H3K122ac, although peptides containing these acetylations appeared at lower frequency (Supplementary Fig. 2a). To examine whether Rtt109-Vps75 showed a preference for the H3 tail, we used the equivalent-length H4 tail peptide (first 20 N-terminal residues) as a substrate and carried out a steady-state rate comparison of H3 and H4 peptides (Supplementary Fig. 2b). In contrast with the H3 peptide, the H4 peptide was acetylated 28-fold slower (Supplementary Fig. 2b). Together, these data indicate that, In vitro, Rtt109-Vps75 can specifically acetylate sites within the H3 tail. Notably, the Rtt109-Asf1 complex showed no significant activity toward the H3 tail peptide (C.E.B. and J.M.D., unpublished data).
Figure 1.

Rtt109-Vps75 acetylates residues within the H3 tail. (a) MS/MS spectrum of histone H3 peptide K(ac)STGGK(ac)APR, residues 9-17, conclusively confirms H3K9 and H3K14 are acetylated within the protein. The observed b and y fragment ions, resulting from collison-induced dissociation of the peptide backbone, are labeled on the peptide sequence and in spectrum with their ion type, position and mass/charge (m/z). (b,c) MATa, bar1 strains of the indicated genotypes were arrested in G1 phase with α factor mating pheromone and released into a synchronous cell cycle in pheromone-free media for the indicated times at 30 °C. Whole-cell extracts were analyzed by immunoblotting with the indicated antibodies. (d) Immunoblot signal from anti-H3K9ac from the mid-S phase 30-min time point in b or c were quantified by chemiluminescence on a LAS-300 (Fujifilm) and calculated relative to the total H3 signal. The average percent acetylation relative to the wild-type (WT) strain for three independent experiments is depicted. (e) Immunoblot signal from anti-H3K14ac. Samples were treated and analyzed as in d. (f) Immunoblot signal from anti-H3K23ac. Samples were treated and analyzed as in d for two independent experiments. Error bars in d and e indicate s.d., and error bars in f indicate average error.
The In vitro acetylation of the H3 tail by Rtt109-Vps75 suggested that H3K9, H3K14 or H3K23 might be targets of the Rtt109-Vps75 complex in vivo. Previous data indicated that these residues are acetylated on newly synthesized molecules, which are most easily detected when cells greatly induce histone expression during S phase33,34. To test for the roles of Rtt109 and Vps75 in modifying these residues, we quantified the levels of these site-specific acetylations in synchronized yeast cells, comparing wild-type, rtt109Δ and vps75Δ cells. Pheromone-synchronized cells were released into pheromone-free media, and we collected samples at 10-min intervals. The synchrony of cultures was analyzed by fluorescence-activated cell sorting (FACS) sorting of propidium iodide-stained aliquots of the cultures, which confirmed that cells of all genotypes were in the middle of S phase at the 30-min time point (Supplementary Fig. 3a online).
To determine the extent of acetylation at H3K9, H3K14 and H3K23, we quantified the chemiluminescence intensities from immunoblots and normalized them to the amount of H3 in each sample. Intensities for each blot were measured at multiple time points, demonstrating that the signals were within the linear range of detection (Supplementary Fig. 3b). In wild-type cells (Fig. 1b-f), the amounts of H3 increase as cells enter S phase, as expected34. Because acetylation of H3K9, H3K14 and H3K23 occurs on newly synthesized molecules, peaks in intensity corresponding to these modifications of the H3 tail are also observed in wild-type cells33. In strains lacking VPS75 or RTT109, the levels of H3K9ac were decreased by ∼60% (remaining acetylation levels: 42 ± 12% for vps75Δ and 39 ± 15% for rtt109Δ), whereas the levels of H3K14ac were largely unaffected by either deletion (98 ± 14% for vps75Δ and 93 ± 10% for rtt109Δ). H3K23ac was also substantially reduced in the deletion strains (remaining acetylation levels: 72 ± 2% for vps75Δ and 54 ± 11% for rtt109Δ), although this effect was less dramatic than the loss of H3K9ac.
To ensure the observed changes in the acetylation patterns were independent of the changes in histone level that occur during S phase, we reloaded the same samples on blots, normalizing for H3 levels, and repeated the quantitative analyses of the modifications (Supplementary Fig. 3c). This manipulation led to the same conclusion. Taken together, these data suggest that Rtt109-Vps75 is integral during S phase for regulating the acetylation of H3K9 and, to a lesser extent, the acetylation of H3K23.
Rtt109 stimulates histone deposition by Vps75
Although Vps75 stimulates Rtt109 HAT activity, it was unclear whether Rtt109 would affect the intrinsic histone-deposition activity of Vps75 (ref. 24). To examine this, we measured the levels of histone deposition via a standard supercoiling assay24,35. Vps75, in the presence or absence of equimolar amounts of Rtt109, was incubated with Topoisomerase I-relaxed pUC19 plasmid and histones H3 and H4 under single-turnover conditions ([Vps75] or [Rtt109-Vps75] > [histones]). At the indicated times, aliquots were removed and quenched into 1% (w/v) SDS and DNA loading dye. After separating DNA topoisomers via agarose gel electrophoresis and staining with ethidium bromide, the levels of supercoiled DNA were visualized. The data in Figure 2 reveal that addition of equimolar Rtt109 increases the levels of histone deposition by Vps75 at least five-fold. Rtt109 does not have intrinsic deposition activity on its own (data not shown), suggesting that Rtt109 stimulates the activity of Vps75. Rtt109-stimulated histone deposition by Vps75 was independent of Rtt109 HAT activity because preincubation of Rtt109-Vps75 with histones and acetyl-CoA did not substantially affect the levels of histone deposition by Rtt109-Vps75 (data not shown). Thus, these studies have uncovered two activities of the Rtt109-Vps75 complex: histone H3 tail acetylation and stimulated histone deposition.
Figure 2.
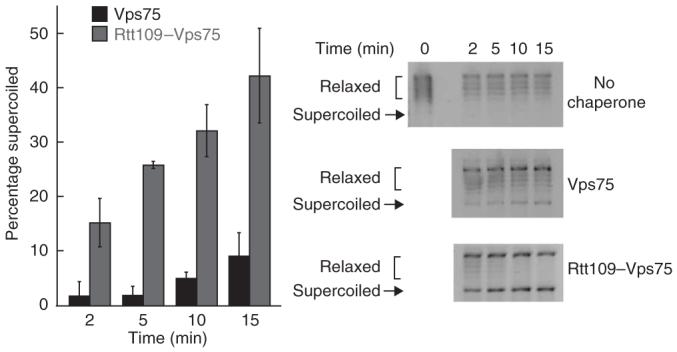
Rtt109 stimulates histone-deposition activity of Vps75. Histone H3 and H4 deposition by Vps75 in the presence or absence of Rtt109 was monitored by supercoiling assay. DNA was unwound before adding to the deposition reaction, and deposition activity was quantified by measuring the density as a percentage of that of the total DNA that was supercoiled in each lane, which corrects for possible variations in loading. Values shown are averages of three independent experiments ± s.d. Shown at the right are representative control assays of no Vps75 or Rtt109 control, Vps75 only or Rtt109-Vps75 from the same gel. The zero time point for each assay used the same stock of relaxed DNA and is shown in the ‘No chaperone’ control. Gel images were inverted for clarity.
X-ray crystal structures of Vps75
The stimulatory activity of Vps75 on Rtt109 prompted us to investigate whether Vps75 adopts a unique structure that permits activation of histone acetylation by Rtt109. We determined the X-ray crystal structures of Vps75 (residues 1-259) in both native and selenomethionine (SeMet)-substituted forms. The SeMet structure was determined to 2.42-Å resolution by multiwavelength anomalous (MAD) phasing, which provided a search model for determining the 2.0-Å resolution structure of native Vps75 (Fig. 3a, Table 2 and Supplementary Fig. 4 online). The two proteins crystallized under different conditions and in nonisomorphic unit cells. Within the asymmetric units of both the native and SeMet crystal forms, two noncrystallographically related Vps75 monomers interact through interdigitation of the loops at the apex of the prominent β-sheet folds that form the core of each monomer. This interface has not been described previously within the Nap1 family, and it is unclear whether it is of functional importance.
Figure 3.

Structure of Vps75. (a) Front and back views of the X-ray crystal structure of native monomer of Vps75. The domain I helix (residues 8-53) is shown in dark blue and domain II (residues 57-221) is shown in green. (b) Structure of Nap1 (PDB 2AYU)36. Domains I and domain II are colored as in a. Structural figures of Vps75 and Nap1 were made using MacPyMOL40.
Table 2. Data collection and refinement statistics.
| Nativea | SeMeta | ||
|---|---|---|---|
| Data collection | |||
| Space group | P212121 | P212121 | |
| Cell dimensions | |||
| a, b, c (Å) | 74.80, 86.08, 85.64 | 79.02, 88.94, 94.26 | |
| Peak | Remote | ||
| Wavelength | 0.9785 | 0.9795 | 0.9686 |
| Resolution (Å)b | 50-2.0 (2.07-2.00) | 65-2.42 (2.55-2.42) | 65-2.42 (2.55-2.42) |
| R sym c | 7.1 (40.8) | 21.7 (150) | 26.6 (256) |
| I / σI | 25.3 (3.0) | 20.6 (1.8) | 21.8 (1.7) |
| Completeness (%) | 99.4 (96.8) | 100 (100) | 100 (100) |
| Redundancy | 4.7 (3.8) | 22.5 (18.4) | 23.4 (20.5) |
| Refinement | |||
| Resolution (Å) | 20-2.0 | 20-2.42 | |
| No. reflections | 35,682 | 24,697 | |
| Rwork / Rfree | 22.9/25.1 | 21.2/25.5 | |
| No. atoms | |||
| Protein | 3,452 | 3,319 | |
| Ligand/ion | - | - | |
| Water | 309 | 279 | |
| B-factors | |||
| Protein | 34.0 | 57.1 | |
| Ligand/ion | - | - | |
| Water | 44.2 | 57.1 | |
| R.m.s. deviations | |||
| Bond lengths (Å) | 0.009 | 0.011 | |
| Bond angles (°) | 0.84 | 0.90 | |
Data statistics from one crystal of native Vps75 and one crystal of SeMet Vps75.
Values in parentheses are for highestresolution shell.
Rsym = ΣΣj|Ij - 〈I〉|/ΣIj, where Ij is the intensity measurement for reflection j and 〈I〉 is the mean intensity for multiply-recorded reflections. The high Rsym values for the ‘Se peak’ and ‘Se remote’ data are due to the high redundancy of these data sets, which were merged data from long and short exposures. Subsets of these data have lower Rsym but insufficient anomalous signal for structure determination.
The overall fold of Vps75 monomers from the native and SeMet structures was quite similar (r.m.s. deviation = 1.7 Å for 199 Cα atoms); however, there were notable differences between the two structures, described in the following section. The Vps75 structures bear substantial similarity to two previously determined histone chaperones, Nap1 and INHAT (compare Figure 3a and Supplementary Figure 4b to Figure 3b)36,37. DALI searches indicated that Vps75 monomers superimpose well with INHAT (r.m.s. deviation 2.2 Å for 145 Cα atoms and 3.3 Å for 154 Cα for the SeMet and native monomers, respectively) and NAP1 family proteins (r.m.s. deviation = 3.7 Å for 173 Cα atoms and 4.0 Å for 183 Cα for the SeMet and native monomers, respectively). Previous structural studies of Nap1 and INHAT indicated that these proteins form dimers through a long N-terminal helix (α1 in Vps75) termed domain I36. Vps75 has a similar N-terminal helix comprising of 45 residues (residues 8-53), although identification of this helix was possible only by determination of the Vps75 structure because of the low overall sequence identity (∼20%) of Vps75 and Nap1 or INHAT (Supplementary Fig. 4c). Vps75 domain I pairs with a symmetrically related domain I from an adjacent Vps75 monomer in both the native and SeMet crystals (Fig. 4a). The structure of the helix is ‘kinked’ by Pro42, which creates a hockey stick-like structure. In total, 16 residues (8 from each monomer) are buried between the two helices, creating a hydrophobic interface (Fig. 4a,b). In addition, there are seven apparent hydrogen-bonding, electrostatic or π-stacking interactions formed between the helices (Fig. 4b). Vps75 buries 18% of the total residues from the domain I helix between the two monomers (8 of 45 residues), a smaller percentage than that reported for Nap1 and INHAT, which bury 43% and 31% of the residues, respectively, within the domain I helix at the interaction site36,37.
Figure 4.

Dimerization between domain I of Vps75 monomers. (a) Vps75 monomers interact with symmetry mates in adjacent asymmetric units creating a dimer through domain I helix (residues 8-53). Residues are colored to indicate those involved in forming the dimer interface and type of interaction with hydrophobic interactions in brown, hydrogen-bonding in green and π-stacking in blue. Domain II is shown in gray. (b) Cartoon of interaction surface created by domain I of Vps75. Residues are colored according to interaction type as in a. Gray sections are not resolved in the current structural models of Vps75. Residues labeled in bold are altered in the Vps75ESEE construct (containing C21E V25S I28E V32E substitutions). (c) Spectra from analytical ultracentrifugation of wild-type Vps75 (black squares) and Vps75ESEE (blue circles).
To determine whether domain I is essential for Vps75 dimer formation in solution, we compared the migration of native Vps75 and Vps75 containing C21E V25S I28E V32E substitutions (Vps75ESEE) in analytical ultracentrifugation (AUC) experiments38. Compared to native Vps75, the domain I mutant Vps75ESEE showed a lower sedimentation rate, consistent with a monomer state for Vps75ESEE and a dimeric state for native Vps75 (Fig. 4c). Corroborating a native dimer, size-exclusion chromatography revealed an apparent molecular weight of 68 ± 1.5 kDa, which is similar to the theoretical molecular weight (63 kDa) of a His-tagged Vps75 dimer (data not shown). These data support a role for domain I as the main interface between monomers of Vps75 in solution, as revealed in the two Vps75 X-ray structures.
Distinct conformations of SeMet and native dimers
As noted above, the structures of SeMet and native Vps75 show substantial differences in the conformation of domain I and domain II (Supplementary Fig. 5 online). In the native structure, domain I is kinked ×15° more sharply than in the SeMet structure (Supplementary Fig. 5c). This reduces the distances between domain II from each monomer by 2-9 Å in the native structure and also rotates domain II toward domain I by 11°. This increase in the kink of domain I offsets the two helices of domain I by a greater degree. The interaction surface between domain I of Vps75 monomers is substantially different to the interfaces described in Nap1 or INHAT and may allow for the conformational freedom observed in the Vps75 dimers (Fig. 4b)36,37. We suggest that these two conformational states of Vps75 may be intimately linked to its ability to activate Rtt109 through enhanced catalytic turnover. Although it could be argued that the SeMet substitutions induce an unnatural conformation of Vps75, two observations suggest that this is unlikely. First, the SeMet-substituted Vps75 was completely functional as an activator of the Rtt109 HAT (data not shown). Second, only one of the three SeMet residues (SeMet40) is within domain I, and this residue is not among those forming the hydrophobic interface (Fig. 4a,b and Supplementary Fig. 5). Thus, we propose that the two structures reflect intrinsic conformational flexibility of the protein, which may be linked to the enhanced catalytic cycle of the Rtt109-Vps75 complex. Below, we offer additional biochemical support for this model.
Distinct charged surfaces within domain II of Vps75
The Vps75 dimer structures reveal two sets of distinctly charged surfaces: a large patch of positive charge on each monomer spanning residues 147 to 177 and a large, negatively charged cavity formed within domain II of each monomer containing a high degree of acidic and polar residues (Fig. 5a and Supplementary Fig. 4c). Six basic residues are found in the basic patches in addition to several polar residues (Arg164, Lys169, Lys170, Lys171, Arg173, Lys177, Ser151, Tyr152, Ser154, Ser165 and Tyr172). Additionally, residues Arg73, Ser75, Arg184, Lys189 and Lys192 are within 12 Å of Lys171. The overall structure consists of a type I β-turn (residues 150 to 153), a loop (residues 154 to 168) and an α-helix (residues 169 to 177). Notably, there is no equivalent basic patch or structural motif in Nap1 or INHAT, and structural alignment of Vps75 with Nap1 indicates that this region in Nap1 contains a β-hairpin implicated in homo-oligomer formation (Fig. 5b)36,39. The analogous region of INHAT is not resolved in the previously reported crystal structure37.
Figure 5.

Comparison of surface electrostatics of the Nap1 family of histone chaperones. (a) Native Vps75 (left) and SeMet Vps75 (right). The acidic cavity and basic patch are indicated for reference. (b) Nap1 (ref. 36; left) and INHAT37 (right). Areas of negative charge are shown in red, and areas of basic charge are shown in blue.
The Vps75 dimer forms a cavity between domain II from each monomer and contains numerous acidic and polar residues. The surface of this cavity is composed of residues 22-37 of domain I and residues 57-76 and 187-221 of domain II (Fig. 5a,b and Supplementary Fig. 4c). The cavity measures 18-21 Å across, 40 Å long and 14 Å from the bottom of domain II to the bottom of the domain I helix. The ∼11° rotation of domain II toward domain I observed in the native Vps75 structure brings the acidic and polar residues from domains I and II into closer proximity compared with the SeMet structure (compare Figure 5a to Figure 5b). This rotation of domain II suggested by the two crystal structures is exemplified by residues Asp136, Asp198 and Glu207 within domain II of the SeMet structure, which are 3-5 Å closer to domain I in the structure of the native Vps75. The conformational motion brings acidic and polar residues into closer proximity and increases the charge density of the acidic cavity. This unique feature of Vps75, not apparent in other NAP1 family members, may be linked to its ability to activate Rtt109. We note that recombinant yeast Nap1 bound Rtt109 poorly and was incapable of activating Rtt109 (Supplementary Fig. 1c and data not shown), demonstrating that Vps75 has a specific interface for interaction with Rtt109.
Acidic cavity mutations in Vps75 reduce Rtt109 activation
The distinct conformational states of Vps75 suggest that intrinsic motion within the acidic cavity may be a crucial step in the catalytic cycle for enhanced acetylation by Rtt109. We tested this hypothesis by creating three triple mutants of Vps75 and assessing their ability to activate Rtt109. We generated two acidic cavity mutants with three alanine substitutions at Asp198, Ser199 Ser202 or Ser205 Glu206 Glu207, referred to as Vps75p and Vps75q, respectively. For comparison, we also generated a triple-alanine mutant (G181A R183A T185A, referred to as Vps75o) in which three residues on the opposite face of domain II from the acidic cavity are altered (Supplementary Fig. 4c). We chose these mutants in particular because previous work on INHAT identified the structurally similar ‘O’, ‘P’ and ‘Q’ mutants as defective in chaperone activity and histone binding37. Particularly notably, Glu207, which is altered in Vps75q, moves ∼5 Å between the SeMet and native structures. Using MacPyMOL, we visualized the effects on the predicted surface electrostatics of the Vps75p and Vps75q in the native Vps75 structure40. Figure 6a-c shows the differences in the electrostatics of the acidic cavity for native Vps75, Vps75p and Vps75q, revealing that the Q mutation of Vps75 substantially reduces the negative-charge density of the acidic cavity, whereas the P mutation has a less dramatic effect. These observations suggested that the Q mutant would disable the catalytic activity of the HAT-chaperone complex to a much greater degree than the O or P mutant. This hypothesis was examined biochemically.
Figure 6.

Characterization of Vps75 acidic cavity mutants. Depictions of the surface electrostatics of the acidic cavity for native Vps75 (a), Vps75p with alanine substitutions at residues 198, 199 and 202 (b) and for Vps75q with alanine substitutions at residues 205, 206 and 207 (c). Negative charge is shown in red and positive charge in blue. (d) Saturation curves of H3-H4 for Vps75o (boxes), Vps75p (open circles) and Vps75q (filled circles). Reactions were performed as described in the Supplementary Methods with full steady-state kinetics shown in Supplementary Table 1. For Rtt109-Vps75q, the kcat/KM value was (8.3 ± 3) × 103 M-1 s-1. For Rtt109-Vps75o, the kcat/KM value was (3.8 ± 1) × 104 M-1 s-1. For Rtt109-Vps75p, the kcat/KM 4.2 ± 2 × 104 M-1 s-1. Assays were performed in duplicate with the average rate from the two experiments shown. Error bars indicate average error. (e) H3 binding curves for Vps75 and Vps75q. Kd was calculated from four independent experiments, and the average Kd ± s.d. is shown. (f) Representative western blot of H3K9ac in wild-type (WT) and Vps75q yeast cells. Cells were treated, prepared for immunoblotting and analyzed as described in Figure 1b. The average percentage of acetylation relative to that of the wild-type strain for each of two independent experiments is shown (designated 1 and 2). P-values were calculated from three measurements of the chemiluminescence in each experiment using the ANOVA function in Kaleidagraph.
As observed for the native Rtt109-Vps75 complex, both Vps75q and Vps75o mutants copurified with Rtt109 in a 1:1 complex, indicating that the triple-alanine mutations did not substantially alter the affinity for Rtt109 (Supplementary Fig. 1c). Using H3-H4 as the substrate, we performed steady-state substrate-saturation kinetics to determine the catalytic competence of H3-H4 acetylation by Rtt109, activated with either Vps75q, Vps75p or Vps75o (Fig. 6d and Supplementary Table 1 online). Data analysis revealed a ten-fold decrease in the kcat/KM value for Rtt109-Vps75q compared to the wild-type complex ((8.3 ± 3) × 103 M-1 s-1 versus (8.4 ± 2) × 104 M-1 s-1). In contrast, Rtt109-Vps75o showed a modest decrease of 2.2-fold in the kcat/KM value for acetylation of H3-H4 ((3.8 ± 1.0) × 104 M-1 s-1) (Fig. 6d and Supplementary Table 1). Similarly, Rtt109-Vps75p showed only a two-fold decrease in the kcat/KM value ((4.2 ± 2) 104 M-1 s-1) compared to the wild type (Fig. 6d and Supplementary Table 1). The fact that the Rtt109-Vps75q complex showed a 4.1-fold change in kcat and a 10-fold change in the kcat/KM suggests that catalysis and not histone affinity was affected primarily (Supplementary Table 1).
To confirm that loss of histone binding affinity was not the basis for the ten-fold loss in catalytic efficiency for the Vps75q mutant, we compared the Kd of native Vps75 and Vps75q for histone H3. We titrated Vps75 or Vps75q into a solution of fluorescein-labeled histone H3, measured the resulting change in fluorescence polarization and determined the Kd from binding curves. The Kd for H3 was 56 ± 8 nM for wild-type Vps75 and 29 ± 7 nM for Vps75q (Fig. 6e), indicating that the Vps75q mutant showed similar, if not slightly better, affinity for histone compared to native Vps75.
To provide further support for the functional role of the Vps75 acidic patch during histone H3 acetylation by Rtt109, a vps75q allele was used to replace the wild-type allele, and we assessed the acetylation efficiency of this strain as in Figure 1 and Supplementary Fig. 3. The levels of H3K9ac in immunoblotting experiments were quantified and normalized to total H3. From two independent experiments, there were small (11% and 10.2%) but statistically significant (P = 0.0013 and P = 0.0016) reductions in H3K9ac in the vps75q cells compared to wild type. As expected, the loss of H3K9ac in the vps75q strain was modest, because Vps75q-stimulated Rtt109 activity is still ten-fold higher than that of Rtt109 alone (Fig. 6d, Table 1 and Supplementary Table 1). Collectively, these data are consistent with the importance of these acidic residues (Ser205, Glu206 and Glu207) within the structurally dynamic acidic cavity and suggest that this feature is crucial for the Vps75-dependent activation of Rtt109.
DISCUSSION
H3K56ac and H3K9ac are associated with nucleosome assembly during DNA replication and transcription5,9,33,41. Recently, both genetic and biochemical evidence indicated that the newly described HAT Rtt109 catalyzed H3K56 acetylation, but also required the histone chaperone Asf1 (refs. 12,13,25). In vitro studies revealed that Vps75, a Nap1 homolog, could also stimulate the Rtt109-dependent acetylation of H3K56. In fact, the Rtt109-Vps75 complex was approximately 20-fold more efficient than the Asf1-stimulated Rtt109 activity13. Although Vps75 and Rtt109 seem to interact tightly both In vitro and in vivo, cells lacking VPS75 showed little defect in H3K56 acetylation13,17,26,42. Collectively, these observations suggested that the Rtt109-Vps75 complex harbors distinct but unknown functions. Here we explored these activities and determined that Rtt109-Vps75 is a dual-functioning complex, stimulating Rtt109-dependent H3 acetylation by an approximately 100-fold increase in catalysis and enhancing Vps75-dependent H3-H4 deposition onto DNA (Fig. 2 and Table 1). Unexpectedly, Rtt109-Vps75 catalyzes efficient acetylation of H3K9, H3K14 and H3K23 In vitro, and molecular genetic analysis provided in vivo evidence for the link between this complex and the acetylation of H3K9 and H3K23 during S phase (Fig. 1). Increased acetyltransferase and histone deposition activity of Rtt109-Vps75 and the enhanced specificity for H3K9 reveal a HAT-chaperone complex that combines histone modification with nucleosome assembly.
Two structurally distinct X-ray structures of Vps75 revealed a Nap1-like fold with several unique features. Notably, the two Vps75 conformers revealed differences in the density of negative charge within the acidic cavity of Vps75. We propose that this intrinsic motion, which alters the charge density, is intimately linked to catalytic turnover by Rtt109. In support of this, the Q mutant of Rtt109-Vps75 (S205A, E206A and E207A) showed a ten-fold decrease in the catalytic efficiency for H3-H4 acetylation compared to the wild-type complex (Fig. 6d and Supplementary Table 1). The drop in catalytic efficiency was not caused by decreased affinity for histone, as the ability of Vps75q to bind histones was not reduced (Fig. 6e). Indeed, modeling these mutations onto the native Vps75 structure revealed significant elimination of negative charge on both sides of the acidic cavity (compare Figure 6a to Figure 6c). Moreover, the Q mutation decreased H3K9ac levels in vivo (Fig. 6f).
Fillingham et al. have recently provided convincing genetic support for the Vps75-dependent acetylation of H3K9 by Rtt109 (ref. 43). Here we present genetic evidence for the Rtt109-Vps75-dependent acetylation of H3K9 and H3K23, as well as further structural and biochemical data that reveal new insights into the molecular mechanism of Rtt109-Vps75 function. Structural studies of Asf1 revealed significant rearrangements in acidic and hydrophobic residues within the histone binding surface and proximal loops upon interaction with histone ligands44-47. In the case of Rtt109-Vps75, the intrinsic motions of Vps75 may reflect functional conformers that mediate accessibility to acetylation sites on histone H3, enhancing the catalytic efficiency of Rtt109. In this study, we provide structural, biochemical and genetic evidence for a HAT-chaperone complex linking histone acetylation and nucleosome assembly.
METHODS
Reagents
Tris buffer, DTT, Tris(2-carboxylethyl) phosphine hydrochloride (TCEP), imidazole, sodium chloride and other chemicals were purchased from Sigma-Aldrich or Fisher. All reagents were of the highest quality and used without further purification.
Peptide synthesis
H3 tail (ARTKQTARKSTGGKAPRKQL) and H4 tail (SGRGKGGKGLGKGGAKRHRK) peptides were synthesized at the University of Wisconsin-Madison Biotechnology Center and further purified via C18 reverse-phase HPLC. Peptide concentration was determined using the BCA assay from Pierce Biotechnology using BSA as a standard.
Histone purification
Recombinant X. laevis histones were purified as described previously by Berndsen and co-workers (plasmids were a previous gift from K. Luger)48. Histone concentration was determined using the extinction coefficients described previously49.
Reconstitution of histone tetramers
Histone tetramers were reconstituted following a previously described method50.
Purification and truncation of Vps75
Vps75 was initially purified using nickel-affinity chromatography as described previously13. Fractions containing Vps75 were pooled and dialyzed into 20 mM Tris, pH 7.3, 1 mM β-mercaptoethanol, 40 mM NaCl, 1 mM EDTA, 7% (v/v) glycerol and 0.01% (v/v) NP-40 (buffer A) overnight at 4 °C. The total concentration of protein was then determined by Bradford assay and 1/120th the mass of trypsin added. The reaction was allowed to incubate at 25 °C for 40 min, then a 1:1 (w/w) amount of soybean trypsin inhibitor was added to stop the digestion reaction, and the reaction was cooled on ice. Vps75 was then purified via anion-exchange chromatography and eluted using a linear gradient from 40 mM to 600 mM NaCl in buffer A. Fractions containing truncated Vps75 as assessed by SDS-PAGE were dialyzed in buffer A with 0.05% (w/v) PMSF. Protein concentration was determined using the Bradford assay with BSA as a protein standard. Selenomethionine (SeMet)-incorporated Vps75 was expressed and purified as described above except that TCEP was substituted for β-mercaptoethanol.
Purification of Rtt109-Vps75 complexes and Rtt109
Rtt109-Vps75 was initially purified using nickel-affinity chromatography as described13. Fractions from the nickel-affinity column containing Rtt109-Vps75 or Rtt109 were pooled and dialyzed into buffer A overnight at 4 °C. Protein was loaded onto a Hi-Trap SP column (GE Healthcare) and eluted using a linear gradient of 40-490 mM NaCl in buffer A. Fractions containing Rtt109-Vps75 in 1:1 stoichiometry or Rtt109 only were pooled and dialyzed against buffer A overnight at 4 °C. Protein concentration was determined from the Bradford assay using BSA as a protein standard and activity assays.
Crystallization of Vps75
Native Vps75 (27 mg ml-1) truncated to residues 1-259 was dialyzed against 20 mM Tris, pH 7.5, at 25 °C, 40 mM NaCl overnight at 4 °C. Crystals were formed by hanging-drop vapor diffusion by suspending 1 μl drops of Vps75 and mother liquor (50 mM Tris, pH 7.4, 10 mM MgCl2, 100 mM KCl and 35% (v/v) PEG 400) at a 1:1 ratio above 1 ml of mother liquor. Crystals were cryoprotected in the mother liquor before being flash-frozen in liquid nitrogen.
SeMet-incorporated Vps75 (20 mg ml-1) truncated to residues 1-259 was dialyzed against 20 mM Tris, pH 7.5, at 25 °C, 40 mM NaCl overnight at 4 °C. Crystals were formed by hanging-drop vapor diffusion by suspending 1 μl drops of Vps75 and mother liquor (0.2 M potassium formate, 20% (w/v) PEG 3350) at a 1:1 ratio above 1 ml of mother liquor. Crystals were cryoprotected in 0.1 M potassium formate, 22% (w/v) PEG 3350 and 25% (w/v) ethylene glycol before being flash-frozen in liquid nitrogen.
Data collection and refinement methods are described in the Supplementary Methods online. For SeMet Vps75, 95.4% of residues fall in the most favored Ramachandran category, with 4.6% in the allowed category. For the native Vps75, 94.0% of residues fall in the most favored Ramachandran category, with 6.0% in the allowed category. Further refinement statistics are shown in Table 2.
Supercoiling assays of histone deposition
The reaction to relax the DNA for the supercoiling assay contained 12 U of calf thymus topoisomerase I (Invitrogen), in 20 mM Tris, pH 8.0, 1 mM DTT, 4 mM MgCl2, 50 mM KCl, 300 fmol pUC19 DNA and 0.07 mg ml-1 BSA in 100 μl. Each unwinding reaction was incubated for 1 h at 37 °C before adding 20 μl to a deposition reaction. Deposition reaction conditions were 20 mM Tris, pH 8.0, 1 mM DTT, 50 mM KCl, 15 μM Vps75 or Rtt109-Vps75 complex, 60 fmol of DNA, 6 μM H3 and 6 μM H4. Aliquots were removed and quenched into 1% (w/v) SDS, 10 mM EDTA and 1 × DNA loading dye at appropriate time points. Aliquots were then loaded onto 0.8% (w/v) TBE agarose gel and run at 22 to 27 °C and 50 V. DNA within the gel was visualized by ethidum bromide staining. Data were analyzed using Labworks version 4.6 (Labworks).
Mutagenesis
Oligonucleotide-encoded mutations in the bacterial Vps75-His6 expression plasmid13 were selected using DpnI restriction endonuclease as described51. To create the Vps75q in S. cerevisiae, the vps75q allele was inserted into pRS406 and integrated at its natural locus by two-step gene replacement as described52,53.
Analytical ultracentrifugation
Solutions of Vps75 or Vps75 with the C21E V25S I28E V32E substitutions with an absorbance at 280 nm (A280) of 0.5 were dialyzed overnight at 4 °C against two changes of 4 liters of 20 mM Tris, pH 7.5, 40 mM NaCl and 0.2 mM TCEP. Samples were flash-frozen in liquid nitrogen until use in centrifugation experiments. After pelleting any precipitated material, undiluted samples were spun at 123,012g at 20 °C in velocity sedimentation experiments. Absorbance was monitored at 280 nm. Data were analyzed in Ultrascan using the van Holde-Weishat method54,55.
Accession codes
Protein Data Bank: Atomic coordinates and structure factors for the native and SeMet-substituted Vps75 have been deposited with the accession codes 3C9D and 3C9B, respectively.
Supplementary Material
ACKNOWLEDGMENTS
We wish to thank S. Anderson at the Life Sciences Collaborative Access Team (LS-CAT) for assistance in data collection on the native Vps75. We also thank K. Satyshur for assistance in crystal screening, and we thank M. Foley and K. Crowley for assistance with the AUC experiment. Finally, we thank members of the Denu and Keck laboratories for helpful comments and critique of the work, especially K. Arnold, B. Smith, L. Li and M. Killoran. This work was supported in part by a predoctoral fellowship from the American Heart Association to C.E.B., US National Institutes of Health (NIH) grant GM059785 to J.M.D., a Shaw award from the Greater Milwaukee Foundation to J.L.K. and NIH grant GM055712 to P.D.K.
References
- 1.Shahbazian MD, Grunstein M. Functions of site-specific histone acetylation and deacetylation. Annu. Rev. Biochem. 2007;76:75–100. doi: 10.1146/annurev.biochem.76.052705.162114. [DOI] [PubMed] [Google Scholar]
- 2.Avvakumov N, Cote J. The MYST family of histone acetyltransferases and their intimate links to cancer. Oncogene. 2007;26:5395–5407. doi: 10.1038/sj.onc.1210608. [DOI] [PubMed] [Google Scholar]
- 3.Baker SP, Grant PA. The SAGA continues: expanding the cellular role of a transcriptional co-activator complex. Oncogene. 2007;26:5329–5340. doi: 10.1038/sj.onc.1210603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yang XJ, Ullah M. MOZ and MORF, two large MYSTic HATs in normal and cancer stem cells. Oncogene. 2007;26:5408–5419. doi: 10.1038/sj.onc.1210609. [DOI] [PubMed] [Google Scholar]
- 5.Masumoto H, Hawke D, Kobayashi R, Verreault A. A role for cell-cycle-regulated histone H3 lysine 56 acetylation in the DNA damage response. Nature. 2005;436:294–298. doi: 10.1038/nature03714. [DOI] [PubMed] [Google Scholar]
- 6.Xu F, Zhang K, Grunstein M. Acetylation in histone H3 globular domain regulates gene expression in yeast. Cell. 2005;121:375–385. doi: 10.1016/j.cell.2005.03.011. [DOI] [PubMed] [Google Scholar]
- 7.Hyland EM, et al. Insights into the role of histone H3 and histone H4 core modifiable residues in Saccharomyces cerevisiae. Mol. Cell. Biol. 2005;25:10060–10070. doi: 10.1128/MCB.25.22.10060-10070.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Celic I, et al. The sirtuins Hst3 and Hst4p preserve genome integrity by controlling histone H3 lysine 56 deacetylation. Curr. Biol. 2006;16:1280–1289. doi: 10.1016/j.cub.2006.06.023. [DOI] [PubMed] [Google Scholar]
- 9.Rufiange A, Jacques PE, Bhat W, Robert F, Nourani A. Genome-wide replication-independent histone H3 exchange occurs predominantly at promoters and implicates H3 K56 acetylation and Asf1. Mol. Cell. 2007;27:393–405. doi: 10.1016/j.molcel.2007.07.011. [DOI] [PubMed] [Google Scholar]
- 10.Garcia BA, et al. Organismal differences in post-translational modifications in histones H3 and H4. J. Biol. Chem. 2007;282:7641–7655. doi: 10.1074/jbc.M607900200. [DOI] [PubMed] [Google Scholar]
- 11.Xhemalce B, et al. Regulation of histone H3 lysine 56 acetylation in Schizosaccharomyces pombe. J. Biol. Chem. 2007;282:15040–15047. doi: 10.1074/jbc.M701197200. [DOI] [PubMed] [Google Scholar]
- 12.Schneider J, Bajwa P, Johnson FC, Bhaumik SR, Shilatifard A. Rtt109 is required for proper H3K56 acetylation: a chromatin mark associated with the elongating RNA polymerase II. J. Biol. Chem. 2006;281:37270–37274. doi: 10.1074/jbc.C600265200. [DOI] [PubMed] [Google Scholar]
- 13.Tsubota T, et al. Histone H3-K56 acetylation is catalyzed by histone chaperonedependent complexes. Mol. Cell. 2007;25:703–712. doi: 10.1016/j.molcel.2007.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Han J, et al. Rtt109 acetylates histone H3 lysine 56 and functions in DNA replication. Science. 2007;315:653–655. doi: 10.1126/science.1133234. [DOI] [PubMed] [Google Scholar]
- 15.Driscoll R, Hudson A, Jackson SP. Yeast Rtt109 promotes genome stability by acetylating histone H3 on lysine 56. Science. 2007;315:649–652. doi: 10.1126/science.1135862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Allis CD, et al. New nomenclature for chromatin-modifying enzymes. Cell. 2007;131:633–636. doi: 10.1016/j.cell.2007.10.039. [DOI] [PubMed] [Google Scholar]
- 17.Han J, Zhou H, Li Z, Xu RM, Zhang Z. The Rtt109-Vps75 histone acetyltransferase complex acetylates non-nucleosomal histone H3. J. Biol. Chem. 2007;282:14158–14164. doi: 10.1074/jbc.M700611200. [DOI] [PubMed] [Google Scholar]
- 18.DeKoning L, Corpet A, Haber JE, Almouzni G. Histone chaperones: an escort network regulating histone traffic. Nat. Struct. Mol. Biol. 2007;14:997–1007. doi: 10.1038/nsmb1318. [DOI] [PubMed] [Google Scholar]
- 19.Eitoku M, Sato L, Senda T, Horikoshi M. Histone chaperones: 30 years from isolation to elucidation of the mechanisms of nucleosome assembly and disassembly. Cell.Mol.LifeSci. 2008;65:414–444. doi: 10.1007/s00018-007-7305-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chang L, et al. Histones in transit: cytosolic histone complexes and diacetylation of H4 during nucleosome assembly in human cells. Biochemistry. 1997;36:469–480. doi: 10.1021/bi962069i. [DOI] [PubMed] [Google Scholar]
- 21.Smith S, Stillman B. Stepwise assembly of chromatin during DNA replication in vitro. EMBO J. 1991;10:971–980. doi: 10.1002/j.1460-2075.1991.tb08031.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Verreault A, Kaufman PD, Kobayashi R, Stillman B. Nucleosome assembly by a complex of CAF-1 and acetylated histones H3/H4. Cell. 1996;87:95–104. doi: 10.1016/s0092-8674(00)81326-4. [DOI] [PubMed] [Google Scholar]
- 23.Glowczewski L, Waterborg JH, Berman JG. Yeast chromatin assembly complex 1 protein excludes nonacetylatable forms of histone H4 from chromatin and the nucleus. Mol. Cell. Biol. 2004;24:10180–10192. doi: 10.1128/MCB.24.23.10180-10192.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Selth L, Svejstrup JQ. Vps75, a new yeast member of the NAP histone chaperone family. J. Biol. Chem. 2007;282:12358–12362. doi: 10.1074/jbc.C700012200. [DOI] [PubMed] [Google Scholar]
- 25.Han J, Zhou H, Li Z, Xu RM, Zhang Z. Acetylation of lysine 56 of histone H3 catalyzed by RTT109 and regulated by ASF1 is required for replisome integrity. J. Biol. Chem. 2007;282:28587–28596. doi: 10.1074/jbc.M702496200. [DOI] [PubMed] [Google Scholar]
- 26.Krogan NJ, et al. Global landscape of protein complexes in the yeast Saccharomyces cerevisiae. Nature. 2006;440:637–643. doi: 10.1038/nature04670. [DOI] [PubMed] [Google Scholar]
- 27.Ito T, Ikehara T, Nakagawa T, Kraus WL, Muramatsu M. p300-mediated acetylation facilitates the transfer of histone H2A-H2B dimers from nucleosomes to a histone chaperone. Genes Dev. 2000;14:1899–1907. [PMC free article] [PubMed] [Google Scholar]
- 28.Asahara H, et al. Dual roles of p300 in chromatin assembly and transcriptional activation in cooperation with nucleosome assembly protein 1 in vitro. Mol. Cell. Biol. 2002;22:2974–2983. doi: 10.1128/MCB.22.9.2974-2983.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Seo SB, et al. Regulation of histone acetylation and transcription by INHAT, a human cellular complex containing the set oncoprotein. Cell. 2001;104:119–130. doi: 10.1016/s0092-8674(01)00196-9. [DOI] [PubMed] [Google Scholar]
- 30.Karetsou Z, Martic G, Sflomos G, Papamarcaki T. The histone chaperone SET/ TAF-Iβ interacts functionally with the CREB-binding protein. Biochem. Biophys. Res. Commun. 2005;335:322–327. doi: 10.1016/j.bbrc.2005.06.210. [DOI] [PubMed] [Google Scholar]
- 31.Park YJ, Chodaparambil JV, Bao Y, McBryant SJ, Luger K. Nucleosome assembly protein 1 exchanges histone H2A-H2B dimers and assists nucleosome sliding. J. Biol. Chem. 2005;280:1817–1825. doi: 10.1074/jbc.M411347200. [DOI] [PubMed] [Google Scholar]
- 32.Park YJ, Luger K. Structure and function of nucleosome assembly proteins. Biochem. Cell Biol. 2006;84:549–558. doi: 10.1139/o06-088. [DOI] [PubMed] [Google Scholar]
- 33.Kuo MH, et al. Transcription-linked acetylation by Gcn5p of histones H3 and H4 at specific lysines. Nature. 1996;383:269–272. doi: 10.1038/383269a0. [DOI] [PubMed] [Google Scholar]
- 34.Adkins MW, Carson JJ, English CM, Ramey CJ, Tyler JK. The histone chaperone anti-silencing stimulates the acetylation of newly synthesized histone H3 in S-phase. J. Biol. Chem. 2007;282:1334–1340. doi: 10.1074/jbc.M608025200. [DOI] [PubMed] [Google Scholar]
- 35.Eckey M, Hong W, Papaioannou M, Baniahmad A. The nucleosome assembly activity of NAP1 is enhanced by Alien. Mol. Cell. Biol. 2007;27:3557–3568. doi: 10.1128/MCB.01106-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Park YJ, Luger K. The structure of nucleosome assembly protein 1. Proc. Natl. Acad. Sci. USA. 2006;103:1248–1253. doi: 10.1073/pnas.0508002103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Muto S, et al. Relationship between the structure of SET/TAF-Iβ/INHAT and its histone chaperone activity. Proc. Natl. Acad. Sci. USA. 2007;104:4285–4290. doi: 10.1073/pnas.0603762104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Miyaji-Yamaguchi M, Okuwaki M, Nagata K. Coiled-coil structure-mediated dimerization of template activating factor-I is critical for its chromatin remodeling activity. J. Mol. Biol. 1999;290:547–557. doi: 10.1006/jmbi.1999.2898. [DOI] [PubMed] [Google Scholar]
- 39.Park YJ, McBryant SJ, Luger K. A β-hairpin comprising the nuclear localization sequence sustains the self-associated states of nucleosome assembly protein 1. J. Mol. Biol. 2008;375:1076–1085. doi: 10.1016/j.jmb.2007.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.DeLano WL. MacPyMOL: a PyMOL-nased Molecular Graphics Application for MacOS X. DeLano Scientific LLC; South San Francisco: 2005. [Google Scholar]
- 41.Recht J, et al. Histone chaperone Asf1 is required for histone H3 lysine 56 acetylation, a modification associated with S phase in mitosis and meiosis. Proc. Natl. Acad. Sci. USA. 2006;103:6988–6993. doi: 10.1073/pnas.0601676103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Collins SR, et al. Functional dissection of protein complexes involved in yeast chromosome biology using a genetic interaction map. Nature. 2007;446:806–810. doi: 10.1038/nature05649. [DOI] [PubMed] [Google Scholar]
- 43.Fillingham J, et al. Chaperone control of the activity and specificity of the histone H3 acetyltransferase Rtt109. Mol. Cell. Biol. 2008;28:4342–4353. doi: 10.1128/MCB.00182-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mousson F, et al. Structural basis for the interaction of Asf1 with histone H3 and its functional implications. Proc. Natl. Acad. Sci. USA. 2005;102:5975–5980. doi: 10.1073/pnas.0500149102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.English CM, Adkins MW, Carson JJ, Churchill ME, Tyler JK. Structural basis for the histone chaperone activity of Asf1. Cell. 2006;127:495–508. doi: 10.1016/j.cell.2006.08.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Natsume R, et al. Structure and function of the histone chaperone CIA/ASF1 complexed with histones H3 and H4. Nature. 2007;446:338–341. doi: 10.1038/nature05613. [DOI] [PubMed] [Google Scholar]
- 47.Malay AD, Umehara T, Matsubara K, Padmanabhan B, Yokoyama S. Crystal structures of fission yeast histone chaperone Asf1 complexed with the Hip1 B domain or the Cac2 C-terminus. J. Biol. Chem. 2008;283:14022–14031. doi: 10.1074/jbc.M800594200. [DOI] [PubMed] [Google Scholar]
- 48.Berndsen CE, et al. Nucleosome recognition by the Piccolo NuA4 histone acetyl-transferase complex. Biochemistry. 2007;46:2091–2099. doi: 10.1021/bi602366n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ. Crystal structure of the nucleosome core particle at 2.8Å resolution. Nature. 1997;389:251–260. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- 50.Tanaka Y, et al. Expression and purification of recombinant human histones. Methods. 2004;33:3–11. doi: 10.1016/j.ymeth.2003.10.024. [DOI] [PubMed] [Google Scholar]
- 51.Sambrook J, Russell DW. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 2001. [Google Scholar]
- 52.Sikorski RS, Hieter P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kaiser C, Michaelis S, Mitchell A. Methods in Yeast Genetics: A Cold Spring Harbor Laboratory Course Manual. vii 234. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 1994. [Google Scholar]
- 54.Schuck P, Perugini MA, Gonzales NR, Howlett GJ, Schubert D. Sizedistribution analysis of proteins by analytical ultracentrifugation: strategies and application to model systems. Biophys. J. 2002;82:1096–1111. doi: 10.1016/S0006-3495(02)75469-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Demeler B, van Holde KE. Sedimentation velocity analysis of highly heterogeneous systems. Anal. Biochem. 2004;335:279–288. doi: 10.1016/j.ab.2004.08.039. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.