

Abstract
The crystal structure of the uncharacterized protein SO2946 from Shewanella oneidensis MR-1 was determined with single-wavelength anomalous diffraction (SAD) and refined to 2.0 Å resolution. The SO2946 protein consists of a short helical N-terminal domain and a large C-terminal domain with the “jelly-roll” topology. The protein assembles into a propeller consisting of three C-terminal blades arranged around a central core formed by the N-terminal domains. The function of SO2946 could not be inferred from the sequence since the protein represents an orphan with no sequence homologs, but the protein’s structure bears a fold similar to that of proteins containing carbohydrate-binding modules. Features such as fold conservation, the presence of a conserved groove and a metal binding region are indicative that SO2946 may be an enzyme and could be involved in binding carbohydrate molecules.
Keywords: Jelly-roll topology, Carbohydrate-binding modules, Orphan protein, Magnesium binding, SAD phasing, Singleton
Introduction
The genomes of many bacterial species are being sequenced, revealing many protein genes with no sequence similarity to previously characterized proteins. Shewanella oneidensis MR-1, whose genome has been sequenced, is a Gram-negative facultative aerobe capable of utilizing many different electron acceptors for respiration. S. oneidensis uses oxygen as an electron acceptor during aerobic respiration, yet while under anaerobic conditions this organism shows high flexibility in utilizing a large range of terminal electron acceptors. These acceptors include fumarate, nitrate, trimethylamine N-oxide, dimethyl sulfoxide, sulfite, thiosulfate, elemental sulfur and metals including Mn(III and IV), Fe(III), Cr(VI), and U(VI) [1–3]. The ability of S. oneidensis to reduce toxic metal ions and radionuclides is of great interest due to potential applications in bioremediation. The Shewanella Federation (http://www.shewanella.org) was created in order to understand the biology of this versatile species at the organism level. To enhance structural coverage of the S. oneidensis genome we have selected this organism for structural studies. Here we describe cloning, expression, purification, crystallization and the crystal structure of the SO2946 protein determined using the SAD method and the semi-automated high-throughput protein structure determination pipeline of the Midwest Center for Structural Genomics (MCSG).
Material and methods
Gene cloning and protein expression
The ORF of SO2946 was amplified by PCR from S. oneidensis MR-1 genomic DNA (ATCC) with KOD DNA polymerase (Novagen, Madison, WI). The gene was cloned into a pMCSG7 vector using a modified ligation-independent cloning protocol (Dieckman et al. 2002). The resulting expression clone produces the SO2946 protein as a fusion protein with an N-terminal His6-tag and a TEV protease recognition site (ENLYFQ↓S). The fusion protein was over-produced in an E. coli BL21-derivative that harbored a plasmid pMAGIC encoding three rare E. coli tRNAs (Arg [AGG/AGA] and Ile [ATA]). A selenomethionine (Se-Met) derivative of the expressed protein was prepared as described previously [4]. The transformed BL21 cells were grown in enriched M9 medium at 37°C [5]. After OD600 reached 1.2, 0.01% (w/v) each of leucine, isoleucine, lysine, phenylalanine, threonine, and valine was added to inhibit the metabolic pathway of methionine and encourage Se-Met incorporation [6]. 60 mg of Se-Met was added to one liter of culture and 15 min later protein expression was induced by 1 mM isopropyl-β-d-thiogalactoside (IPTG). The cells were then incubated at 20°C overnight. The harvested cells were resuspended in lysis buffer (500 mM NaCl, 5% glycerol, 50 mM HEPES pH 8.0, 10 mM imidazole and 10 mM 2-mercaptoethanol), and stored at −20°C.
Protein purification and crystallization
The Se-Met labeled protein was purified according to standard protocol [7]. The harvested cells were re-suspended in lysis buffer and lysozyme at 1 mg/ml and 100 µl of protease inhibitor cocktail (Sigma, P8849) were added per 2 g of wet cells, which were kept on ice for 20 min before sonication. The lysate was clarified by centrifugation at 36,000 × g for 1 h and filtered through a 0.22 µm filter. It was then applied to a 5 ml HiTrap Ni-NTA column (GE Health Systems) on the AKTA Express (GE Health Systems). His6-tagged protein was eluted using buffer containing a higher concentration of imidazole (500 mM NaCl, 5% glycerol, 50 mM HEPES, pH 8.0, 250 mM imidazole, 10 mM 2-mercaptoethanol), and the His6-tag was cleaved from the protein by treatment with recombinant His6-tagged TEV protease. A second Ni-NTA affinity chromatography was performed manually to remove the His6-tag and His6-tagged TEV protease.
The protein was dialyzed against crystallization buffer (200 mM NaCl, 20 mM HEPES pH 8.0, 2 mM DTT) and was concentrated using Centricon centrifugal concentrators with 5 k MW cutoff (Amicon) to 130 mg/ml. The molecular mass of the SO2946 protein in solution was evaluated by size-exclusion chromatography according to the procedure described previously [8].
Crystallization was performed by the hanging drop vapor diffusion method at 18°C. Colorless, needle-shaped crystals were grown by mixing 1 µl of the protein with 1 µl of the mother liquor consisting of (0.2 M magnesium chloride, 0.1 M Tris pH 7.0, 25% Peg3350) and equilibrated against 500 µl of this solution. The crystals reached the size 0.1 × 0.1 × 0.7 mm within 4–5 days. Prior to data collection, crystals were rinsed in a cryoprotectant solution consisting of a 20% glycerol in the crystal mother liquor, and then flash-frozen in liquid nitrogen. The crystals belonged to primitive monoclinic space group P21 with cell dimensions of a = 56.57 Å, b = 53.06 Å, c = 112.65 Å, β = 95.65°.
Data collection and structure determination
An X-ray florescence spectrum was recorded from the sample prior to data collection, which identified the presence of Se in the protein crystal. Diffraction data were collected using SBC-Collect software at 100 K at the 19BM beamline of the Structural Biology Center at the Advanced Photon Source, Argonne National Laboratory. Diffraction data were recorded near selenium absorption edge wavelength (λ1 = 0.9794 Å) from a single crystal of Se-Met labeled protein. Data sets were integrated, reduced and scaled with the HKL2000 suite [9]. Data statistics are summarized in Table 1.
Table 1.
Data collection and refinement statistics
| Data set | |
| Beamline | APS, 19-BM |
| Wavelength (Å) | 0.9794 |
| Space group | P21 |
| a,b,c (Å) | 53.06, 56.57, 112.65 |
| β (°) | 95.65 |
| Resolution (Å) | 30–2.0 |
| (2.02–2.09) | |
| No. observations/ | 3,37,222 |
| No. unique reflections | 87,860 |
| Completeness (%) | 99.6 (99.5) |
| Redundancy | 3.8 (3.5) |
| Rmarge (%)a | 11.0 (30.0) |
| I/σb | 11.9 (3.1) |
| Refinement | |
| Protein molecules | 3 |
| Solvent/Mg ions | 674/3 |
| Rcryst (%)c | 16.70 |
| Rfree (%)d | 20.30 |
| RMSD bonds (Å) | 0.009 |
| RMSD angles (°) | 1.158 |
| RMSD chiral centers | 0.078 |
| B average protein/water (Å2) | 35.3/39.6 |
| Ramachandran plot (%)e favored/allowed/outlierse | 95.7/100/0 |
Values in parentheses indicate values for the highest resolution shell
Rsym = (Σ|Ihkl − <I>|)/ΣIhkl, where the average intensity <I> is taken over all symmetry equivalent measurements and Ihkl is the measured intensity for any given reflection.
I/σ is the mean reflection intensity divided by the average estimated error.
Rcryst = ||Fo| − |Fc||/|Fo|, where Fo and Fc are the observed and calculated structure factor amplitudes, respectively.
Rfree is equivalent to Rcryst but is calculated for 5% of the reflections chosen at random and omitted from the refinement process. r.m.s.d., root mean square deviation.
Validated using MolProbity server [17]
Structure refinement
The structure was determined by the SAD method using peak data set to 2.0 Å resolution. The initial phases were determined and density modification performed using the HKL3000 software package [10] which uses SHELXD [11] to find sites, and RESOLVE [12] to carry out density modification. The initial experimental map was of good quality and was used to autotrace model with the ARP/wARP programs [13]. The initial model generated by ARP/wARP covered 85% of total residues in a single polypeptide and was further extended manually using the program COOT [14]. Electron density map calculated at 1 σ was well connected, except for the N-terminal residues 1–23 (1–18 in chain B), which are disordered in the crystal structure. Cycles of manual corrections of the model and refinement with REFMAC 5.0 [15] were repeated at the preliminary stages of refinement. 5% randomly selected reflections were used to calculate Rfree to monitor bias in model building and refinement. At the final stage, TLS refinement was applied with each monomer defined as a TLS group [16]. The final model was refined against all data reflections in the resolution range of 20.0–2.0 Å. Refinement statistics are shown in Table 1.
Quality of the model
The final model consists of three monomers in the asymmetric unit containing 722 residues out of a possible 795 residues (including 3 residues per monomer, a cloning artifact from the expression vector), 674 water molecules and 3 magnesium ions. The protein chain was modeled from residues 24–262 in chains A and B and residues 19–262 in chain C. Structure refinement was performed without imposing the 3-fold non-crystallographic symmetry. Overall, the electron density maps were of high quality and allowed reliable modeling of all three monomers. The quality of the structure was evaluated with the validation tools included in the programs COOT [14] and MOLPROBITY [17].
Coordinates
The atomic coordinates and structure factors of SO2946 have been deposited in the Protein Data Bank as 2A5Z.
Results and discussion
Overall structure
The protein SO2946 forms a tight homotrimer about 78 Å wide and long (Fig. 1a) resembling a three-blade propeller. In the asymmetric unit, the subunits of the trimer have identical topologies and are superimposed with root-mean-square deviation (r.m.s.d.) values of 0.39 Å (chains A/B), 0.48 Å (chains A/C), and 0.44 Å (chains B/C). The SO2946 monomer has dimensions of 39 Å × 40 Å × 63 Å and consists of two structural domains, a small N-terminal α-helical domain (residues 19–45) and a large C-terminal, anti-parallel β-pleated sheet sandwich domain (residues 46–262) (Fig. 1b). The N-terminal domain, which creates the ~20 Å long “handle” of the propeller, is strongly positively charged and is responsible for trimer formation and stabilization. The C-terminal domain displays the classic “jelly-roll” topology and is composed of two anti-parallel β-sheets, several long loops, one long beta hairpin and two short alpha-helical turns H2 (108–112) and H3 (114–117). These regions also contribute to the formation of the trimer, especially the loop formed by residues 54–65 and the beta hairpin (residues 75–97). The core of the C-terminal domain consists of two central, closely packed β-sheets (A and B). The sheet A is composed of 6 strands (S1↓, S4↓, S10↑, S11↓, S12↑, S15↑) and sheet B is composed of 9 strands (S2↓, S3↑, S5↑, S6↑, S7↓, S8↑, S9↓, S13↓, S14↓), (Fig. 1b). This arrangement forms a large groove on the concave site of sheet B that is partially covered by the loop between S5 and S6 (residues 143–149).
Fig. 1.

(a) Ribbon diagrams of the homotrimer of SO2946 viewed down the non-crystallographic threefold showing subunit organization in the trimer. The box highlights the metal interaction region in one of the subunits in trimer. (b) Stereo ribbon diagram of a monomer of SO2946. Helical regions are colored blue, while β-strands and loops regions are shown in orange. The magnesium ion is shown as a green sphere. (c) A close-up view of the metal-binding region at the N-terminus of molecule A of SO2946. The images were generated with the program Pymol (http://pymol.sourceforge.net/)
The analysis of crystal structure strongly suggests that the protein is a trimer. A calculation of accessible surface area using the PISA server [18] indicates that the trimer is very likely a biologically-relevant oligomeric form. An extensive interaction between tightly associated subunits of the trimer buries ~3,200 Å2 of the solvent-accessible surface per monomer, which corresponds to ~ 26% of the total solvent-accessible surface area. This oligomeric state is consistent with the size-exclusion chromatography experiment results showing a molecular mass of 82.7 kDa (calculated mass of trimer 82.5 kDa). The electrostatic potential analysis indicates that the trimeric “jelly-roll” domains form a large, partially positively charged surface with a small negatively charged chamber (volume of ~400 Å3) in the center of the trimer formed on the interface between the C-terminal domains. The handle-like region of the trimer is composed of alternating positively and negatively charged patches with a highly positively charged handle region.
Functional searches
A comparison of the functional and evolutionary relationships between protein sequences, performed using BLASTP and PSI-BLAST tools [19], did not identify any statistically significant match to the sequence of SO2946, indicating that it represents a novel singleton protein.
Analysis of the genomic neighborhood of SO2946 shows that it is surrounded by other genes which encode proteins of unknown function. In turn, these genes are flanked by the genes which encode prophage lambda So tail assembly proteins (Q8ED31, Q8ED30, Q8ED23, Q8ED22-uniprot code) suggesting that SO2946 might interact in prophage lambda So tail assembly. However, only prophage lambda So (Q8ED31), host specific protein J has assigned function. It is linked to the hydrolysis of O-glycosyl compounds, indicating the presence of proteins involved in carbohydrate binding/hydrolysis in this region [20].
Structural comparisons of SO2946 with the structures deposited in the PDB using the DALI server [21], identified several structural homologs. The top hits were carbohydrate binding proteins (PDB code 2D3S, Z-score 12, RMSD for 174 Cα atoms 3.1 Å, sequence identity 13%) [22], sialidase (PDB code 1MZ5, Z-score 10.7, RMSD for 172 Cα atoms 5.2 Å, sequence identity 7%), and polyguluronate lyase (PDB code 1UAI, Z-score 10.5, RMSD for 155 Cα atoms 3.0 Å, sequence identity 7%) [23]. Other significant homologs include favin, the glucose and mannose-binding lectin (PDB code 2B7Y, Z-score 7.2, RMSD for 129 Cα atoms 2.9 Å, sequence identity 9%) [24], alpha-l-arabinofuranosidase, protein binding arabinose (PDB code 1WD3, Z-score 10.5, RMSD for 155 Cα atoms 3.0 Å, sequence identity 7%) [25].
Nearly all of these structural homologs of SO2946 are classified as carbohydrate binding proteins or are involved in carbohydrates processing. Similar results have been obtained using the SSM program [18] which identified several peanut lectin proteins and porcine pancreatic alpha-amylase as highest secondary structure matches [26, 27]. These results indicate clearly that SO2946 has similar structure to numerous proteins containing carbohydrate-binding modules (CBM). All these proteins include the “jelly-roll” fold.
Similarity to carbohydrate-binding modules
The results of the structural comparisons indicate strong fold similarity of SO2946 to carbohydrate-binding module (CBM) containing proteins, suggesting that this protein may bind carbohydrates. Searches of enzyme families that degrade, modify, or create glycosidic bonds in the Carbohydrate-Active enzyme database (http://www.cazy.org/CAZY/) revealed a large group of enzymes clustered into 51 families based on sequence similarity and fold topology. Based on the structure of the ligand binding sites, CBM proteins can be categorized into two groups: those that bind the polysaccharides at a planar-hydrophobic carbohydrate-binding site (type A) and CBMs that bind single polysaccharide chains at a structurally conserved cleft (type B) [28]. 14 protein families with structurally characterized CBMs share the “jelly-roll” fold [29, 30].
Analysis of the SO2946 structure indicates the presence of 30 Å × 15 Å × 10 Å deep elongated grooves (type B-like) located on the concave sites of sheet B in each subunit of the trimer. These grooves are lined with hydrophobic, hydrophilic and charged residues providing rich ligand binding potential for polycarbohydrate bonds. One end of this groove leads directly to an open cavity in the center of the trimer with His58, Ser69 and His227 (supplied by the second subunit) at the entrance to the cavity. This groove has been identified as a carbohydrate-binding surface in many proteins which contain CBMs [29, 30]. Located near the middle of the groove, there are charged residues Glu156 and Arg150 which point from the opposite sides toward the middle of the groove (Fig. 2). One the other end there is F161 which sits on the bottom of the groove. These residues could possibly participate in substrate binding as mixture of the aromatic and charged residues are frequently observed to interact with carbohydrates in CBMs. Super-imposition of the groove region of SO2946 on cleft-like motifs of the above listed enzymes shows that it is well-conserved within β-sheet region, while the loop orientation differs between structures. The significant difference at the solvent-exposed loop region could be explained by the fact that this region plays an important role in substrate recognition and could undergo movements in order to reflect the conformation of ligand. Comparison of the SO2946 groove region with other structurally characterized CBMs of the type B did not reveal a close homologue allowing categorization of this protein to the specific family of CBMs. This may suggest that SO2946 may represent a new family of CBMs.
Fig. 2.
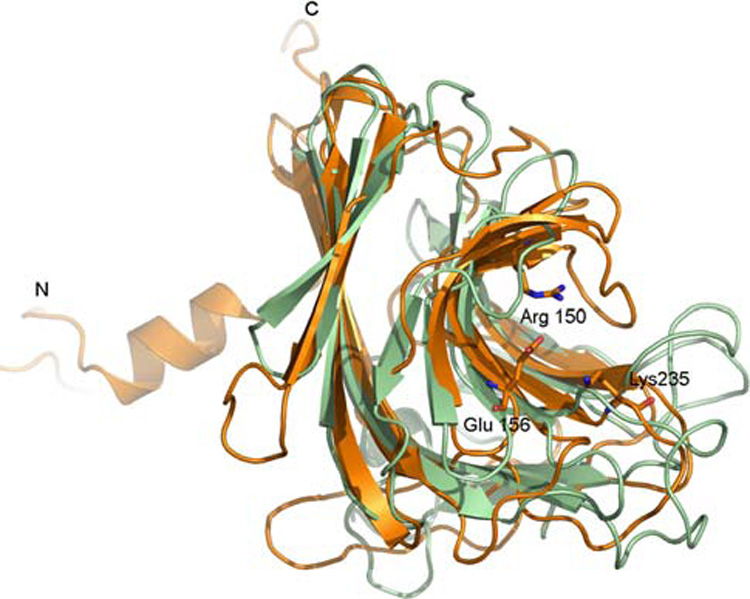
Ribbon diagram of the superposition of a monomer of SO2946 (orange) with a monomer of the favin, glucose- and mannose-binding lectin (green) (PDB code 1UAI). Structures are shown such that the deep groove region is seen from the side. Charged residues in SO2946 pointing towards the center of the cleft are labeled and shown as sticks
Metal binding
A common feature attributed to some proteins containing CBM is the presence of divalent metal cations bound to the N-terminus of the protein. Typically these metal ions bind in the loop region, providing rigidity and adding to the stability of the protein fold. However, it was also shown that in some members of CBM-containing families metal plays an important role in carbohydrate recognition [29]. CBMs have been reported to bind either one (Ca+2) or two different metals (Ca+2 and Mn+2) [29–31]. In the early stages of the structure refinement of SO2946 the experimental Fo – Fc map showed significant positive peaks of electron density indicating the presence of metal ions or other transition metals. These sites were initially treated as water molecules. Based on the X-ray fluorescence measurement, which did not revealed the presence of Ca+2 and Mn+2 ions in the protein crystal, these sites were assumed to be Mg+2 ions due to the presence of 0.2 M magnesium chloride in the crystallization solution. Each of the magnesium ions is positioned at the N-terminal β-pleated sheet A (at the extensive loop region) at the similar position often observed to be occupied by calcium atoms in other CBMs (Fig. 1a, c). The three-fold related metals ions are positioned ~28 Å apart and assume an octahedral geometry arrangement with six oxygen atoms provided by the protein residues at the typical magnesium-oxygen bond distance. Among these residues, side chain oxygens of Asp54 (OD1, 2.16 Å ) and Asp247 (OD1, 2.18 Å ) act as mono-dentate ligands of the magnesium, whereas remaining coordination is provided by the carbonyl oxygen atoms of Asp54 (O, 2.17 Å ), Phe88 (O, 2.17 Å), Gly89 (O, 2.16 Å) and Asp247 (O, 2.17 Å) (Fig. 1c). The role of the metal ion is most-likely to stabilize the protein.
Conclusions
The protein SO2946 is annotated as an uncharacterized orphan protein with no sequence similarity to any known or predicted protein. However, structure analysis of SO2946 shows that it assumes the “jelly roll” fold and closely resembles proteins known to bind or process carbohydrates but share very low sequence similarity. Structural Comparison of SO2946 with well characterized proteins containing similar CBMs shows that they share conserved structural features such as a metal binding region and a substrate-binding groove known to bind carbohydrate molecules. These features suggest that SO2946 may interact with carbohydrates using the conserved groove regions in the trimer. Further biochemical and biophysical studies are needed to confirm if SO2946 does indeed play a role in carbohydrate-binding.
Acknowledgements
The authors wish to thank all members of the Structural Biology Center at Argonne National Laboratory for their help in conducting the experiments. Authors are grateful to Dr. S. Rajan for comments and helpful discussion, R. Jedrzejczak for help with the size-exclusion chromatography experiment and M. Chodkiewicz-Nocek for assistance with figures. This work was supported by National Institutes of Health grant GM074942 and by the U.S. Department of Energy, Office of Biological and Environmental Research, under contract DE-AC02-06CH11357.
Footnotes
The submitted manuscript has been created by UChicago Argonne, LLC, Operator of Argonne National Laboratory (“Argonne”). Argonne, a U.S. Department of Energy Office of Science laboratory, is operated under Contract No. DE-AC02-06CH11357. The U.S. Government retains for itself, and others acting on its behalf, a paid-up nonexclusive, irrevocable worldwide license in said article to reproduce, prepare derivative works, distribute copies to the public, and perform publicly and display publicly, by or on behalf of the Government.
Contributor Information
B. Nocek, Midwest Center for Structural Genomics and Structural Biology Center, Biosciences Division, Argonne National Laboratory, 9700 South Cass Avenue, Building 202, Argonne, IL 60439, USA
L. Bigelow, Midwest Center for Structural Genomics and Structural Biology Center, Biosciences Division, Argonne National Laboratory, 9700 South Cass Avenue, Building 202, Argonne, IL 60439, USA
J. Abdullah, Midwest Center for Structural Genomics and Structural Biology Center, Biosciences Division, Argonne National Laboratory, 9700 South Cass Avenue, Building 202, Argonne, IL 60439, USA
A. Joachimiak, Email: andrzejj@anl.gov, Midwest Center for Structural Genomics and Structural Biology Center, Biosciences Division, Argonne National Laboratory, 9700 South Cass Avenue, Building 202, Argonne, IL 60439, USA.
References
- 1.Bouhenni R, Gehrke A, Saffarini D. Identification of genes involved in cytochrome c biogenesis in Shewanella oneidensis, using a modified mariner transposon. Appl Environ Microbiol. 2005;71:4935–4937. doi: 10.1128/AEM.71.8.4935-4937.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Heidelberg JF, Paulsen IT, Nelson KE, Gaidos EJ, Nelson WC, Read TD, Eisen JA, Seshadri R, Ward N, Methe B, et al. Genome sequence of the dissimilatory metal ion-reducing bacterium Shewanella oneidensis. Nat Biotechnol. 2002;20:1118–1123. doi: 10.1038/nbt749. [DOI] [PubMed] [Google Scholar]
- 3.Venkateswaran K, Dollhopf ME, Aller R, Stackebrandt E, Nealson KH. Shewanella amazonensis sp. nov., a novel metal-reducing facultative anaerobe from Amazonian shelf muds. Int J Syst Bacteriol. 1998;48(Pt 3):965–972. doi: 10.1099/00207713-48-3-965. [DOI] [PubMed] [Google Scholar]
- 4.Walsh MA, Dementieva I, Evans G, Sanishvili R, Joachimiak A. Taking MAD to the extreme: ultrafast protein structure determination. Acta Crystallogr D Biol Crystallogr. 1999;55(Pt 6):1168–1173. doi: 10.1107/s0907444999003698. [DOI] [PubMed] [Google Scholar]
- 5.Donnelly MI, Zhou M, Millard CS, Clancy S, Stols L, Eschen-feldt WH, Collart FR, Joachimiak A. An expression vector tailored for large-scale, high-throughput purification of recombinant proteins. Protein Expr Purif. 2006;47(2):446–454. doi: 10.1016/j.pep.2005.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.van den Ent F, Lockhart A, Kendrick-Jones J, Löwe J. Crystal structure of the N-terminal domain of MukB: a protein involved in chromosome partitioning. Structure. 1999;7(10):1181–1187. doi: 10.1016/s0969-2126(00)80052-0. [DOI] [PubMed] [Google Scholar]
- 7.Kim Y, Dementieva I, Zhou M, Wu R, Lezondra L, Quartey P, Joachimiak G, Korolev O, Li H, Joachimiak A. Automation of protein purification for structural genomics. J Struct Funct Genomics. 2004;5:111–118. doi: 10.1023/B:JSFG.0000029206.07778.fc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nocek B, Chang C, Li H, Lezondra L, Holzle D, Collart F, Joachimiak A. Crystal structures of delta1-pyrroline-5-carboxylate reductase from human pathogens Neisseria meningitides and Streptococcus pyogenes. J Mol Biol. 2005;354:91–106. doi: 10.1016/j.jmb.2005.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Macromol Crystallogr Pt A. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 10.Minor W, Cymborowski M, Otwinowski Z, Chruszcz M. HKL-3000: the integration of data reduction and structure solution - from diffraction images to an initial model in minutes. Acta Crystallogr D Biol Crystallogr. 2006;62:859–866. doi: 10.1107/S0907444906019949. [DOI] [PubMed] [Google Scholar]
- 11.Schneider TR, Sheldrick GM. Substructure solution with SHELXD. Acta Crystallogr D Biol Crystallogr. 2002;58:1772–1779. doi: 10.1107/s0907444902011678. [DOI] [PubMed] [Google Scholar]
- 12.Terwilliger TC. SOLVE and RESOLVE: automated structure solution and density modification. Methods Enzymol. 2003;374:22–37. doi: 10.1016/S0076-6879(03)74002-6. [DOI] [PubMed] [Google Scholar]
- 13.Perrakis A, Morris R, Lamzin VS. Automated protein model building combined with iterative structure refinement. Nat Struct Biol. 1999;6:458–463. doi: 10.1038/8263. [DOI] [PubMed] [Google Scholar]
- 14.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 15.Bailey The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 16.Painter J, Merritt EA. A molecular viewer for the analysis of TLS rigid-body motion in macromolecules. Acta Crystallogr D Biol Crystallogr. 2005;61:465–471. doi: 10.1107/S0907444905001897. [DOI] [PubMed] [Google Scholar]
- 17.Lovell SC, Davis IW, Arendall WB, III, de Bakker PI, Word JM, Prisant MG, Richardson JS, Richardson DC. Structure validation by Calpha geometry: phi, psi and Cbeta deviation. Proteins. 2003;15:437–450. doi: 10.1002/prot.10286. [DOI] [PubMed] [Google Scholar]
- 18.Krissinel E, Henrick K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr D Biol Crystallogr. 2004;60:2256–2268. doi: 10.1107/S0907444904026460. [DOI] [PubMed] [Google Scholar]
- 19.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 20.Laskowski RA, Watson JD, Thornton JM. Protein function prediction using local 3D templates. J Mol Biol. 2005;351:614–626. doi: 10.1016/j.jmb.2005.05.067. [DOI] [PubMed] [Google Scholar]
- 21.Holm L, Sander C. Protein structure comparison by alignment of distance matrices. J Mol Biol. 1993;233:123–138. doi: 10.1006/jmbi.1993.1489. [DOI] [PubMed] [Google Scholar]
- 22.Kulkarni KA, Sinha S, Katiyar S, Surolia A, Vijayan M, Suguna K. Structural basis for the specificity of basic winged bean lectin for the Tn-antigen: a crystallographic, thermodynamic and modelling study. FEBS Lett. 2005;579:6775–6780. doi: 10.1016/j.febslet.2005.11.011. [DOI] [PubMed] [Google Scholar]
- 23.Osawa T, Matsubara Y, Muramatsu T, Kimura M, Kakuta Y. Crystal structure of the alginate (poly alpha-l-guluronate) lyase from Corynebacterium sp. at 1.2 A resolution. J Mol Biol. 2005;345:1111–1118. doi: 10.1016/j.jmb.2004.10.081. [DOI] [PubMed] [Google Scholar]
- 24.Reeke GN, Jr, Becker JW. Three-dimensional structure of favin: saccharide binding-cyclic permutation in leguminous lectins. Science. 1986;234:1108–1111. doi: 10.1126/science.3775378. [DOI] [PubMed] [Google Scholar]
- 25.Miyanaga A, Koseki T, Matsuzawa H, Wakagi T, Shoun H, Fushinobu S. Crystal structure of a family 54 alpha-L-arabinofuranosidase reveals a novel carbohydrate-binding module that can bind arabinose. J Biol Chem. 2004;279:44907–44914. doi: 10.1074/jbc.M405390200. [DOI] [PubMed] [Google Scholar]
- 26.Kundhavai Natchiar S, Arockia Jeyaprakash A, Ramya TN, Thomas CJ, Suguna K, Surolia A, Vijayan M. Structural plasticity of peanut lectin: an X-ray analysis involving variation in pH, ligand binding and crystal structure. Acta Crystallogr D Biol Crystallogr. 2004;60(pt 2):211–219. doi: 10.1107/S090744490302849X. [DOI] [PubMed] [Google Scholar]
- 27.Bompard-Gilles C, Rousseau P, Rouge P, Payan F. Substrate mimicry in the active center of a mammalian alpha-amylase: structural analysis of an enzyme-inhibitor complex. Structure. 1996;4:1441–1452. doi: 10.1016/s0969-2126(96)00151-7. [DOI] [PubMed] [Google Scholar]
- 28.Henrissat B, Claeyssens M, Tomme P, Lemesle L, Mornon JP. Cellulase families revealed by hydrophobic cluster analysis. Gene. 1989;81:83–95. doi: 10.1016/0378-1119(89)90339-9. [DOI] [PubMed] [Google Scholar]
- 29.Boraston AB, Bolam DN, Gilbert HJ, Davies GJ. Carbohydrate-binding modules: fine-tuning polysaccharide recognition. Biochem J. 2004;382:769–781. doi: 10.1042/BJ20040892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Carvalho AL, Goyal A, Prates JA, Bolam DN, Gilbert HJ, Pires VM, Ferreira LM, Planas A, Romao MJ, Fontes CM. The family 11 carbohydrate-binding module of Clostridium thermocellum Lic26A-Cel5E accommodates beta-1, 4- and beta-1, 3-1, 4-mixed linked glucans at a single binding site. J Biol Chem. 2004;279:34785–34793. doi: 10.1074/jbc.M405867200. [DOI] [PubMed] [Google Scholar]
- 31.Notenboom V, Boraston AB, Chiu P, Freelove AC, Kilburn DG, Rose DR. Recognition of cello-oligosaccharides by a family 17 carbohydrate-binding module: an X-ray crystallographic, thermodynamic and mutagenic study. J Mol Biol. 2001;314:797–806. doi: 10.1006/jmbi.2001.5153. [DOI] [PubMed] [Google Scholar]