

Abstract
The expression of the cholesterol transporter ATP-binding cassette transporter A1 (ABCA1) in the brain and its role in the lipidation of apolipoproteins indicate that ABCA1 may play a critical role in brain cholesterol metabolism. To investigate the role of ABCA1 in brain cholesterol homeostasis and trafficking, we characterized mice that specifically lacked ABCA1 in the CNS, generated using the Cre/loxP recombination system. These mice showed reduced plasma high-density lipoprotein (HDL) cholesterol levels associated with decreased brain cholesterol content and enhanced brain uptake of esterified cholesterol from plasma HDL. Increased levels of HDL receptor SR-BI in brain capillaries and apolipoprotein A-I in brain and CSF of mutant mice were evident. Cholesterol homeostasis changes were mirrored by disturbances in motor activity and sensorimotor function. Changes in synaptic ultrastructure including reduced synapse and synaptic vesicle numbers were observed. These data show that ABCA1 is a key regulator of brain cholesterol metabolism and that disturbances in cholesterol transport in the CNS are associated with structural and functional deficits in neurons. Moreover, our findings also demonstrate that specific changes in brain cholesterol metabolism can lead to alterations in cholesterol uptake from plasma to brain.
Introduction
Cholesterol is required for myelination (Saher et al., 2005), dendrite differentiation (Goritz et al., 2005) and synaptic activity (Mauch et al., 2001). Disturbances in CNS cholesterol homeostasis are implicated in neurodegenerative diseases including Alzheimer's (Vance et al., 2005) and Huntington disease (Valenza et al., 2005).
Normally, plasma cholesterol is not taken up by the brain because of the tight junctions between brain capillary endothelial cells (BCECs) (Björkhem and Meaney, 2004; Dietschy and Turley, 2004). Brain cholesterol is synthesized in situ, and excretion of excess cholesterol occurs primarily after conversion to 24S-OH cholesterol by the brain-specific enzyme CYP46A1 (Dietschy and Turley, 2004). CNS cholesterol biosynthesis is high during development but declines after myelination is complete (Quan et al., 2003). It is postulated that adult neurons depend on glia-secreted cholesterol transported on apolipoprotein E (ApoE)-containing HDL-like particles (Mauch et al., 2001) and taken up by neurons via ApoE receptors (Vance et al., 2005).
ATP-binding cassette transporter A1 (ABCA1) mediates the transport of intracellular cholesterol to extracellular lipid-free apolipoprotein A-I (ApoA-I) and ApoE (Oram and Heinecke, 2005), a process critical for HDL synthesis (Brooks-Wilson et al., 1999). ABCA1 is expressed in neurons and glia (Wellington et al., 2002; Hirsch-Reinshagen et al., 2004; Tachikawa et al., 2005). The most important apolipoproteins in brain lipid homeostasis are ApoE, synthesized and secreted by astrocytes, and ApoA-I, derived from plasma or BCECs (Pitas et al., 1987; Weiler-Güttler et al., 1990). ABCA1 mediates cholesterol transport from glia to ApoE and ApoA-I and is critical for efficient secretion and lipidation of brain ApoE (Hirsch-Reinshagen et al., 2004; Wahrle et al., 2004). Despite the role of CNS cholesterol in synaptic function and neurodegenerative disorders, mechanisms regulating its transport and homeostasis are not well understood. Moreover, not much is known about how specific changes in CNS lipid metabolism impact plasma cholesterol.
In mice lacking ABCA1 globally, the specific role of brain ABCA1 cannot be studied without concurrent changes in whole body cholesterol homeostasis. Abca1−/− mice have very low plasma HDL and ApoA-I (Oram and Heinecke, 2005) diminishing the pool of ApoA-I available for transport into the CNS and lack ABCA1 in BCECs, each of which can have separate effects on CNS cholesterol homeostasis. Indeed, previous studies of specific deletion of ABCA1 in pancreas (Brunham et al., 2007) and intestine (Brunham et al., 2006) have unmasked tissue specific functions of ABCA1 not readily apparent in Abca1−/− mice.
To examine the exclusive role of brain ABCA1 in CNS cholesterol homeostasis and behavioral function, we developed mice lacking ABCA1 in neurons and glia (Abca1−B/−B). These mice exhibit reduced plasma HDL cholesterol levels and increased brain uptake of esterified cholesterol from plasma HDL. Locomotor activity and sensorimotor function are altered in Abca1−B/−B mice and the number of cortical excitatory synapses and synaptic vesicles are decreased.
These findings demonstrate that ABCA1 plays a critical role in brain cholesterol homeostasis and regulates behavioral function and synaptic development. Our results also show that perturbed brain cholesterol balance can alter the uptake of cholesterol from plasma to brain.
Materials and Methods
Animals.
Mice lacking ABCA1 selectively in the brain were generated by crossing ABCA1 floxed mice (Timmins et al., 2005) with nestin-Cre mice (Jackson ImmunoResearch Laboratories). ABCA1 floxed (control mice) and ABCA1 floxed, nestin-Cre positive (Abca1−B/−B) littermates at 3 months of age were used in the experiments unless stated otherwise. Mice were housed under 12 h light/dark cycles and received a standard laboratory chow diet. All animal procedures were approved by the University of British Columbia Animal Care Committee.
Brain and plasma sterol analysis.
Mice were killed by anesthetic overdose, blood was collected and cortex and hippocampus were dissected. Sterols were extracted from brain tissues as well as plasma using chloroform-methanol. The levels of each sterol were determined after conversion to the corresponding trimethylsilyl ethers by gas chromatography-flame ionization detection and gas chromatography-mass spectrometry as reported previously (Lütjohann et al., 2002). Tissue dry weight was determined after overnight drying in a SpeedVac ultracentrifuge dryer (ThermoFisher Scientific). Plasma cholesterol, HDL-cholesterol, phospholipids and triglycerides were measured by standard enzymatic assays (Wako Pure Chemical Industries).
Brain HDL and cholesteryl ester uptake.
Analysis of HDL metabolism was performed as previously described (Pittman and Taylor, 1986). HDL (density = 1.063 − 1.21 g/ml) was isolated from wild-type C57BL/6J mice by centrifugation and labeled with 125I-tyramine cellobiose (125I-TC) in its protein and 3H-cholesteryl oleyl ether (3H-CEt) (GE Healthcare Bio-Sciences) in its cholesteryl ester (CE) moiety. Mice were injected with 125I-TC- and 3H-CEt double-labeled HDL via the tail vein, blood samples were collected at different time points and plasma decay rates (fractional catabolic rate, FCR) for 125I and 3H were calculated. Mice were killed 24 h after injection and brain levels of HDL-associated 125I-TC as well as 3H-CEt were determined. Brain uptake rates of 125I-TC and 3H-CEt are expressed as organ FCR and are calculated as follows: Brain FCR = plasma FCR × fraction of total body tracer recovered in brain (Pittman and Taylor, 1986).
Selective HDL-CE uptake is calculated as the difference in brain FCRs for 3H-CEt and 125I-TC, because 3H-CEt accumulation represents both the selective uptake of CE and HDL-associated CE and uptake of 125I-TC traces the HDL holo-particle uptake.
Western analysis.
Brain regions were homogenized in 10 mm Tris-HCl, 1% (v/v) Triton X-100, 40 μm phenylmethylsulfonyl fluoride and complete protease inhibitors (Roche Diagnostics Corp.). Protein concentration in brain tissue and capillaries isolated from brains was determined by the Lowry assay. Equal amounts of total protein were separated by SDS-PAGE, transferred to polyvinylidene difluoride (PVDF) membranes, and probed with either an anti-ABCA1(Wellington et al., 2002), anti-SR-BI (Novus Biologicals), anti-ApoE (Millipore Bioscience Research Reagents), anti-ApoA-I (Biodesign), anti-CYP46A1 (a gift from Dr. D. Russell, University of Texas Southwestern Medical Center, Dallas, TX), anti-GAPDH (Millipore Bioscience Research Reagents), anti-actin or anti-tubulin (Sigma) antibody. Densitometric analysis was performed using NIH ImageJ software.
CSF collection.
CSF was isolated from the cisterna magna as previously described (DeMattos et al., 2002). Briefly, the meninges overlying the cisterna magna were exposed and cleaned in anesthetized animals. A 30G1/2 needle was used to puncture the arachnoid membrane covering the cisterna, and the CSF was collected with a polypropylene narrow bore pipette over a period of 2 min.
ApoA-I, ApoE and albumin analysis in the CSF.
Three microliters of CSF per sample were mixed with 10× nondenaturing loading dye (bromophenol blue, 40% glycerol and 1 m Tris, pH 6.8) and separated by 6% native-PAGE, transferred to PVDF membranes and then probed with either 1:500 anti-ApoE (Santa Cruz) or 1:500 anti-ApoA-I (Biodesign) antibody for 2 h. Membranes were washed with PBS-T (PBS with 0.1% Tween 20) four times for 8 min each and incubated for 1 h with 1:1000 horseradish peroxidase (HRP)-labeled anti-goat secondary antibody (Santa Cruz) or 1:1000 anti-rabbit antibody (Santa Cruz), respectively. Sizes of particles were determined by comparison with proteins of known molecular diameter in the GE Healthcare Bio-Science native high molecular weight marker.
CSF and blood were collected from control and Abca1−B/−B mice. Albumin levels were analyzed using the mouse albumin quantitation kit from Bethyl Laboratories.
Capillary depletion.
Brain capillaries were isolated using a modified depletion method from Triguero et al.(Triguero et al., 1990). Mice were anesthetized and perfused with ice-cold PBS for 2 min. Brains were dissected out quickly and meninges were removed. Pooled brains from control and Abca1−B/−B mice were homogenized in a glass homogenizer in 2 volumes of physiological buffer (10 mm HEPES, 141 mm NaCl, 4 mm KCl, 1 mm NaH2PO4, 2.8 mm CaCl2, 1 mm MgSO4, 10 mm d-glucose, protease inhibitors, pH 7.4) with 10 strokes on ice. Then 1.5 volumes of cold 26% dextran were added for a final dextran concentration of 16% and the tissue was homogenized with 3 more strokes. The homogenate was centrifuged at 1200 × g for 30 min at 4°C in a swinging-bucket rotor (Beckman Coulter). The supernatant containing brain tissue and the pellet containing the capillaries were carefully separated. To ensure that the capillary pellet was not contaminated by brain tissue, the levels of the platelet endothelial cell adhesion molecule-1 (PECAM-1), a BCECs marker, were analyzed by Western blotting.
Behavioral analysis.
All animal procedures were conducted in agreement with the guidelines of the Animal Care Committee at the University of British Columbia. Locomotor activity in the open-field apparatus (Med Associates) was analyzed in male control and Abca1−B/−B mice at 3 months. Mice were transferred to the procedure room and allowed to acclimatize for 1 h. Each mouse was then placed in the middle of the open-field box and distance traveled, rearing, stereotypic counts and the speed of movement were recorded in a 1 h session.
Acoustic startle (AS) and prepulse inhibition (PPI) of AS were analyzed at 6 months of age using the startle response system (SR-Lab, San Diego Instruments). Mice were placed into startle chambers and given a 5 min acclimation period with background noise alone (70 dB). Mice were then presented with 100 startle stimuli of noise intensities ranging from background to 120 dB with a duration of 50 ms, divided into 10 blocks of 10 trials. Each block contained individual trials of each intensity and the trials were pseudorandomized within each block. Intertrial interval was randomized and set between 8 and 32 s. During the PPI test, after the 5 min acclimation period, mice were presented with eight trial blocks. The first and last block consisted of six pulse-alone (120 dB) trials. The other six blocks consisted of six trials each including a no-pulse (background), pulse (120 dB) and four prepulse + pulse trials with prepulse intensities set at 72, 74, 78 or 86 dB for a duration of 20 ms and preceding pulse by 20 ms. Percentage PPI was calculated as [1 − (average prepulse startle amplitude − no pulse)/(average pulse startle amplitude − no pulse)] × 100.
Confocal microscopy.
Mice were deeply anesthetized and perfused with 4% paraformaldehyde (PFA). Brains were dissected and left overnight in 4% PFA before being transferred into 30% sucrose solution. Brains were frozen and sliced on the cryostat into 25 μm coronal sections. Free floating sections were blocked in 1% goat serum in PBS with 0.3% Triton X-100 for 1 h. Sections were incubated with primary antibodies (rabbit anti-CYP46A1, gift from Dr. D. Russell, 1:200), rat anti-glial fibrillary acidic protein (GFAP) (Zymed, 1:50) or rabbit anti-synaptotagmin (Stressgen, 1:200) in 1% goat serum, PBS and 0.3% Triton X-100 overnight at room temperature. Sections were then washed three times in PBS for 10 min and incubated with appropriate secondary fluorescent antibodies (goat anti-rabbit Alexa Fluor 488 or goat anti-rat Alexa Fluor 594, 1:1000) in PBS for 1 h, followed by three 10 min washes in PBS. Sections were dried, mounted and imaged on a Zeiss 510 Meta laser scanning confocal microscope.
Electron microscopy.
Five mice per group were anesthetized and injected with 15 μl of heparin intracardially. Mice were perfused with 4% PFA and 0.125% glutaraldehyde for 20 min at a rate of 6 ml/min. Brains were dissected, left overnight in 4% PFA and 0.125% glutaraldehyde and cut into 400 μm sections on a vibratome. Postfixing, embedding, sectioning and staining were performed at the University of British Columbia BioImaging facility. Briefly, 1 mm2 tissue blocks from layers five and six of the motor cortex (1.1 mm from bregma) were cut out, rinsed in 0.1 m sodium cacodylate buffer and secondary fixed in 1% osmium tetroxide with 1.5% potassium ferricyanide in 0.1 m sodium cacodylate for 2 h. Tissue was washed two times in distilled water and stained en bloc with 2% uranyl acetate for 45 min. Samples were dehydrated in a series of ethanol dilutions and transferred to 100% propylene oxide for 15 min, followed by graded resin infiltration and embedding. Ultrathin sections were prepared on a Leica Ultracut T using 45 degree diamond histoknife. Thin sections were counterstained with Sato's lead. Images were taken using a Hitachi H7600 Transmission Electron Microscope and analyzed using Image J. Synapse numbers were identified at a magnification of 15,000× in a total area of 2000 μm2 per animal. Synapses were identified based on synaptic vesicle and postsynaptic density morphology as either asymmetric (excitatory synapses, with round vesicles and a thick postsynaptic density) or symmetric (inhibitory synapses, with presence of flattened vesicles and no thickening of postsynaptic membrane). Synaptic vesicle numbers were counted manually at 50,000× and were classified either as docked (in close proximity to the presynaptic membrane) or reserve pool. A total of 150 excitatory synapses per each mouse were counted.
Statistical analysis.
All data are presented as means ± SEM. Differences between groups were analyzed using Student's t test. Correlation between fraction of plasma 3H-CEt and brain 3H-CEt uptake was analyzed using linear regression. AS and PPI data were analyzed by two-way repeated measures ANOVA with genotype as a between-subjects factor and AS or PPI as repeated-measures factor. Least-squares differences post hoc comparison was used to detect differences at the p < 0.05 level.
Results
Brain ABCA1 impacts plasma cholesterol levels
Abca1−B/−B mice were generated by crossing loxP-flanked (floxed) ABCA1 mice (Timmins et al., 2005) with transgenic mice expressing Cre recombinase under the control of the neuronal and glial specific nestin promoter (Tronche et al., 1999). Abca1−B/−B mice were born at expected Mendelian frequencies, were fertile and did not differ in weight from littermate controls (floxed ABCA1 mice). Immunoblot analysis showed an absence of ABCA1 protein in all regions of the brain that were examined (Fig. 1A). Nestin expression has been reported in some progenitor cells such as liver oval cells serving as hepatocyte precursors in response to liver injury (Gleiberman et al., 2005). Because hepatic as well as intestinal ABCA1 are critical contributors to plasma HDL levels (Timmins et al., 2005; Brunham et al., 2006), we analyzed ABCA1 levels in these organs and found that they were normal (Fig. 1A). ABCG1 and ABCG4 are other cholesterol transporters expressed in the brain. No change in ABCG1 and ABCG4 mRNA levels were found in the brains of Abca1−B/−B mice (supplemental Fig. 1, available at www.jneurosci.org as supplemental material).
Figure 1.

Brain and plasma cholesterol levels in Abca1−B/−B mice. A, Selective loss of ABCA1 in the brains of Abca1−B/−B mice. Tissue lysates from different brain regions, liver, and small intestine from control (+/+) and Abca1−B/−B mice were analyzed by immunoblotting using a polyclonal antibody for ABCA1. GAPDH was used as a loading control. Densitometry analysis of the liver and intestine blot is shown (means ± SEM, n = 4). B, Cholesterol levels in cortex and hippocampus were analyzed by gas-liquid chromatographic mass spectrometry. **p < 0.01 versus control littermates, n = 6 per group. C, Levels of total cholesterol (TC), HDL-cholesterol (HDL-C), phospholipids (PL), and triglycerides (TG) in plasma measured by enzymatic assays. *p < 0.05, versus control littermates, n = 12–20 per group. Ctx, Cortex; Hipp, hippocampus; Cb, cerebellum; BG, basal ganglia.
To examine the specific impact of brain ABCA1 on central and peripheral cholesterol homeostasis, we measured cholesterol levels in cortex and hippocampus, areas of high ABCA1 expression (Tachikawa et al., 2005) and in plasma of Abca1−B/−B mice. Reduced levels of cholesterol were found in Abca1−B/−B cortex compared with control littermates (Fig. 1B). No difference in brain weight was observed (data not shown). Interestingly, this was associated with a significant reduction in plasma cholesterol levels, including total cholesterol and HDL-cholesterol (Fig. 1C). Consistent with decreased HDL cholesterol, plasma phospholipid levels were reduced in Abca1−B/−B mice. The levels of triglycerides, which are associated with larger lipoprotein particles and chylomicrons, remained unchanged (Fig. 1C). This demonstrates that selective loss of brain ABCA1 activity reduced the levels of circulating cholesterol by ∼25% or 170 μg per ml of plasma.
Brain cholesterol and plant sterol uptake from plasma are increased in Abca1−B/−B mice
Because the brain normally contributes very little to plasma cholesterol levels (Dietschy and Turley, 2004), a reduction in cholesterol transport from the brain because of the absence of ABCA1 was not expected to result in a significant decrease in plasma total and HDL cholesterol levels. We therefore hypothesized that uptake of circulating cholesterol to brain may be increased in Abca1−B/−B mice to compensate for cholesterol transport deficits in the CNS, ultimately resulting in decreased plasma cholesterol levels.
To further investigate this hypothesis, we used a radioisotope labeling technique that allows HDL metabolism to be monitored in vivo (Pittman and Taylor, 1986). This technique uses nonhydrolyzable radioactive tracers which become trapped in tissues after uptake. This allows for the measurement of catabolism of HDL by specific tissues without the release of free label into plasma (Glass et al., 1983a,b; Pittman et al., 1983). HDL cholesterol is taken up by tissues either through HDL holo-particle internalization or selective uptake of esterified cholesterol (cholesteryl ester, CE) via scavenger receptor BI (SR-BI) (Acton et al., 1996). Mice were injected with murine HDL particles double labeled with 125I-TC and 3H-CEt. 125I-TC labels HDL apolipoproteins and 3H-CEt traces the CE moieties of HDL (Pittman and Taylor, 1986). 125I-TC traces the metabolism of HDL holo-particles and the difference between uptake rates of 3H-CEt and 125I-TC reflects tissue-specific selective uptake of CE from HDL particle (Pittman et al., 1987).
The fraction of 3H activity (cpm) remaining in plasma 24 h after injection of labeled HDL was reduced by 28% in Abca1−B/−B compared with control mice (Fig. 2A), suggesting that uptake of 3H-CEt labeled CE from plasma was higher in Abca1−B/−B mice. Interestingly, this decrease in plasma 3H-CEt labeled CE parallels the 27% reduction in HDL cholesterol observed in Abca1−B/−B mice (Fig. 1C). There was no difference in the plasma fraction of 125I cpm at 24 h indicating that increased 3H-CEt clearance in Abca1−B/−B mice was due to selective 3H-CEt uptake and not increased HDL holo-particle uptake.
Figure 2.

Brain selective uptake of CE from plasma HDL. Control (n = 4) and Abca1−B/−B (n = 5) mice were injected with HDL particles labeled with 125I-TC in the apolipoprotein and 3H-CEt in the CE moiety, respectively, and plasma clearance and tissue uptake of 125I-TC and 3H-CEt were measured. Data are shown as means ± SEM. A, Fraction of 3H and 125I activity remaining in plasma at 24 h. B, Brain uptake rate, measured by brain FCR, of 3H-CEt and 125I-TC. C, Selective brain 3H-CE uptake rate. D, Regression analysis of correlation between the fraction of 3H cpm left in plasma at 24 h and brain 3H-CE uptake (black boxes: control mice, gray triangles: Abca1−B/−B mice). *p < 0.05, **p < 0.01 compared with control mice.
Because the tracer activity found in the brain is normally very low (Brundert et al., 2005), we measured 125I-TC and 3H-CEt uptake in whole brains instead of discrete regions to detect the maximum radioactivity. Brain uptake rate of total 3H-CEt, measured by brain FCR (Pittman and Taylor, 1986), was higher in ABCA1−B/−B than in control mice (Fig. 2B). In contrast, no change in brain FCR for 125I-TC was observed in Abca1−B/−B mice confirming that HDL holo-particle uptake was not affected. The rate of selective uptake of 3H-CEt labeled CE was increased by >2-fold in Abca1−B/−B compared with control mice (Fig. 2C). We observed a significant negative correlation (r2 = 0.71, p = 0.01) between the fraction of 3H activity found in plasma 24 h after injection and brain uptake of 3H-CEt (Fig. 2D), indicating that reduced plasma 3H-CEt levels were associated with increased brain 3H-CEt uptake. Importantly, these changes in cholesterol uptake were specific to the brain as no differences in rates of 3H-CEt and 125I-TC uptake were observed between control and Abca1−B/−B mice in other tissues including the liver (2.2 ± 0.6 vs 2.1 ± 0.1 pools/d, p = 0.9) and adrenals (0.018 ± 0.002 vs 0.012 ± 0.002 pools/d, p = 0.1). These findings demonstrate that the metabolism of plasma HDL is altered in Abca1−B/−B mice, such that selective uptake of HDL-associated CE is increased specifically in the brains of these mice.
To further validate these findings, we investigated whether brain uptake of another sterol was elevated in Abca1−B/−B mice. The plant sterol sitosterol is a C-24-alkyl homolog of cholesterol (Berge, 2003). Sitosterol is absorbed in the intestine by the Niemann-Pick C1 Like 1 transporter and although 95% of sitosterol is resecreted into the intestinal lumen (Berge, 2003), the rest is transported in plasma on lipoproteins including HDL (Jansen et al., 2006). Although very low levels of plant sterols are found in the brain, sitosterol is transported to the brain specifically on HDL particles (Jansen et al., 2006) and thus represents an independent marker for brain sterol uptake from plasma HDL. We therefore measured sitosterol levels in brains of Abca1−B/−B mice which exhibited significantly higher levels than control mice in cortex and hippocampus (Fig. 3). Increased brain sitosterol in Abca1−B/−B mice was likely due to an increase in selective uptake from plasma HDL and not a result of a general increase in intestinal absorption as there were no significant differences in plasma sitosterol levels between Abca1−B/−B (3.1 ± 0.5 μg/ml) and control mice (2.7 ± 0.3 μg/ml, p = 0.5). It should be noted that the increase in brain sitosterol uptake is not reflected by a detectable reduction in plasma sitosterol because of the 100-fold higher sitosterol levels in plasma compared with brain.
Figure 3.

Brain sitosterol levels. Sitosterol levels were analyzed by GC-MS in cortex (Ctx) and hippocampus (Hipp) of control and Abca1−B/−B mice. Data are shown as means ± SEM. *p < 0.05, ***p < 0.001 versus control mice; n = 6 per group.
To exclude global blood–brain barrier disruption as a possible cause for increased brain cholesterol and sitosterol uptake in Abca1−B/−B mice, the albumin CSF/plasma ratio was measured. CSF albumin is derived from plasma and increased permeability of the blood–brain barrier would be expected to increase the albumin CSF/plasma concentration quotient (Bowman et al., 2007). There was no difference in the CSF/plasma albumin ratio between control and Abca1−B/−B mice (0.0023 ± 0.0003 vs 0.0018 ± 0.0002, p = 0.18), indicating that the integrity of the blood–brain barrier was not compromised.
Abca1−B/−B mice have increased levels of SR-BI in brain capillaries and ApoA-I in brain and CSF
SR-BI mediates the selective cellular uptake of CE from HDL particles (Acton et al., 1996). Because functional SR-BI is expressed in BCECs (Goti et al., 2001), SR-BI may facilitate increased brain CE uptake in Abca1−B/−B mice. Western blot analysis of capillaries isolated from whole brains demonstrated a twofold increase in SR-BI protein levels in Abca1−B/−B mice (Fig. 4B). ABCA1 also increases flux of cholesterol across membranes, but we did not observe a change in the levels of ABCA1 in the BCECs of Abca1−B/−B mice (Fig. 4B) suggesting that upregulation of ABCA1 at the blood–brain barrier is not required for increased brain cholesterol uptake.
Figure 4.

Levels of SR-BI in brain capillaries from Abca1−B/−B mice. Brain capillaries were isolated from control and Abca1−B/−B mice. A, A representative of a Western blot for the BCEC marker PECAM-1. PECAM-1 signal was found in the isolated capillaries (lanes 5–8) in control and Abca1−B/−B mice but not in the capillary-depleted brain tissue (lanes 1–4). GAPDH was used as loading control. B, Capillaries were immunoblotted for SR-BI and ABCA1 (n = 4 per group). Actin was used as a loading control. Graph shows means ± SEM of densitometric measurements of SR-BI and ABCA1 signal intensity. Blots are representative of several experiments. **p < 0.01 compared with control mice.
We measured the levels of brain ApoE and ApoA-I to determine whether these apolipoproteins are involved in increased brain cholesterol uptake in Abca1−B/−B mice. In agreement with the role for ABCA1 in the secretion of ApoE from astrocytes and the regulation of brain ApoE levels (Hirsch-Reinshagen et al., 2004; Wahrle et al., 2004), Western blot analysis demonstrated reduced levels of ApoE in the brains of Abca1−B/−B mice (Fig. 5A). In contrast, the levels of brain ApoA-I were significantly elevated. Because ApoA-I is not synthesized in the CNS (Elshourbagy et al., 1985) and we found no detectable levels of ApoA-I mRNA in brains of Abca1−B/−B mice (data not shown), this increase is either due to increased ApoA-I uptake from peripheral circulation or secretion from BCECs. However, as shown in Figure 2B, there was no change in the uptake of 125I-TC labeled HDL apolipoprotein in Abca1−B/−B mice. Considering that most of the apolipoprotein content in HDL is ApoA-I, this suggests that the major source of elevated brain ApoA-I in Abca1−B/−B mice are likely BCECs. However, we were unable to detect a consistent and statistically significant increase in ApoA-I protein levels in BCECs by Western blotting (data not shown). This may be due to rapid secretion of ApoA-I from BCECs and different rates of secretion in individual mice.
Figure 5.

ApoE and ApoA-I levels in the brain and CSF of Abca1−B/−B mice. A, Immunoblots of brain tissue from control and Abca1−B/−B mice using ApoE and ApoA-I specific antibodies. Tubulin was used as a loading control. Densitometry analysis demonstrated a 40% decrease in ApoE and a 250% increase in ApoA-I signal intensity in Abca1−B/−B mice compared with control mice. Graph shows means ± SEM (*p < 0.05, **p < 0.01, n = 3 per group). B, CSF samples (3 μl) were run on a nondenaturing gel and probed for ApoE (left) and ApoA-I (right).
In mice, ApoE and ApoA-I in the CSF associate with HDL particles 10–17 and 10 nm in diameter, respectively (Wahrle et al., 2004). We analyzed the levels and size distribution of CSF ApoE and ApoA-I containing lipoproteins by nondenaturing gel electrophoresis. As expected, ApoE levels were reduced in CSF of Abca1−B/−B mice (Fig. 5B). The levels of CSF ApoA-I, however, were considerably elevated. ApoA-I was associated with HDL particles 8–10 nm in diameter, similar to those in control animals and as reported in wild-type mice (Wahrle et al., 2004). This demonstrates that although ApoA-I levels are significantly increased in the brain and CSF of Abca1−B/−B mice, this ApoA-I is predominately associated with small, less lipidated HDL particles. Notably, the total levels of cholesterol in the CSF were similar in control (9.5 ± 0.5 μg/ml) and Abca1−B/−B mice (8.7 ± 0.7 μg/ml, p = 0.4).
Brain cholesterol turnover is increased in Abca1−B/−B mice
Cholesterol transport across the basolateral membrane of cultured BCECs can be induced in vitro by the brain cholesterol-specific metabolite and nuclear liver-X-receptor agonist 24S-OH cholesterol (Panzenboeck et al., 2006). This suggests that elevated brain 24S-OH cholesterol production is a potential mechanism for activating cholesterol transport across the blood–brain barrier in Abca1−B/−B mice.
Supporting this hypothesis is our observation that Abca1−B/−B mice displayed an increased ratio of 24S-OH cholesterol to cholesterol (Fig. 6A, left) compared with control mice in both cortex and hippocampus. Plasma levels of 24S-OH cholesterol were also increased in Abca1−B/−B mice (Fig. 6A, right).
Figure 6.
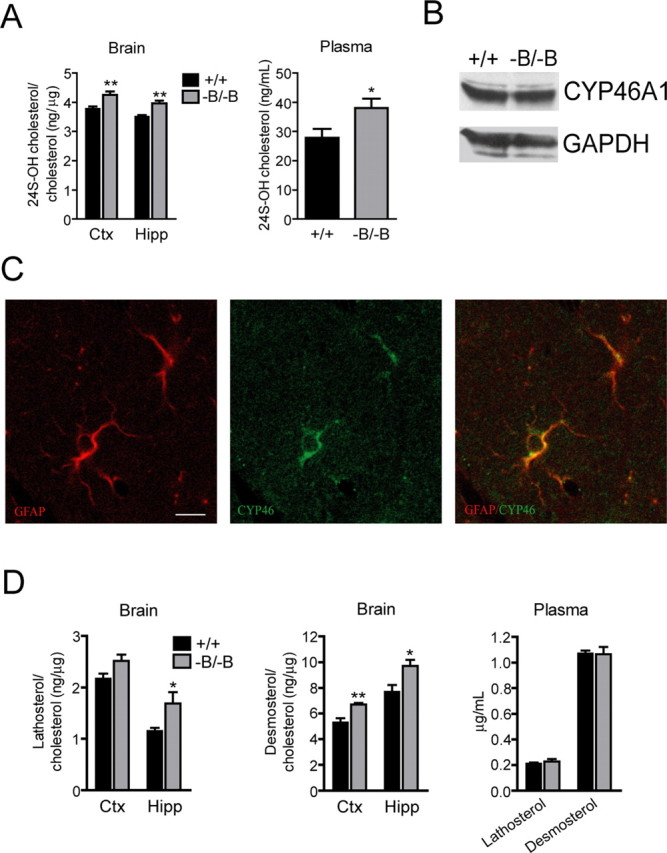
Brain cholesterol turnover in Abca1−B/−B mice. Brain and plasma levels of cholesterol metabolite 24S-OH cholesterol and cholesterol precursors lathosterol and desmosterol were analyzed by GC-MS (n = 6 per group, means ± SEM). A, Left, The ratio of 24S-OH cholesterol to cholesterol in cortex and hippocampus of control and ABCA1−B/−B mice. A, Right, Plasma levels of 24S-OH cholesterol. B, Western blot shows cortical CYP46A1 levels (representative of several experiments). C, Confocal images of hippocampal sections from a wild-type mouse show colocalization of the astrocytic marker GFAP (red) and CYP46A1 (green). Scale bar, 10 μm. D, Graphs show ratios of lathosterol to cholesterol and desmosterol to cholesterol in hippocampus and cortex as well as plasma lathosterol and desmosterol levels. Ctx, Cortex; Hipp, hippocampus. *p < 0.05, **p < 0.01 versus control mice.
There was no change in CYP46A1 mass in the brains of Abca1−B/−B mice (Fig. 6B), suggesting that increased 24S-OH cholesterol production is either a result of increased neuronal CYP46A1 enzymatic activity or reciprocal changes in CYP46A1 expression in neurons and glia. Although CYP46A1 is a neuron-specific enzyme (Lund et al., 1999), glial CYP46A1 expression was reported in brains of Alzheimer patients (Brown, III et al., 2004). Immunofluorescence analysis of brain sections from wild-type mice indicated that in addition to distribution in neurons (data not shown), CYP46A1 colocalizes with the astrocytic marker GFAP (Fig. 6C) indicating that glial cells can also express CYP46A1 and may therefore contribute to brain 24S-OH cholesterol production.
Brain cholesterol homeostasis is under tight regulatory control whereby changes in cholesterol metabolism are reflected in changes in synthesis (Lund et al., 2003). Because of the exquisite regulation of brain cholesterol balance and the findings of reduced cortical cholesterol levels and increased metabolism to 24S-OH cholesterol, we next determined whether this was accompanied by alterations in cholesterol synthesis in Abca1−B/−B mice. To assess this, the levels of cholesterol biosynthetic intermediates (Lütjohann et al., 2002) were measured in brain and plasma. The lathosterol to cholesterol ratio was significantly increased in Abca1−B/−B mice compared with control mice in hippocampus (Fig. 6D, left). Higher ratios of desmosterol to cholesterol were observed in ABCA1−B/−B compared with control mice in cortex and hippocampus (Fig. 6D, middle). Plasma lathosterol and desmosterol levels were unchanged, indicating that the increase in cholesterol synthesis was specific to the brain (Fig. 6D, right). Lanosterol is the first sterol precursor in the cholesterol biosynthetic pathway (Lütjohann et al., 2002). No change in the lanosterol to cholesterol ratio in the brain was observed in Abca1−B/−B mice (data not shown), suggesting that cholesterol biosynthesis was selectively increased in post-lanosterol steps, when the biosynthetic pathway diverges and continues via the lathosterol and desmosterol routes (Lütjohann et al., 2002).
We considered the possibility that increased brain sterol precursor to cholesterol levels could be a result of reduced conversion of lathosterol and desmosterol to cholesterol. However, reduced desmosterol conversion would be associated with reduced levels of 3beta-hydroxysterol Delta24-reductase (DHCR24) also known as seladin-1 and increased levels of all precursors carrying a double bond at C-24 including lanosterol (Waterham et al., 2001). We also measured the levels of dihydrolanosterol, the post-lanosterol step and a lathosterol precursor (Lindenthal et al., 2002) and found no changes in Abca1−B/−B cortex or hippocampus (data not shown). This suggests that the increase in cholesterol precursors is not due to their decreased degradation.
Abca1−B/−B mice exhibit abnormal behavior and synaptic structure
In view of the critical role of cholesterol in brain development, synaptogenesis and synaptic activity (Mauch et al., 2001; Dietschy and Turley, 2004), changes in brain cholesterol homeostasis were postulated to influence behavioral function and synaptic development in Abca1−B/−B mice. We assessed a wide range of behaviors in control and Abca1−B/−B mice. Spontaneous locomotor activity was attenuated in Abca1−B/−B mice in the open-field test (Fig. 7A), including a reduction in horizontal (distance traveled) and vertical (number of rears) activity. Stereotypic behavior and movement velocity were also reduced in Abca1−B/−B mice (stereotypic counts: 10,060 ± 329 in control vs 6896 ± 378 in Abca1−B/−B mice, p < 0.001; velocity: 47.55 ± 2.6 cm/min in control vs 35.2 ± 1.4 cm/min in Abca1−B/−B mice, p < 0.0001). Although motor coordination measured by rotarod performance was normal at 3 months of age, Abca1−B/−B mice showed reduced latency to fall at 9 months (data not shown). No changes in spatial learning in the Morris water maze were observed between control and Abca1−B/−B mice (data not shown).
Figure 7.
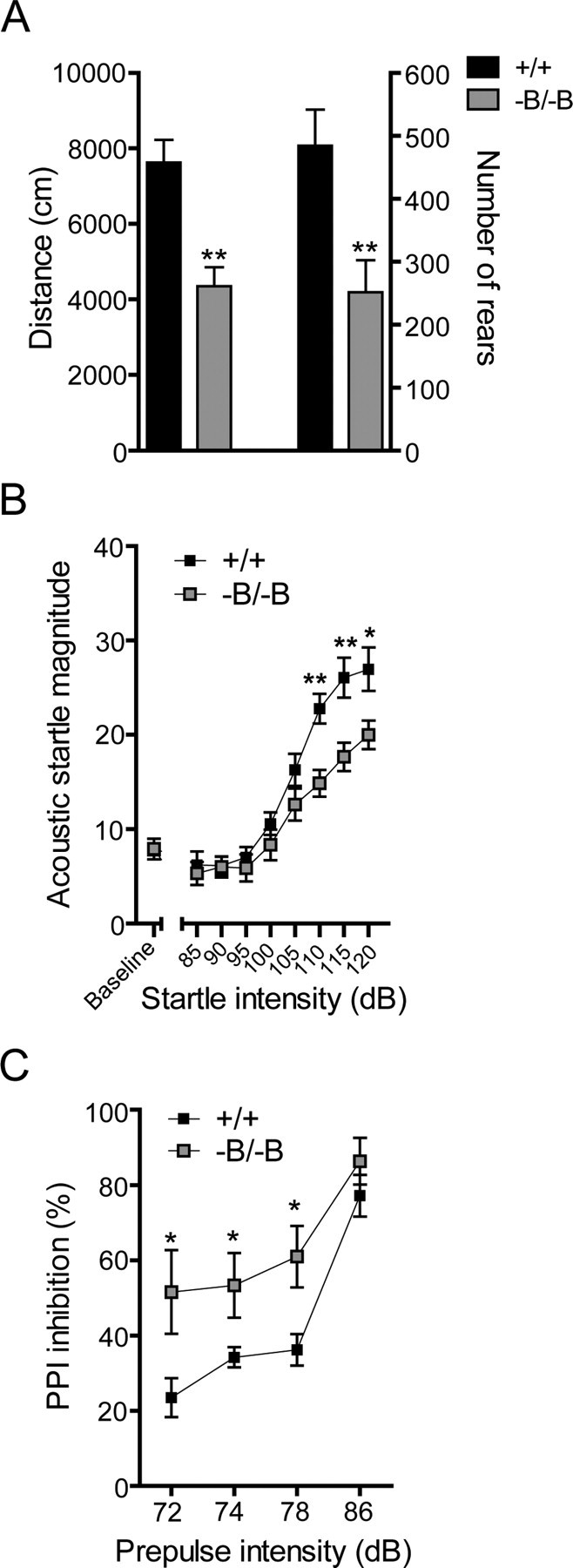
Behavioral function in Abca1−B/−B mice. A, Spontaneous locomotor activity was measured in 3-month-old control and Abca1−B/−B mice during a 1 h session in an open field chamber. Specific aspects of locomotor activity including horizontal (distance) and vertical (number of rears) activity were analyzed (n = 11–12). Data show means ± SEM B, Acoustic startle response at various startle intensities. C, PPI of acoustic startle measured at prepulse intensities of 72, 74, 78, and 86 dB (n = 11 per group). *p < 0.05, **p < 0.01, ***p < 0.001 compared with control mice.
Sensorimotor function was assessed with the AS and the PPI of AS tests. A significant reduction in AS magnitude was observed in Abca1−B/−B mice at high intensity startle stimuli (110 dB-120 dB) (Fig. 7B). Conversely, PPI was increased in Abca1−B/−B mice at 2 dB, 4 dB and 8 dB prepulse intensities (Fig. 7C). The behavioral data indicate that loss of brain ABCA1 affects motor and sensorimotor functions that are mainly controlled by forebrain structures.
We next investigated whether synaptic alterations in the cortex are associated with disturbances in motor and sensorimotor function in the absence of brain ABCA1. Immunofluorescence analysis of synaptotagmin, a presynaptic protein associated with synaptic vesicles, revealed changes in neuronal distribution including reduced staining intensity of synaptic puncta in cortex of Abca1−B/−B mice (Fig. 8A) suggesting a possible presynaptic deficit. We therefore examined the synaptic ultrastructure in the cortex using electron microscopy to analyze the numbers of synapses and synaptic vesicles (Fig. 8B). Quantitative ultrastructural analysis revealed a 16% decrease in the number of synapses in Abca1−B/−B mice (Fig. 8C), which could be attributed to the reduction in the number of excitatory synapses. Synapses with perforated postsynaptic densities represent an intermediate between a continuous synapse and a divided synapse that has formed new synaptic contacts on two separate spines of the same dendrite (Carlin and Siekevitz, 1983). The proportion of synapses containing a perforated postsynaptic density was 20% in control mice but only 12% in Abca1−B/−B mice (Fig. 8D). Abca1−B/−B mice also had an 18% decrease in the average number of synaptic vesicles per synapse in the reserve vesicle pool and a reduction in the number of docked vesicles (Fig. 8E).
Figure 8.
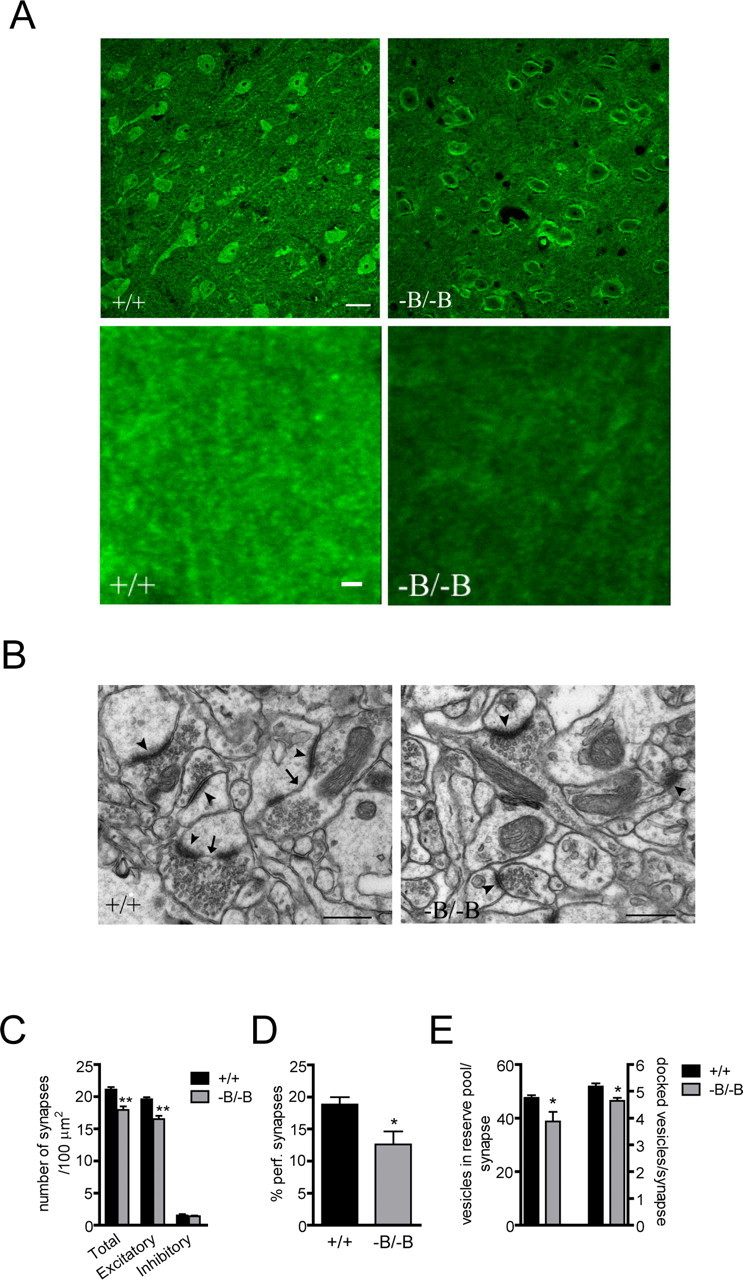
Morphological and ultrastructural synaptic analysis in Abca1−B/−B mice. A, Confocal images showing neuronal synaptotagmin immunofluorescence in the cortex of control and Abca1−B/−B mice. Top images show neuronal synaptotagmin distribution and bottom images show synaptic puncta staining at high magnification. Scale bar, top images: 10 μm; bottom images: 2 μm. B, Transmission electron microscope images of synapses in the cortex of control (left) and Abca1−B/−B (right) mice. A synapse was identified by a well defined postsynaptic density and at least three docked vesicles (arrowheads). C, The number of synapses was counted in a total area of 2000 μm2. Synapses were identified either as excitatory or inhibitory based on morphology (see Materials and Methods). D, The number of perforated synapses, i.e., synapses containing segmented postsynaptic density (arrows). E, The numbers of docked vesicles and the vesicles in the reserve pool were counted in 150 excitatory synapses from each mouse. Data show means ± SEM. *p < 0.05 and **p < 0.01 compared with control mice. n = 5 per group. Scale bar, 500 nm.
Hence, attenuated locomotor activity and altered sensorimotor function in Abca1−B/−B mice is associated with reduced number of excitatory synapses and vesicles in the cortex.
Discussion
We report that changes in brain cholesterol homeostasis induced by loss of brain ABCA1 activity can lead to alterations in cholesterol transport across the blood–brain barrier. Abca1−B/−B mice demonstrate increased brain uptake of esterified cholesterol from plasma HDL which is associated with reduced plasma HDL cholesterol levels. Inactivation of brain ABCA1 also leads to motor and sensorimotor behavioral and synaptic changes. These findings implicate ABCA1 as a key regulator of brain cholesterol homeostasis in addition to its established role in the periphery and demonstrate a role for ABCA1 in the regulation of neuronal function.
The brain cholesterol pool separates from the rest of the body during the formation of the blood–brain barrier (Dietschy and Turley, 2004). CNS relies on in situ cholesterol synthesis and efficient transport and recycling mechanisms (Dietschy and Turley, 2004). A key unexpected finding is that Abca1−B/−B mice have reduced plasma HDL cholesterol levels. Impaired cholesterol efflux from the CNS cannot explain this reduction because of the negligible contribution of the brain to circulating cholesterol concentration (Björkhem and Meaney, 2004; Dietschy and Turley, 2004). We previously demonstrated that the liver and intestine are major contributors to plasma HDL (Timmins et al., 2005; Brunham et al., 2006) and hepatic and intestinal ABCA1 levels were unaffected in Abca1−B/−B mice. The reduced fraction of labeled CE in plasma in Abca1−B/−B mice after 24 h points to increased HDL CE uptake from plasma. We hypothesized that there is increased brain cholesterol uptake from plasma HDL in Abca1−B/−B mice to compensate for the reduction in brain cholesterol concentration highlighting the evolutionary requirement to maintain normal cholesterol homeostasis in the brain. There is no significant brain cholesterol uptake from plasma under conditions of normal brain cholesterol metabolism (Dietschy and Turley, 2004) despite the fact that BCEC basolateral and apical membranes express proteins involved in cholesterol and lipoprotein transport including ABCA1, SR-BI and low density lipoprotein (LDL) receptor (Dehouck et al., 1997; Goti et al., 2001; Panzenboeck et al., 2002). Uptake and transport of HDL-associated esterified cholesterol (Goti et al., 2001) and LDL (Dehouck et al., 1997) by cultured BCECs were reported. Most ABCA1 is expressed in the basolateral membrane in BCECs rather than the apical membrane whereas most SR-BI is expressed in the apical membrane (Panzenboeck et al., 2006). This orientation of ABCA1 and SR-BI implies the existence of a mechanism for plasma-to-brain cholesterol flux. Interestingly, 24S-OH cholesterol stimulates cholesterol transport across the basolateral membrane of BCECs in vitro (Panzenboeck et al., 2002, 2006) and the CSF-facing membrane of the choroid plexus epithelial cells constituting the blood-CSF barrier (Fujiyoshi et al., 2007). Hence, barriers of the CNS contain a mechanism for mediating a potential increase in cholesterol uptake.
Loss of brain ABCA1 is associated with increased 24S-OH cholesterol production and increased brain selective uptake of radioisotope-labeled esterified cholesterol from plasma HDL. No increase in CE uptake was found in other organs and lower plasma levels of labeled CE correlated with its higher levels in the brain, suggesting that increased brain CE uptake is the primary mechanism underlying the reduced plasma HDL cholesterol levels. Although CE selective uptake rate in the whole brain was increased by two-fold in Abca1−B/−B mice, it was still considerably lower than in the liver or adrenals. ∼80% of brain cholesterol in the mouse is in myelin and is subject to slow turnover rates (Dietschy and Turley, 2004). In fact, as little as 2% of total brain cholesterol may be present in metabolically active neurons where turnover is high (Dietschy and Turley, 2004). Hence, although the relative uptake rate of CE in whole brain is low, a two-fold increase in uptake in Abca1−B/−B mice could significantly impact the cholesterol pool and metabolism in neurons.
SR-BI levels are increased in brain capillaries from Abca1−B/−B mice, suggesting that upregulation of SR-BI at the blood–brain barrier mediates the increased brain CE uptake. Although SR-BI knock-out mice do not exhibit changes in brain cholesterol levels or synthesis rate (Quan et al., 2003), reduced brain selective CE uptake was observed in mice lacking SR-BI (Brundert et al., 2005). Elevated brain sitosterol levels support the finding of increased brain sterol uptake. HDL sitosterol is esterified by lecithin:cholesterol acyltransferase almost as efficiently as cholesterol (Temel et al., 2003), suggesting that, once esterified, it may be taken up from HDL in a manner similar to that of CE.
After uptake by BCECs, CE is presumably hydrolyzed to remove the ester bond to allow the efficient transport of free cholesterol to an apolipoprotein acceptor. We observed a marked increase in ApoA-I levels in brain tissue and CSF of Abca1−B/−B mice. Lipid-free ApoA-I is lipidated exclusively by ABCA1 (Oram and Heinecke, 2005). Therefore, in the absence of ABCA1 in brain parenchyma, ApoA-I is most likely lipidated by ABCA1 in the BCECs. We propose that levels of ApoA-I are elevated to facilitate the increase in cholesterol transport from plasma into the brain.
The reason underlying the need for increased brain cholesterol uptake in Abca1−B/−B mice may be a deficit in cholesterol transport between glial cells and neurons. Glia-derived cholesterol carried by ApoE plays a critical role in synaptic activity (Mauch et al., 2001). Because ABCA1 mediates cholesterol transport from glia to apoE (Hirsch-Reinshagen et al., 2004; Wahrle et al., 2004), a reduction in brain ABCA1 activity may limit the neuronal cholesterol supply from glia. In fact, the absence of ABCA1 inhibits cholesterol efflux from cultured glial cells leading to lipid accumulation (Hirsch-Reinshagen et al., 2004).
Another way to deal with neuronal cholesterol deficit could be to increase synthesis as observed in Abca1−B/−B mice. Elevated production of 24S-OH cholesterol could reflect increased cholesterol turnover in glia such as astrocytes. Although CYP46A1 is expressed predominantly in neurons (Lund et al., 1999), glial CYP46A1 expression was reported in AD patients (Brown, III et al., 2004). CYP46A1 is expressed in GFAP-positive cells of control and Abca1−B/−B mice. Hence, astrocytes may express small levels of CYP46A1 that could be activated in response to cholesterol accumulation.
Brain cholesterol uptake is not affected by fluctuations in plasma cholesterol levels (Quan et al., 2003; Dietschy and Turley, 2004). Increased cholesterol uptake in Abca1−B/−B mice implies a mechanism for altered plasma to brain HDL cholesterol transport regulated specifically by the CNS. Hence, if brain cholesterol balance is perturbed as seen in Abca1−B/−B mice, the CNS may initiate a sequence of events to increase cholesterol transport across the BCECs in an attempt to compensate for neuronal cholesterol deficits.
Cholesterol impacts synaptic activity, vesicle release (Mauch et al., 2001) and neurotransmitter receptor and transporter function (Butchbach et al., 2004; Magnani et al., 2004; Ponce et al., 2008). Cholesterol is implicated in the regulation of motor and cognitive function because of its role in myelination (Saher et al., 2005) and isoprenoid synthesis (Kotti et al., 2006). Brain ABCA1 may affect behavioral function via its regulation of brain cholesterol transport and metabolism. Behavioral and synaptic abnormalities observed in Abca1−B/−B mice suggest that ABCA1 activity is important for normal neuronal function. Reduced PPI is seen in neuropsychiatric and neurological disorders including schizophrenia, Huntington disease and Tourette's syndrome (Castellanos et al., 1996; Geyer et al., 2002). Antipsychotic drugs reduce locomotor activity and increase PPI in mouse models of schizophrenia (Ralph et al., 2001; Maehara et al., 2008). Reduced activity and increased PPI in Abca1−B/−B mice implicate a specific role for brain ABCA1 in sensorimotor gating and suggest that ABCA1 may affect behaviors associated with neuropsychiatric disorders.
The attenuation in the number of cortical synapses and synaptic vesicles in Abca1−B/−B mice supports the hypothesis that loss of brain ABCA1 likely leads to a deficit in cholesterol trafficking from glia to neurons which may affect synaptogenesis (Barres and Smith, 2001). The reduction in perforated synapses suggests that ABCA1 is involved in the regulation of synaptic plasticity in the cortex. ABCA1 activity may also impact neuronal membrane lipid composition which could have presynaptic and postsynaptic effects on neurotransmitter release and signaling. Moreover, ApoE receptors are involved in neuronal signaling and synaptic plasticity (Herz and Chen, 2006) and attenuated brain ApoE levels may contribute to functional deficits in Abca1−B/−B mice.
Our findings raise the important question of whether cholesterol transport across the blood–brain barrier is altered in neurodegenerative diseases. ABCA1 gene polymorphisms, including loss-of-function mutations, have been associated with an increased risk for Alzheimer's disease (AD) (Chu et al., 2007; Rodríguez-Rodríguez et al., 2007). Reduced plasma HDL levels are reported in AD (Merched et al., 2000) and patients with memory decline (Singh-Manoux et al., 2008). Interestingly, elevated 24S-OH cholesterol levels in plasma and CSF (Lütjohann et al., 2000; Papassotiropoulos et al., 2002; Schönknecht et al., 2002) have also been observed in AD. These data, in addition to the reports suggesting that overexpression of ABCA1 in the brain decreases amyloid deposition (Wahrle et al., 2008) and improves neuronal function (Jiang et al., 2008), all point to significant disturbances in CNS cholesterol homeostasis that may be influenced at least in part by ABCA1 function. Together, these data encourage further evaluation of brain ABCA1 as a therapeutic target in neurodegenerative disease.
Footnotes
This work was supported by the Canadian Institutes of Health Research (M.R.H.) and National Institutes of Health Grant HL 49373 (J.S.P.). J.M.K. holds postdoctoral fellowships from the Heart and Stroke Foundation of Canada and the Michael Smith Foundation for Health Research. M.R.H. holds a Canada Research Chair in Human Genetics. We thank Silvia Friedrichs, Anja Kerksiek for technical assistance, Rochelle Hines for advice with the electron microscopy experiment, and Dr. David Russell for the CYP46A1 antibody.
References
- Acton S, Rigotti A, Landschulz KT, Xu S, Hobbs HH, Krieger M. Identification of scavenger receptor SR-BI as a high density lipoprotein receptor. Science. 1996;271:518–520. doi: 10.1126/science.271.5248.518. [DOI] [PubMed] [Google Scholar]
- Barres BA, Smith SJ. Neurobiology. Cholesterol—making or breaking the synapse. Science. 2001;294:1296–1297. doi: 10.1126/science.1066724. [DOI] [PubMed] [Google Scholar]
- Berge KE. Sitosterolemia: a gateway to new knowledge about cholesterol metabolism. Ann Med. 2003;35:502–511. doi: 10.1080/07853890310014588. [DOI] [PubMed] [Google Scholar]
- Björkhem I, Meaney S. Brain cholesterol: long secret life behind a barrier. Arterioscler Thromb Vasc Biol. 2004;24:806–815. doi: 10.1161/01.ATV.0000120374.59826.1b. [DOI] [PubMed] [Google Scholar]
- Bowman GL, Kaye JA, Moore M, Waichunas D, Carlson NE, Quinn JF. Blood-brain barrier impairment in Alzheimer disease: stability and functional significance. Neurology. 2007;68:1809–1814. doi: 10.1212/01.wnl.0000262031.18018.1a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks-Wilson A, Marcil M, Clee SM, Zhang LH, Roomp K, van Dam M, Yu L, Brewer C, Collins JA, Molhuizen HO, Loubser O, Ouelette BF, Fichter K, Ashbourne-Excoffon KJ, Sensen CW, Scherer S, Mott S, Denis M, Martindale D, Frohlich J, et al. Mutations in ABC1 in Tangier disease and familial high-density lipoprotein deficiency. Nat Genet. 1999;22:336–345. doi: 10.1038/11905. [DOI] [PubMed] [Google Scholar]
- Brown J, 3rd, Theisler C, Silberman S, Magnuson D, Gottardi-Littell N, Lee JM, Yager D, Crowley J, Sambamurti K, Rahman MM, Reiss AB, Eckman CB, Wolozin B. Differential expression of cholesterol hydroxylases in Alzheimer's disease. J Biol Chem. 2004;279:34674–34681. doi: 10.1074/jbc.M402324200. [DOI] [PubMed] [Google Scholar]
- Brundert M, Ewert A, Heeren J, Behrendt B, Ramakrishnan R, Greten H, Merkel M, Rinninger F. Scavenger receptor class B type I mediates the selective uptake of high-density lipoprotein-associated cholesteryl ester by the liver in mice. Arterioscler Thromb Vasc Biol. 2005;25:143–148. doi: 10.1161/01.ATV.0000149381.16166.c6. [DOI] [PubMed] [Google Scholar]
- Brunham LR, Kruit JK, Iqbal J, Fievet C, Timmins JM, Pape TD, Coburn BA, Bissada N, Staels B, Groen AK, Hussain MM, Parks JS, Kuipers F, Hayden MR. Intestinal ABCA1 directly contributes to HDL biogenesis in vivo. J Clin Invest. 2006;116:1052–1062. doi: 10.1172/JCI27352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunham LR, Kruit JK, Pape TD, Timmins JM, Reuwer AQ, Vasanji Z, Marsh BJ, Rodrigues B, Johnson JD, Parks JS, Verchere CB, Hayden MR. Beta-cell ABCA1 influences insulin secretion, glucose homeostasis and response to thiazolidinedione treatment. Nat Med. 2007;13:340–347. doi: 10.1038/nm1546. [DOI] [PubMed] [Google Scholar]
- Butchbach ME, Tian G, Guo H, Lin CL. Association of excitatory amino acid transporters, especially EAAT2, with cholesterol-rich lipid raft microdomains: importance for excitatory amino acid transporter localization and function. J Biol Chem. 2004;279:34388–34396. doi: 10.1074/jbc.M403938200. [DOI] [PubMed] [Google Scholar]
- Carlin RK, Siekevitz P. Plasticity in the central nervous system: do synapses divide? Proc Natl Acad Sci U S A. 1983;80:3517–3521. doi: 10.1073/pnas.80.11.3517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellanos FX, Fine EJ, Kaysen D, Marsh WL, Rapoport JL, Hallett M. Sensorimotor gating in boys with Tourette's syndrome and ADHD: preliminary results. Biol Psychiatry. 1996;39:33–41. doi: 10.1016/0006-3223(95)00101-8. [DOI] [PubMed] [Google Scholar]
- Chu LW, Li Y, Li Z, Tang AY, Cheung BM, Leung RY, Yik PY, Jin DY, Song YQ. A novel intronic polymorphism of ABCA1 gene reveals risk for sporadic Alzheimer's disease in Chinese. Am J Med Genet B Neuropsychiatr Genet. 2007;144B:1007–1013. doi: 10.1002/ajmg.b.30525. [DOI] [PubMed] [Google Scholar]
- Dehouck B, Fenart L, Dehouck MP, Pierce A, Torpier G, Cecchelli R. A new function for the LDL receptor: transcytosis of LDL across the blood-brain barrier. J Cell Biol. 1997;138:877–889. doi: 10.1083/jcb.138.4.877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeMattos RB, Bales KR, Parsadanian M, O'Dell MA, Foss EM, Paul SM, Holtzman DM. Plaque-associated disruption of CSF and plasma amyloid-beta (Abeta) equilibrium in a mouse model of Alzheimer's disease. J Neurochem. 2002;81:229–236. doi: 10.1046/j.1471-4159.2002.00889.x. [DOI] [PubMed] [Google Scholar]
- Dietschy JM, Turley SD. Thematic review series: brain Lipids. Cholesterol metabolism in the central nervous system during early development and in the mature animal. J Lipid Res. 2004;45:1375–1397. doi: 10.1194/jlr.R400004-JLR200. [DOI] [PubMed] [Google Scholar]
- Elshourbagy NA, Boguski MS, Liao WS, Jefferson LS, Gordon JI, Taylor JM. Expression of rat apolipoprotein A-IV and A-I genes: mRNA induction during development and in response to glucocorticoids and insulin. Proc Natl Acad Sci U S A. 1985;82:8242–8246. doi: 10.1073/pnas.82.23.8242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiyoshi M, Ohtsuki S, Hori S, Tachikawa M, Terasaki T. 24S-hydroxycholesterol induces cholesterol release from choroid plexus epithelial cells in an apical- and apoE isoform-dependent manner concomitantly with the induction of ABCA1 and ABCG1 expression. J Neurochem. 2007;100:968–978. doi: 10.1111/j.1471-4159.2006.04240.x. [DOI] [PubMed] [Google Scholar]
- Geyer MA, McIlwain KL, Paylor R. Mouse genetic models for prepulse inhibition: an early review. Mol Psychiatry. 2002;7:1039–1053. doi: 10.1038/sj.mp.4001159. [DOI] [PubMed] [Google Scholar]
- Glass C, Pittman RC, Weinstein DB, Steinberg D. Dissociation of tissue uptake of cholesterol ester from that of apoprotein A-I of rat plasma high density lipoprotein: selective delivery of cholesterol ester to liver, adrenal, and gonad. Proc Natl Acad Sci U S A. 1983a;80:5435–5439. doi: 10.1073/pnas.80.17.5435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass CK, Pittman RC, Keller GA, Steinberg D. Tissue sites of degradation of apoprotein A-I in the rat. J Biol Chem. 1983b;258:7161–7167. [PubMed] [Google Scholar]
- Gleiberman AS, Encinas JM, Mignone JL, Michurina T, Rosenfeld MG, Enikolopov G. Expression of nestin-green fluorescent protein transgene marks oval cells in the adult liver. Dev Dyn. 2005;234:413–421. doi: 10.1002/dvdy.20536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goritz C, Mauch DH, Pfrieger FW. Multiple mechanisms mediate cholesterol-induced synaptogenesis in a CNS neuron. Mol Cell Neurosci. 2005;29:190–201. doi: 10.1016/j.mcn.2005.02.006. [DOI] [PubMed] [Google Scholar]
- Goti D, Hrzenjak A, Levak-Frank S, Frank S, van der Westhuyzen DR, Malle E, Sattler W. Scavenger receptor class B, type I is expressed in porcine brain capillary endothelial cells and contributes to selective uptake of HDL-associated vitamin E. J Neurochem. 2001;76:498–508. doi: 10.1046/j.1471-4159.2001.00100.x. [DOI] [PubMed] [Google Scholar]
- Herz J, Chen Y. Reelin, lipoprotein receptors and synaptic plasticity. Nat Rev Neurosci. 2006;7:850–859. doi: 10.1038/nrn2009. [DOI] [PubMed] [Google Scholar]
- Hirsch-Reinshagen V, Zhou S, Burgess BL, Bernier L, McIsaac SA, Chan JY, Tansley GH, Cohn JS, Hayden MR, Wellington CL. Deficiency of ABCA1 impairs apolipoprotein E metabolism in brain. J Biol Chem. 2004;279:41197–41207. doi: 10.1074/jbc.M407962200. [DOI] [PubMed] [Google Scholar]
- Jansen PJ, Lütjohann D, Abildayeva K, Vanmierlo T, Plösch T, Plat J, von Bergmann K, Groen AK, Ramaekers FC, Kuipers F, Mulder M. Dietary plant sterols accumulate in the brain. Biochim Biophys Acta. 2006;1761:445–453. doi: 10.1016/j.bbalip.2006.03.015. [DOI] [PubMed] [Google Scholar]
- Jiang Q, Lee CY, Mandrekar S, Wilkinson B, Cramer P, Zelcer N, Mann K, Lamb B, Willson TM, Collins JL, Richardson JC, Smith JD, Comery TA, Riddell D, Holtzman DM, Tontonoz P, Landreth GE. ApoE promotes the proteolytic degradation of Abeta. Neuron. 2008;58:681–693. doi: 10.1016/j.neuron.2008.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotti TJ, Ramirez DM, Pfeiffer BE, Huber KM, Russell DW. Brain cholesterol turnover required for geranylgeraniol production and learning in mice. Proc Natl Acad Sci U S A. 2006;103:3869–3874. doi: 10.1073/pnas.0600316103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindenthal B, Sudhop T, Schiedermaier P, Agnan M, Sauerbruch T, von Bergmann K. Serum plant sterols and biliary cholesterol secretion in humans: studies with ursodeoxycholic acid. J Lipid Res. 2002;43:1072–1077. doi: 10.1194/jlr.m100438-jlr200. [DOI] [PubMed] [Google Scholar]
- Lund EG, Guileyardo JM, Russell DW. cDNA cloning of cholesterol 24-hydroxylase, a mediator of cholesterol homeostasis in the brain. Proc Natl Acad Sci U S A. 1999;96:7238–7243. doi: 10.1073/pnas.96.13.7238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund EG, Xie C, Kotti T, Turley SD, Dietschy JM, Russell DW. Knockout of the cholesterol 24-hydroxylase gene in mice reveals a brain-specific mechanism of cholesterol turnover. J Biol Chem. 2003;278:22980–22988. doi: 10.1074/jbc.M303415200. [DOI] [PubMed] [Google Scholar]
- Lütjohann D, Papassotiropoulos A, Björkhem I, Locatelli S, Bagli M, Oehring RD, Schlegel U, Jessen F, Rao ML, von Bergmann K, Heun R. Plasma 24S-hydroxycholesterol (cerebrosterol) is increased in Alzheimer and vascular demented patients. J Lipid Res. 2000;41:195–198. [PubMed] [Google Scholar]
- Lütjohann D, Brzezinka A, Barth E, Abramowski D, Staufenbiel M, von Bergmann K, Beyreuther K, Multhaup G, Bayer TA. Profile of cholesterol-related sterols in aged amyloid precursor protein transgenic mouse brain. J Lipid Res. 2002;43:1078–1085. doi: 10.1194/jlr.m200071-jlr200. [DOI] [PubMed] [Google Scholar]
- Maehara S, Hikichi H, Satow A, Okuda S, Ohta H. Antipsychotic property of a muscarinic receptor agonist in animal models for schizophrenia. Pharmacol Biochem Behav. 2008;91:140–149. doi: 10.1016/j.pbb.2008.06.023. [DOI] [PubMed] [Google Scholar]
- Magnani F, Tate CG, Wynne S, Williams C, Haase J. Partitioning of the serotonin transporter into lipid microdomains modulates transport of serotonin. J Biol Chem. 2004;279:38770–38778. doi: 10.1074/jbc.M400831200. [DOI] [PubMed] [Google Scholar]
- Mauch DH, Nägler K, Schumacher S, Göritz C, Müller EC, Otto A, Pfrieger FW. CNS synaptogenesis promoted by glia-derived cholesterol. Science. 2001;294:1354–1357. doi: 10.1126/science.294.5545.1354. [DOI] [PubMed] [Google Scholar]
- Merched A, Xia Y, Visvikis S, Serot JM, Siest G. Decreased high-density lipoprotein cholesterol and serum apolipoprotein AI concentrations are highly correlated with the severity of Alzheimer's disease. Neurobiol Aging. 2000;21:27–30. doi: 10.1016/s0197-4580(99)00103-7. [DOI] [PubMed] [Google Scholar]
- Oram JF, Heinecke JW. ATP-binding cassette transporter A1: a cell cholesterol exporter that protects against cardiovascular disease. Physiol Rev. 2005;85:1343–1372. doi: 10.1152/physrev.00005.2005. [DOI] [PubMed] [Google Scholar]
- Panzenboeck U, Balazs Z, Sovic A, Hrzenjak A, Levak-Frank S, Wintersperger A, Malle E, Sattler W. ABCA1 and scavenger receptor class B, type I, are modulators of reverse sterol transport at an in vitro blood-brain barrier constituted of porcine brain capillary endothelial cells. J Biol Chem. 2002;277:42781–42789. doi: 10.1074/jbc.M207601200. [DOI] [PubMed] [Google Scholar]
- Panzenboeck U, Kratzer I, Sovic A, Wintersperger A, Bernhart E, Hammer A, Malle E, Sattler W. Regulatory effects of synthetic liver X receptor- and peroxisome-proliferator activated receptor agonists on sterol transport pathways in polarized cerebrovascular endothelial cells. Int J Biochem Cell Biol. 2006;38:1314–1329. doi: 10.1016/j.biocel.2006.01.013. [DOI] [PubMed] [Google Scholar]
- Papassotiropoulos A, Lütjohann D, Bagli M, Locatelli S, Jessen F, Buschfort R, Ptok U, Björkhem I, von Bergmann K, Heun R. 24S-hydroxycholesterol in cerebrospinal fluid is elevated in early stages of dementia. J Psychiatr Res. 2002;36:27–32. doi: 10.1016/s0022-3956(01)00050-4. [DOI] [PubMed] [Google Scholar]
- Pitas RE, Boyles JK, Lee SH, Hui D, Weisgraber KH. Lipoproteins and their receptors in the central nervous system. Characterization of the lipoproteins in cerebrospinal fluid and identification of apolipoprotein B,E(LDL) receptors in the brain. J Biol Chem. 1987;262:14352–14360. [PubMed] [Google Scholar]
- Pittman RC, Taylor CA., Jr Methods for assessment of tissue sites of lipoprotein degradation. Methods Enzymol. 1986;129:612–628. doi: 10.1016/0076-6879(86)29094-1. [DOI] [PubMed] [Google Scholar]
- Pittman RC, Carew TE, Glass CK, Green SR, Taylor CA, Jr, Attie AD. A radioiodinated, intracellularly trapped ligand for determining the sites of plasma protein degradation in vivo. Biochem J. 1983;212:791–800. doi: 10.1042/bj2120791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pittman RC, Knecht TP, Rosenbaum MS, Taylor CA., Jr A nonendocytotic mechanism for the selective uptake of high density lipoprotein-associated cholesterol esters. J Biol Chem. 1987;262:2443–2450. [PubMed] [Google Scholar]
- Ponce J, de la Ossa NP, Hurtado O, Millan M, Arenillas JF, Dávalos A, Gasull T. Simvastatin reduces the association of NMDA receptors to lipid rafts: a cholesterol-mediated effect in neuroprotection. Stroke. 2008;39:1269–1275. doi: 10.1161/STROKEAHA.107.498923. [DOI] [PubMed] [Google Scholar]
- Quan G, Xie C, Dietschy JM, Turley SD. Ontogenesis and regulation of cholesterol metabolism in the central nervous system of the mouse. Brain Res Dev Brain Res. 2003;146:87–98. doi: 10.1016/j.devbrainres.2003.09.015. [DOI] [PubMed] [Google Scholar]
- Ralph RJ, Paulus MP, Fumagalli F, Caron MG, Geyer MA. Prepulse inhibition deficits and perseverative motor patterns in dopamine transporter knock-out mice: differential effects of D1 and D2 receptor antagonists. J Neurosci. 2001;21:305–313. doi: 10.1523/JNEUROSCI.21-01-00305.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez-Rodríguez E, Mateo I, Llorca J, Sánchez-Quintana C, Infante J, Garca-Gorostiaga I, Sánchez-Juan P, Berciano J, Combarros O. Association of genetic variants of ABCA1 with Alzheimer's disease risk. Am J Med Genet B Neuropsychiatr Genet. 2007;144B:964–968. doi: 10.1002/ajmg.b.30552. [DOI] [PubMed] [Google Scholar]
- Saher G, Brügger B, Lappe-Siefke C, Möbius W, Tozawa R, Wehr MC, Wieland F, Ishibashi S, Nave KA. High cholesterol level is essential for myelin membrane growth. Nat Neurosci. 2005;8:468–475. doi: 10.1038/nn1426. [DOI] [PubMed] [Google Scholar]
- Schönknecht P, Lütjohann D, Pantel J, Bardenheuer H, Hartmann T, von Bergmann K, Beyreuther K, Schröder J. Cerebrospinal fluid 24S-hydroxycholesterol is increased in patients with Alzheimer's disease compared to healthy controls. Neurosci Lett. 2002;324:83–85. doi: 10.1016/s0304-3940(02)00164-7. [DOI] [PubMed] [Google Scholar]
- Singh-Manoux A, Gimeno D, Kivimaki M, Brunner E, Marmot MG. Low HDL cholesterol is a risk factor for deficit and decline in memory in midlife: the Whitehall II study. Arterioscler Thromb Vasc Biol. 2008;28:1556–1562. doi: 10.1161/ATVBAHA.108.163998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tachikawa M, Watanabe M, Hori S, Fukaya M, Ohtsuki S, Asashima T, Terasaki T. Distinct spatio-temporal expression of ABCA and ABCG transporters in the developing and adult mouse brain. J Neurochem. 2005;95:294–304. doi: 10.1111/j.1471-4159.2005.03369.x. [DOI] [PubMed] [Google Scholar]
- Temel RE, Gebre AK, Parks JS, Rudel LL. Compared with Acyl-CoA:cholesterol O-acyltransferase (ACAT) 1 and lecithin:cholesterol acyltransferase, ACAT2 displays the greatest capacity to differentiate cholesterol from sitosterol. J Biol Chem. 2003;278:47594–47601. doi: 10.1074/jbc.M308235200. [DOI] [PubMed] [Google Scholar]
- Timmins JM, Lee JY, Boudyguina E, Kluckman KD, Brunham LR, Mulya A, Gebre AK, Coutinho JM, Colvin PL, Smith TL, Hayden MR, Maeda N, Parks JS. Targeted inactivation of hepatic Abca1 causes profound hypoalphalipoproteinemia and kidney hypercatabolism of apoA-I. J Clin Invest. 2005;115:1333–1342. doi: 10.1172/JCI23915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Triguero D, Buciak J, Pardridge WM. Capillary depletion method for quantification of blood-brain barrier transport of circulating peptides and plasma proteins. J Neurochem. 1990;54:1882–1888. doi: 10.1111/j.1471-4159.1990.tb04886.x. [DOI] [PubMed] [Google Scholar]
- Tronche F, Kellendonk C, Kretz O, Gass P, Anlag K, Orban PC, Bock R, Klein R, Schütz G. Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nat Genet. 1999;23:99–103. doi: 10.1038/12703. [DOI] [PubMed] [Google Scholar]
- Valenza M, Rigamonti D, Goffredo D, Zuccato C, Fenu S, Jamot L, Strand A, Tarditi A, Woodman B, Racchi M, Mariotti C, Di Donato S, Corsini A, Bates G, Pruss R, Olson JM, Sipione S, Tartari M, Cattaneo E. Dysfunction of the cholesterol biosynthetic pathway in Huntington's disease. J Neurosci. 2005;25:9932–9939. doi: 10.1523/JNEUROSCI.3355-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vance JE, Hayashi H, Karten B. Cholesterol homeostasis in neurons and glial cells. Semin Cell Dev Biol. 2005;16:193–212. doi: 10.1016/j.semcdb.2005.01.005. [DOI] [PubMed] [Google Scholar]
- Wahrle SE, Jiang H, Parsadanian M, Legleiter J, Han X, Fryer JD, Kowalewski T, Holtzman DM. ABCA1 is required for normal central nervous system ApoE levels and for lipidation of astrocyte-secreted apoE. J Biol Chem. 2004;279:40987–40993. doi: 10.1074/jbc.M407963200. [DOI] [PubMed] [Google Scholar]
- Wahrle SE, Jiang H, Parsadanian M, Kim J, Li A, Knoten A, Jain S, Hirsch-Reinshagen V, Wellington CL, Bales KR, Paul SM, Holtzman DM. Overexpression of ABCA1 reduces amyloid deposition in the PDAPP mouse model of Alzheimer disease. J Clin Invest. 2008;118:671–682. doi: 10.1172/JCI33622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterham HR, Koster J, Romeijn GJ, Hennekam RC, Vreken P, Andersson HC, FitzPatrick DR, Kelley RI, Wanders RJ. Mutations in the 3beta-hydroxysterol Delta24-reductase gene cause desmosterolosis, an autosomal recessive disorder of cholesterol biosynthesis. Am J Hum Genet. 2001;69:685–694. doi: 10.1086/323473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiler-Güttler H, Sommerfeldt M, Papandrikopoulou A, Mischek U, Bonitz D, Frey A, Grupe M, Scheerer J, Gassen HG. Synthesis of apolipoprotein A-1 in pig brain microvascular endothelial cells. J Neurochem. 1990;54:444–450. doi: 10.1111/j.1471-4159.1990.tb01892.x. [DOI] [PubMed] [Google Scholar]
- Wellington CL, Walker EK, Suarez A, Kwok A, Bissada N, Singaraja R, Yang YZ, Zhang LH, James E, Wilson JE, Francone O, McManus BM, Hayden MR. ABCA1 mRNA and protein distribution patterns predict multiple different roles and levels of regulation. Lab Invest. 2002;82:273–283. doi: 10.1038/labinvest.3780421. [DOI] [PubMed] [Google Scholar]