

Abstract
Elimination of hydrogen sulfide from glutathione (GSH) converts a well-known cellular nucleophile to an electrophilic species, γ-glutamyldehydroalanylglycine (EdAG). We have found that a sulfonium metabolite formed from GSH and busulfan undergoes a facile β-elimination reaction to give EdAG, which is an αβ-unsaturated dehydroalanyl analog of GSH. EdAG was identified as a metabolite of busulfan in a human liver cytosol fraction. EdAG condenses with GSH in a Michael addition reaction to produce a lanthionine thioether (GSG), which is a non-reducible analog of glutathione disulfide (GSSG). EdAG was less cytotoxic than busulfan to C6 rat glioma cells. GSH and EdAG were equally effective in displacing a glutathione S-transferase isozyme (human GSTA1-1) from a GSH-Agarose column. The finding of an electrophilic metabolite of GSH suggests that alteration of cellular GSH concentrations, irreversible non-reducible glutathionylation of proteins, and interference with GST function may contribute to the toxicity of busulfan.
Introduction
Glutathione (GSH) is the most prevalent low molecular weight cellular thiol in mammalian cells. In addition to a major cellular role in oxidative status, there is an important function of GSH in sequestering toxic, electrophilic xenobiotics (Boyland and Chasseaud, 1969; Chasseaud, 1976). Nucleophilic reactivity of GSH is based on conjugation of its thiol group with electrophilic compounds (Commandeur et al., 1995). β-Elimination of hydrogen sulfide from a cysteine residue forms a dehydroalanine residue. Dehydroalanine-containing peptides react as Michael acceptors that link cysteine residues with dehydroalanine groups to generate non-reducible thioethers as a posttranslational modification (Friedman et al., 1977; Dufour et al., 2007; Willey et al., 2007).
The dehydroalanine analog of GSH, γ-glutamyldehydroalanylglycine (EdAG) is shown in Figure 1. To accomplish the β-elimination reaction on GSH, strongly basic conditions are needed to dissociate the relatively non-acidic α-proton that has a pKa projected to be above 21 (Murkin and Tanner, 2002). Lysis of glutathione disulfide (GSSG) with strong alkali produces EdAG as one of the identified products (Jones et al., 1983). The elimination reaction can be facilitated by the presence of a good leaving group in the β-position (Bentley, 2006). For example, EdAG can be readily synthesized from a dinitrophenyl derivative of GSH (Asquith and Carthew, 1972). More recently, Munter and coworkers identified EdAG as a base catalyzed product formed from a chloroprene-GSH adduct (Munter et al., 2003).
Figure 1.
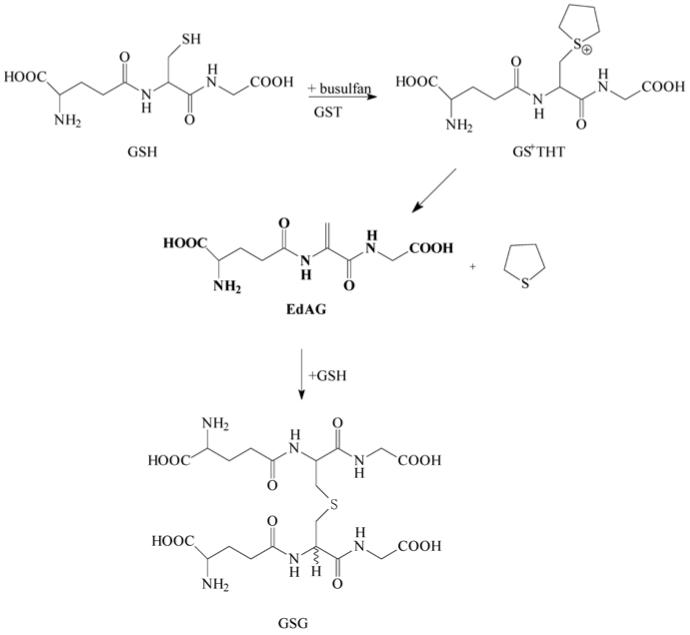
Formation of γ-glutamyldehydroalanylglycine (EdAG) from glutathione and busulfan.
Conjugation with GSH, forming an unstable S-glutathione sulfonium conjugate (GS+THT, Figure 1), is the major elimination pathway of the anticancer drug busulfan (Roberts and Warwick, 1961; Czerwinski et al., 1996; Ritter et al., 1999). Tetrahydrothiophene is released from GS+THT under alkaline conditions (Roberts and Warwick, 1961), or through involvement of the mercapturic acid pathway and β-lyases (Cooper et al., 2008; Marchand et al., 1988; Onkenhout et al., 1986).
In this communication, we report that the other product formed in the β-elimination reaction of GS+THT is EdAG, the dehydroalanine analogue of GSH. We have shown that EdAG reacts as an electrophile in the formation of non-reducible thioether adducts with sulfhydryl-containing compounds, and binds to the glutathione S-transferase (GST) isoform responsible for GS+THT formation.
Methods
Busulfan, L-cysteine, GSH, 1-bromo-4-chlorobutane, deuterium oxide, formic acid, butanol, Dowex-50W, DMSO, d6-DMSO, ammonium hydroxide, 1-chloro-2,4-dinitrobenzene, and GST-Agarose were obtained from Sigma Chemical Company (St. Louis, MO). Acetic acid, sodium bicarbonate, dibasic potassium phosphate, monobasic potassium phosphate, sodium hydroxide, hydrochloric acid, acetonitrile (optima), methanol (HPLC grade), sodium chloride, acetone, Tris, Tween 20, and 2-mercaptoethanol were purchased from Fisher Scientific (Pittsburgh, PA). Pooled human liver cytosol was obtained from Gentest (Woburn, MA). TCA was obtained from Fluka Chemie (Gmbh). Human GSTA1-1 was obtained from Invitrogen (Carlsbad, CA). Electrophoresis reagents polyacrylamide, SDS, ammonium persulfate, bromophenol blue and TEMED were obtained from Biorad Laboratories (Hercules, CA). Coomassie Brilliant Blue 250 was obtained from Life Technologies (Grand Island, NY).
γ-Glutamyl-β-(S-tetrahydrothiophenium)alanyl-glycine(GS+THT) was prepared by a modified method of Marchand et al., (1988). Purification was by semi-preparative HPLC on an Econosphere C8 semi-preparative column (10 × 250 mm), and a UV/VIS detector set at wavelength of 210 nm. Isocratic elution was with 50:50 methanol/water (v/v) at a flow rate of 3.0 mL/min. Solvents were removed under vacuum on a rotary evaporator.
γ-Glutamyldehydroalanylglycine (EdAG) was prepared by the elimination of 2,4-dinitrothiophenolate from S-(2,4-dinitrophenyl)glutathione (Sokolovsky et al., 1963) as described by Asquith and Carthew (1972). Positive ionization electrospray MS: m/z 274 (MH+), MS/MS: m/z 145 (MH+ - pyroglutamic acid); 1H NMR (600 MHz, D2O) δH 2.08 (2H, m), 2.48 (2H, m), 3.46 (1H, m), 3.93 (2H, s), 5.71 (1H, s) and 5.79 (1H, s) ppm.
The stability of GS+THT at pH 7.4 and 8.0 was determined by measuring rates of its disappearance. Stock solutions were made in distilled water (2 mg/mL), and further diluted in 100 mM potassium phosphate buffer (pH 7.4 or pH 8.0) to produce a 0.83 mM final concentration of GS+THT. Solutions were incubated in a shaking water bath at 37 °C. Aliquots (6 μL) removed at 0, 0.25, 0.5, 0.75, 1.0, 1.5, 2, 3, 4, 5, and 6 h were diluted with 1.5 mL distilled water and 50-μL aliquots were injected into a Waters Alliance 2695 separation module equipped with a Waters 996 Photodiode Array Detector, and coupled to a Waters Micromass ZMD mass spectrometer with electrospray ionization (Waters, Milford, MA). The photodiode array detector was programmed to scan between 200 and 300 nm, and a mass spectrometer was programmed to utilize electrospray ionization in a positive ion mode with selected ion recording of the m/z 362 ion. The mobile phase consisted of methanol/water (50/50, v/v) pumped at 0.4 mL/min through an Agilent Zorbax SB-NC C18 150 × 4.6 mm reversed phase column (Agilent, Santa Clara, CA).
Identification of EdAG after incubation of busulfan with human liver cytosol
Busulfan (0-5 mM) was incubated with human liver cytosol (total protein 1.0 mg) in the presence of 1.0 mM GSH in 100 mM Tris (Trizma) buffer (pH 7.4, total volume 1.0 mL). Busulfan was dissolved in DMSO, and the total DMSO concentration was less than 1.0% v/v. The mixture was preincubated for 5 min in a shaking water bath maintained at 37 °C, and reactions were started by the addition of busulfan. The formation of GS+THT, EdAG, and a non-reducible lanthionine thioether, GSG, were monitored by LC-MS using a Thermo Hybercarb column (100 × 2.1 mm, 5 μm, ThermoFisher, Waltham, MA). A gradient of mobile phase A (0.5% formic acid in 90/10, v/v, water/acetonitrile) and mobile phase B (0.1% formic acid in 50/50, v/v, isopropanol/methanol) at 0.2 mL/min was run 0-5 minutes, 100% A; 5-7 minutes, 0→50% B; 7-9 minutes, 50→0% B; and 9-15 minutes, 100% A. A Finnigan LCQ DECA mass spectrometer (ThermoFisher) was operated in the positive ion electrospray ionization mode with full scan mode monitoring GSH, GS+THT, EdAG, and GSG at m/z 308, 362, 274, and 581, respectively. The structure of EdAG was confirmed using LC-MS/MS in a separate run that fragmented m/z 274 to give the representative product ions of m/z 256, 199, and 145 consistent with neutral losses of water, glycine, and pyroglutamic acid, respectively, based on a comparison of the mass spectrum and retention time of the metabolite with that of authentic standard (Figure 2A). The structure of GSG was also confirmed in a separate LC-MS/MS run that fragmented m/z 581 to give the product ions of m/z 563, 452, and 323, which correspond to the neutral losses of water, pyroglutamic acid, and 2 pyroglutamic acids, respectively (Figure 2B).
Figure 2.
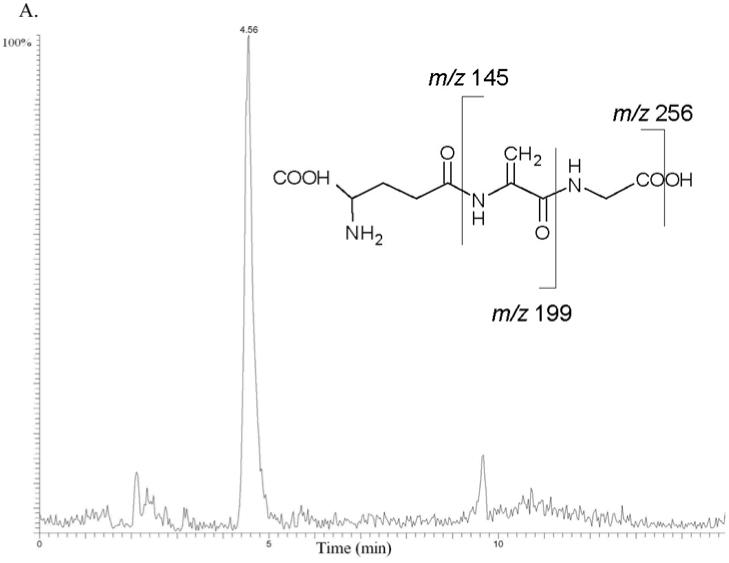
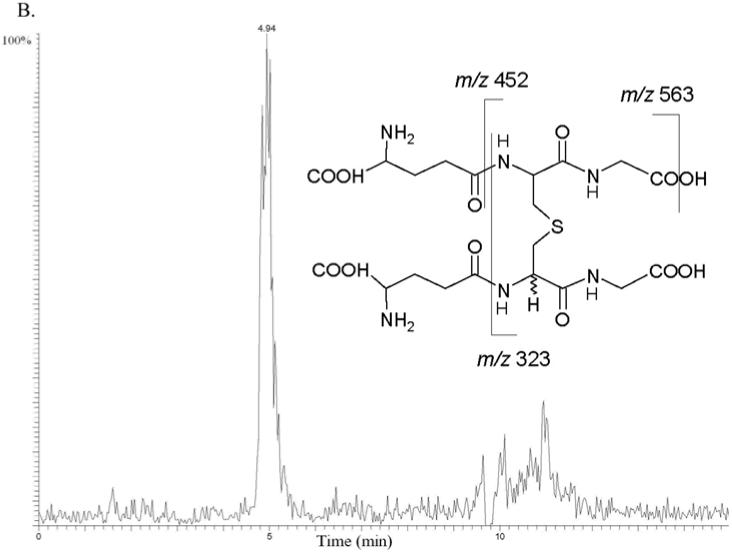
Busulfan metabolism by human liver cytosolic fractions. MS/MS mass chromatograms of EdAG (A) and GSG (B) at 4 hour incubation time. The proposed MS/MS fragmentation patterns for EdAG and GSG are shown in the insets. (A) Neutral losses from the EdAG MH+ ion (m/z 274) are consistent with loss of water (m/z 256), glycine (m/z 199), and pyroglutamic acid (m/z 145). (B) Neutral losses from the MH+ ion (m/z 581) are consistent with loss of water (m/z 563), loss of pyroglutamic acid (m/z 452), and loss of two molecules of pyroglutamic acid (m/z 323).
Electrophilic reactivity of EdAG toward GSH and cysteine
GSH-EdAG adduct (GSG)
GSH (1.0 mM in 100 mM Tris buffer, pH 7.4) was mixed with an aqueous solution of EdAG (1.0 mM) in 100 mM Tris buffer, pH 7.4, and the mixture was heated to 37 °C for 12 h to provide the GSH-EdAG adduct (2-amino-5-[[3-[2-[[4-amino-5-hydroxy-5-oxopentanoyl]amino]-3-(carboxymethylamino)-3-oxopropyl]sulfanyl-1-(carboxymethylamino)-1-oxopropan-2-yl]amino]-5-oxopentanoic acid, GSG). Identification of GSG was made by mass spectral analysis. The mass spectrum of the primary component in the reaction mixture showed a protonated molecular species ion at m/z 581, consistent with molecular mass of this adduct. The MS/MS fragmentation of the adduct MH+ ion in an ion trap mass spectrometer produced three daughter ions of m/z 563, 452, and 323 consistent with the neutral losses of water, pyroglutamic acid, and two molecules of pyroglutamic acid, respectively.
Cysteine-EdAG adduct [S-(β-alanyl)glutathione]
A solution of EdAG in water (1.0 mM) was mixed with a solution of cysteine (1.0 mM in 100 mM Tris buffer, pH 7.4) in 100 mM Tris buffer, pH 7.4 (final volume 1.0 mL). The mixture was stirred overnight in a 37 °C water bath. The mixture was further diluted 200-fold in a solution of 1% formic acid in methanol/water (50/50, v/v), and the product was detected by electrospray mass spectrometry. The mass spectrum of the mixture showed that EdAG had been consumed in the reaction and the major product detected had a protonated molecular species of m/z 396 that was consistent with the molecular weight of the cysteine-EdAG adduct [S-(β-alanyl)glutathione]. MS/MS fragmentation of this adduct produced a daughter ion with m/z 267 consistent with the neutral loss of pyroglutamic acid. Further fragmentation of the daughter ion produced fragments at m/z 249 and 192 corresponding to the loss of water and glycine, respectively.
The reaction was monitored by 1H NMR. EdAG (50 mM) and cysteine (50 mM) were dissolved in 100 mM potassium phosphate buffer prepared in D2O (pD ∼ 8). Disappearance of the vinylic 1H signals at 5.71 and 5.79 ppm of EdAG showed that the reaction was complete in 90 minutes at 37 °C.
Binding of EdAG to human GSTA1-1
Recombinant GSTA1-1 (1 μg) or pooled human liver cytosol (50 μg protein) in TBS-T (10 mM Tris, pH 7.6, 0.14 M NaCl, 0.05% Tween 20) was incubated with 50 μL of GSH-Agarose for 30 min on ice. The GSH-Agarose was washed 3 times with TBS-T to remove unbound material and incubated with 10 mM GSH or EdAG to elute bound material. All samples were precipitated with trichloroacetic acid (20% w/v final concentration). The pellets were washed with acetone, air-dried and resuspended in Laemmli buffer containing 2% w/v SDS and 5% v/v 2-mercaptoethanol. SDS-PAGE analysis was conducted using a 4% stacking gel and a 12% resolving gel at 100V for 2 h using a Biorad minigel apparatus. The gel was stained with a solution containing 0.1% w/v Coomassie Brilliant Blue, 46% v/v methanol, 7.5% w/v acetic acid and destained with a solution containing 5% v/v methanol and 7.5% w/v acetic acid in water.
Cytotoxicity of EdAG In Vitro
C6 rat glioma cells obtained from the American Type Culture Collection (Manassas, VA) were maintained in DMEM-F12 (1:1) medium (Gibco, Grand Island, NY) supplemented with 1% of antibiotic-antimycotic mixture (Gibco, Grand Island, NY) and 10% fetal calf serum (FCS; Sigma, St. Louis, MO) at 37 °C under an atmosphere of 5% CO2-95% air. For toxicity experiments, the cells were seeded into 12-well Primaria cluster dishes (Becton Dickson Labware, Franklin Lakes, NJ) at a density of 5×104 cells/well and incubated in 0.5 mL of DMEM-F12/FCS medium overnight. Subsequently, the cells were exposed to busulfan and EdAG for 24 h. Busulfan was added in DMSO (final concentration in the culture medium of DMSO was <1%) and EdAG was added as an aqueous solution. Sister cultures incubated without any additions or with DMSO served as controls. Cell viability was assessed by the trypan blue exclusion test. Briefly, the medium was removed and the adhering cells were trypsinized and stained with trypan blue. Viable, dye excluding, cells were counted in a hemocytometer.
Results
Conversion of busulfan to EdAG
Busulfan was incubated with human liver cytosol in 100 mM Tris buffer, pH 7.4 at 37 °C in the presence of GSH. EdAG was identified as a metabolite of busulfan (Figure 2A) based on LC/MS data. EdAG was trapped as a glutathione conjugate, GSG (Figure 2B). Figure 1 shows the proposed pathway of EdAG and GSG formation from busulfan, GSH, and GSTs. The proposed intermediate in the conversion of busulfan to EdAG, sulfonium GS+THT, was detected in the incubation mixture by LC/MS (Cooper et al., 2008). GS+THT was proposed to be an intermediate in the reaction because incubation of GS+THT in 100 mM potassium phosphate buffer (pH 7.4) for 6 h at 37 °C resulted in significant loss of GS+THT. These findings are consistent with the report by Roberts and Warwick (1961) showing that GS+THT is unstable at pH 7.4 based on the detection of tetrahydrothiophene as a degradation product. The most likely pathway to the formation of tetrahydrothiophene from GS+THT is through a β-elimination reaction proposed in Figure 1 showing the production of tetrahydrothiophene and EdAG. To confirm the identity of EdAG in the reaction, GS+THT (57 mM) in deuterated 100 mM phosphate buffer (pD ∼8.0) was incubated for 12 hours at 37 °C. The formation of EdAG from GS+THT was detected by monitoring the vinylic proton signals of EdAG in the 1H NMR spectrum.
Reactivity of EdAG toward the nucleophiles, cysteine and GSH
EdAG contains an α,β-unsaturated carbonyl system that adds nucleophiles, such as sufhydryl anions, to form a conjugate or adduct. Condensation of EdAG with cellular nucleophiles, GSH and cysteine, under physiological conditions yielded a glutathione analog with a lanthionine cross linkage (GSG), and S-(β-alanyl)glutathione, respectively. Mass spectral evidence in support of the assigned structure of GSG is shown in Figure 2B, which produced the same MS/MS spectrum as the GSG formed in the busulfan and human liver cytosol incubation. Because dehydroalanine is achiral, the addition of GSH or cysteine to EdAG forms a new chiral center, and there is the probability that GSG consists of a pair of diastereomers. The stereochemistry of synthesized GSG and S-(β-alanyl)glutathione was not determined.
Analysis of binding of EdAG to GSTA1-1
An analysis of the capacity of EdAG to bind to GST was facilitated by the use of GSH-Agarose. As shown in Figure 3, human GSTA1-1 bound to GSH-Agarose was eluted with 10 mM GSH (lane 8) or 10 mM EdAG (lane 9). The band at approximately 70 KDa in lane 7 is most likely aggregated GSTA1.1. GST species present in pooled human liver cytosol and binding to GSH-Agarose were also eluted by 10 mM GSH (lane 4) as well as by 10 mM EdAG (lane 6). This observation confirms that GST can recognize EdAG and further establishes that EdAG can effectively compete with GSH for binding to GST.
Figure 3.
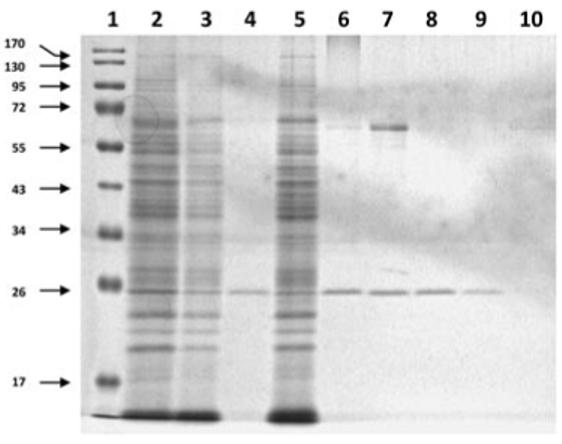
Analysis of binding of EdAG GST. Experimental samples subjected to PAGE as described in Materials and Methods. Molecular wt. standards (Lane 1). Human liver cytosol (lane 2), unbound material from liver cytosol after incubation with GSH-Agarose (lanes 3 and 5), liver protein eluted from GSH-Agarose with GSH (lane 4) and EdAG (lane 6). Purified human GSTA1-1 (lane 7), unbound material after incubation with GSH-Agarose (lane 10), human GSTA1-1 eluted from GSH-Agarose with GSH (lane 8) and with EdAG (lane 9).
The occurence of a single band with Mr ∼24,000 in lanes 4 and 6 corroborates the absence of additional proteins in the liver cytosol capable of binding to GSH-Agarose that can be eluted with GSH or EdAG, respectively. Our method does not test for possible EdAG binding within the liver cytosol, although it is conceivable that EdAG can bind to the sulfhydryl moieties of other proteins, potentially irreversibly forminga mixed sulfide lanthionine linkage.
In Vitro cytotoxicity of EdAG
The exposure of C6 cells to busulfan for 24 h profoundly decreases the number of viable cells. The concentrations of busulfan fall within the concentration ranges recently used by Lanvers-Kaminsky et al. (2006) who found a great variation in cellular susceptibility to this anticancer drug. For example, while growth of leukemia cell lines was inhibited by micromolar concentrations, millimolar concentrations were required to inhibit growth of osteosarcoma cell lines. We found the LD50 value for C6 cells to be approximately 460 μM (Figure 4), indicating that these cells are modestly resistant to the toxic effects of busulfan. EdAG showed a similar cytotoxicity profile. However, twice as high concentrations were required to show the same level of toxicity exhibited by busulfan. The LD50 for EdAG was approximately 880 μM.
Figure 4.
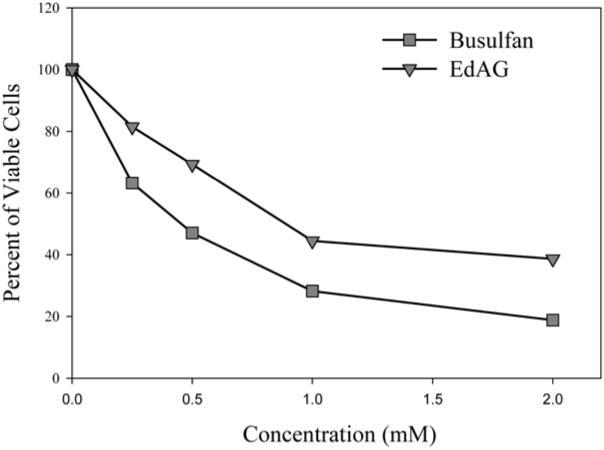
EdAG and busulfan and cytotoxicity against C6 cells. Each point represents the average of three determinations. The LD50 values were approximately 460 μM and 880 μM for busulfan and EdAG, respectively.
Discussion
There is extensive evidence that GSH is a cellular trapping agent of electrophilic compounds, such as alkylating anticancer agents. In the case of busulfan, conjugation with GSH leads initially to the formation of the conjugate GS+THT that is further converted by elimination of tetrahydrothiophene to form the dehydroalanine analog, EdAG. Whereas GS+THT formation is catalyzed in part by GST enzymes, it is not known whether the formation of EdAG from GS+THT is an enzyme-catalyzed process or occurs solely in a non-enzymatic β-elimination reaction. Nevertheless, under physiological conditions (pH 7.4 and 37 °C), GS+THT spontaneously dissociates to tetrahydrothiophene and EdAG. This instability of GS+THT leads to a GSH analogue in which the cysteine residue is converted to an electrophilic dehydroalanine moiety. Addition of a second molecule of GSH provides GSG, which is a glutathione adduct of EdAG.
The detection of both EdAG and GSG by LC-MS and LC-MS/MS in incubations of busulfan with human liver cytosol fortified with GSH suggests that EdAG is a metabolite of busulfan and that GSG is a conjugate of EdAG. The relevance of this busulfan metabolism pathway to medical practice has not been determined, although it is important to note that EdAG is weakly cytotoxic and can be categorized as a reactive metabolite toward cellular nucleophiles. EdAG formed in liver soluble fraction preparations is trapped by GSH. Addition of a sulfhydryl to the double bond of a dehydroalanine residue can occur readily forming a thioether or lanthionine linkage (Asquith and Carthew, 1972), and there are examples of lantibiotic antimicrobial agents that contain lanthionine residues (Chatterjee et al., 2005). Lantibiotics consist of cross-linked or cyclized peptides that are formed though Michael addition of a cysteine group to dehydroalanine residues. Lanthionine residues are associated with aging of the human lens and cataractogenesis (Linetsky and LeGrand, 2005). These lanthionines include non-reducible mixed thioethers with glutathione. Lanthionine thioether linkages are not reducible in comparison with disulfide linkages, such as those found in reversible mixed-disulfide S-glutathionylation of proteins (Reddy et al., 2000). Dehydroalanine residues have been observed as artifacts in proteome analysis (Herbert el al., 2003), as common post-translational modifications in human serum albumin (Bar-Or et al., 2008), and after loss of selenium following oxidation of selenocysteine moieties from selenoproteins (Ma et al., 2003). Heat and alkali treatment of foods result in the formation of dehydroalanyl residues and resulting cross-linked amino acids in proteins, as well as the epimerization of L-amino acid stereoisomers (Friedman, 1999). Electrophilic addition at the α,β-unsaturated moiety of dehydroalanine residues by a variety of nucleophiles is an entry to functionalized proteins (Snow et al., 1976; Friedman et al., 1977; Bernardes et al., 2008). Lanthionine bridges can also form in proteins during derivatization steps prior to mass spectral analysis, presumably through intramolecular condensation of a cysteine residue with a dehydroalanine (Amini and Nilsson, 2008).
The electrophilic reactivity of EdAG may be the cause of certain effects observed in busulfan therapy. Conditioning therapy with cytotoxic drugs, such as busulfan and cyclophosphamide, in patients undergoing bone marrow transplant is the most important factor leading to the development of hepatic venoocclusive disease (HVOD) (Kalayoglu-Besisik et al., 2005). HVOD is caused by the destruction of sinusoidal endothelial cells and the surrounding centriobular hepatocytes (Wadleigh et al., 2003). It was proposed that HVOD associated with conditioning regimens of busulfan in bone transplant patients is related to the metabolites of busulfan rather than to the parent compound (Srivastava et al., 2004). DeLeve and Wang (2000) suggested that this complication can be caused either directly through oxidative stress or indirectly through GSH depletion. We hypothesize that these conditions could be related to the non-enzymatic decomposition of GS+THT to EdAG. Interestingly, it has recently been shown that busulfan treatment of mice resulted in upregulation of glutathione synthesis and increased toxicity of busulfan (Bouligand et al., 2006). We suggest that higher GSH levels lead to higher concentrations of GS+THT. An increase in GS+THT would lead to increased non-enzymatic formation of EdAG, which in turn, would lead to non-reducible glutathionylation of proteins associated with the circulatory system. Thus, increased non-reducible glutathionylation may contribute to busulfan-induced HVOD. Future studies are needed to explore the suggestion that GST catalyzes the formation of GSG by binding both GSH and EdAG.
Acknowledgments
The work reported herein was supported in part by the National Institutes of Health grants RO1 ES08421 (to A.J.L.C.) and NIH-NCI 5-PO1-CA47741 and the Mylan Chair of Pharmacology at WVU Health Sciences Center (to W.P.P.), and by National Institute of Justice grant IJ-CX-K014 (to P.S.C.).
The Department of Basic Pharmaceutical Sciences (School of Pharmacy, West Virginia University, Morgantown, WV) was the primary laboratory of origin.
Abbreviations
- EdAG
γ-glutamyldehydroalanylglycine
- GSH
glutathione
- GST
glutathione S-transferase
- GSSG
glutathione disulfide
- GS+THT
γ-glutamyl-β-(S-tetrahydrothiophenium)alanylglycine
- GSG
(2-amino-5-[[3-[2-[[4-amino-5-hydroxy-5-oxopentanoyl]amino]-3-(carboxymethylamino)-3-oxopropyl]sulfanyl-1-(carboxymethylamino)-1-oxopropan-2-yl]amino]-5-oxopentanoic acid)
- HVOD
hepatic venoocclusive disease
Footnotes
Recommended section assignment: Toxicology
References
- Amini A, Nilsson E. Quantitative analysis of polypeptide pharmaceuticals by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. J Pharm Biomed Anal. 2008;46:411–417. doi: 10.1016/j.jpba.2007.10.028. [DOI] [PubMed] [Google Scholar]
- Anger B, Haug U, Seidler R, Heimpel H. Polycythemia vera. A clinical study of 141 patients. Blut. 1989;59:493–500. doi: 10.1007/BF00329494. [DOI] [PubMed] [Google Scholar]
- Asquith RS, Carthew P. The preparation and subsequent identification of a dehydroalanyl peptide from alkali-treated oxidised glutathione. Biochim Biophys Acta. 1972;285:346–351. doi: 10.1016/0005-2795(72)90319-4. [DOI] [PubMed] [Google Scholar]
- Bar-Or R, Rael LT, Bar-Or D. Dehydroalanine derived from cysteine is a common post-translational modification in human serum albumin. Rapid Commun Mass Spectrom. 2008;22:711–716. doi: 10.1002/rcm.3421. [DOI] [PubMed] [Google Scholar]
- Bentley TW. Additivity rules using similarity models for chemical reactivity: calculation and interpretation of electrofugality and nucleofugality. Chemistry. 2006;25:6514–6520. doi: 10.1002/chem.200600517. [DOI] [PubMed] [Google Scholar]
- Bernardes GJ, Chalker JM, Errey JC, Davis BG. Facile conversion of cysteine and alkyl cysteines to dehydroalanine on protein surfaces: versatile and switchable access to functionalized proteins. J Am Chem Soc. 2008;130:5052–5053. doi: 10.1021/ja800800p. [DOI] [PubMed] [Google Scholar]
- Bouligand J, Deroussent A, Simonnard N, Opolon P, Morizet J, Connault E, Daudigeos E, Re M, Paci A, Vassal G. Induction of glutathione synthesis explains pharmacodynamics of high-dose busulfan in mice and highlights putative mechanisms of drug interaction. Drug Metab Dispos. 2007;35:306–314. doi: 10.1124/dmd.106.012880. [DOI] [PubMed] [Google Scholar]
- Boyland E, Chasseaud LF. The role of glutathione and glutathione S-transferases in mercapturic acid biosynthesis. Advances in Enzymology. 1969;32:173–219. doi: 10.1002/9780470122778.ch5. 1969. [DOI] [PubMed] [Google Scholar]
- Braide SA, Davies TJ. Factors that affect the induction of γ-glutamyltransferase in epileptic patients receiving anti-convulsant drugs. Ann Clin Biochem. 1987;24:391–399. doi: 10.1177/000456328702400408. [DOI] [PubMed] [Google Scholar]
- Brodsky I. Busulfan versus hydroxyurea in the treatment of polycythemia vera (PV) and essential thrombocythemia (ET) Am J Clin Oncol. 1998;21:105–106. doi: 10.1097/00000421-199802000-00024. [DOI] [PubMed] [Google Scholar]
- Chasseaud LF. Conjugation with glutathione and mercapturic acid excretion. In: Arias IM, Jakoby WB, editors. Glutathione: Metabolism and Function. Raven Press; New York: 1976. pp. 77–114. [Google Scholar]
- Chatterjee C, Paul M, Xie L, van der Donk WA. Biosynthesis and mode of action of lantibiotics. Chem Rev. 2005;105:633–684. doi: 10.1021/cr030105v. [DOI] [PubMed] [Google Scholar]
- Commandeur JNM, Stijntjes GJ, Vermeulen NPE. Enzymes and transport systems involved in the formation and disposition of glutathione S-conjugates. Role in bioactivation and detoxication mechanisms of xenobiotics. Pharmacol Rev. 1995;47:271–330. [PubMed] [Google Scholar]
- Cooper AJL, Younis IR, Niatsetskaya ZV, Krasnikov BF, Pinto JT, Petros WP, Callery PS. Metabolism of the cysteine S-conjugate of busulfan involves a β-lyase. Drug Metab Dispos. 2008;36:1546–1552. doi: 10.1124/dmd.108.020768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czerwinski M, Gibbs JP, Slattery JT. Busulfan conjugation by glutathione S-transferases α, μ and π. Drug Metab Dispos. 1996;24:1015–1019. [PubMed] [Google Scholar]
- DeLeve LD, Wang X. Role of oxidative stress and glutathione in busulfan toxicity in cultured murine hepatocytes. Pharmacology. 2000;60:143–154. doi: 10.1159/000028359. [DOI] [PubMed] [Google Scholar]
- Dufour A, Hindré T, Haras D, Le Pennec JP. The biology of lantibiotics from the lacticin 481 group is coming of age. FEMS Microbiol Rev. 2007;31:134–167. doi: 10.1111/j.1574-6976.2006.00045.x. [DOI] [PubMed] [Google Scholar]
- Friedman M. Chemistry, biochemistry, nutrition, and microbiology of lysinoalanine, lanthionine, and histidinoalanine in food and other proteins. J Agric Food Chem. 1999;47:1295–1319. doi: 10.1021/jf981000+. [DOI] [PubMed] [Google Scholar]
- Friedman M, Finley JW, Yeh LS. Reactions of proteins with dehydroalanines. Adv Exp Med Biol. 1977;86B:213–224. doi: 10.1007/978-1-4757-9113-6_15. [DOI] [PubMed] [Google Scholar]
- Hasselbalch H, Berild D. Transition of myelofibrosis to polycythaemia vera. Scand J Haematol. 1983;30:161–166. doi: 10.1111/j.1600-0609.1983.tb01464.x. [DOI] [PubMed] [Google Scholar]
- Herbert B, Hopwood F, Oxley D, McCarthy J, Laver M, Grinyer J, Goodall A, Williams K, Castagna A, Righetti PG. β-Elimination: an unexpected artefact in proteome analysis. Proteomics. 2003;3:826–831. doi: 10.1002/pmic.200300414. [DOI] [PubMed] [Google Scholar]
- Jones AJ, Helmerhorst E, Stokes GB. The formation of dehydroalanine residues in alkali-treated insulin and oxidized glutathione. A nuclear-magnetic-resonance study. Biochem J. 1983;211:499–502. doi: 10.1042/bj2110499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalayoglu-Besisik S, Yenerel MN, Caliskan Y, Ozturk S, Besisik F, Sargin D. Time-related changes in the incidence, severity, and clinical outcome of hepatic veno-occlusive disease in hematopoietic stem cell transplantation patients during the past 10 years. Transplant Proc. 2005;37:2285–2289. doi: 10.1016/j.transproceed.2005.03.025. [DOI] [PubMed] [Google Scholar]
- Lanvers-Kaminsky C, Bremer A, Dirksen U, Jürgens H, Boos J. Cytotoxicity of treosulfan and busulfan on pediatric tumor cell lines. Anticancer Drugs. 2006;17:657–662. doi: 10.1097/01.cad.0000215059.93437.89. [DOI] [PubMed] [Google Scholar]
- Linetsky M, LeGrand RD. Glutathionylation of lens proteins through the formation of thioether bond. Mol Cell Biochem. 2005;272:133–144. doi: 10.1007/s11010-005-6908-1. [DOI] [PubMed] [Google Scholar]
- Ma S, Caprioli RM, Hill KE, Burk RF. Loss of selenium from selenoproteins: Conversion of selenocysteine to dehydroalanine in vitro. J Am Soc Mass Spectrom. 2003;14:593–600. doi: 10.1016/S1044-0305(03)00141-7. [DOI] [PubMed] [Google Scholar]
- Marchand DH, Remmel RP, Abdel-Monem MM. Biliary excretion of a glutathione conjugate of busulfan and 1,4-diiodobutane in the rat. Drug Metab Dispos. 1988;16:85–92. [PubMed] [Google Scholar]
- Munter T, Cottrell L, Golding BT, Watson WP. Detoxication pathways involving glutathione and epoxide hydrolase in the in vitro metabolism of chloroprene. Chem Res Toxicol. 2003;16:1287–1297. doi: 10.1021/tx034107m. [DOI] [PubMed] [Google Scholar]
- Murkin AS, Tanner ME. Dehydroalanine-based inhibition of a peptide epimerase from spider venom. J Org Chem. 2002;67:8389–8394. doi: 10.1021/jo0204653. [DOI] [PubMed] [Google Scholar]
- Onkenhout W, van Loon WM, Buijs W, van der Gen A, Vermeulen NPE. Biotransformation and quantitatitive determination of sulfur-containing metabolites of 1,4-dibromobutane in the rat. Drug Metab Dispos. 1986;14:608–612. [PubMed] [Google Scholar]
- Reddy S, Jones AD, Cross CE, Wong PS, Van Der Vliet A. Inactivation of creatine kinase by S-glutathionylation of the active-site cysteine residue. Biochem J. 2000;347:821–827. [PMC free article] [PubMed] [Google Scholar]
- Ritter CA, Bohnenstengel F, Hofmann U, Kroemer HK, Sperker B. Determination of tetrahydrothiophene formation as a probe of in vitro busulfan metabolism by human glutathione S-transferase A1-1: use of a highly sensitive gas chromatographic-mass spectrometric method. J Chromatogr B Biomed Sci Appl. 1999;730:25–31. doi: 10.1016/s0378-4347(99)00170-x. [DOI] [PubMed] [Google Scholar]
- Roberts JJ, Warwick GP. The mode of action of alkylating agents. III. The formation of 3-hydroxytetrahydrothiophene-1:1-dioxide from 1:4-dimethanesulphonyloxbutane (myleran), S-ß-L-alanyl-tetrahydrothiophenium mesylate, tetrahydrothiophene and tetrahydrothiophene-1:1-dioxide in the rat, rabbit and mouse. Biochem Pharmacol. 1961;6:217–227. [Google Scholar]
- Silver RT. Chronic myeloid leukemia. Hematol Oncol Clin North Am. 2003;17:1159–1173. doi: 10.1016/s0889-8588(03)00088-1. [DOI] [PubMed] [Google Scholar]
- Snow JT, Finley JW, Friedman M. Relative reactivities of sulfhydryl groups with N-acetyl dehydroalanine and N-acetyl dehydroalanine methyl ester. Int J Pept Protein Res. 1976;8:57–64. doi: 10.1111/j.1399-3011.1976.tb02481.x. [DOI] [PubMed] [Google Scholar]
- Sokolovsky M, Sadeh T, Patchornik A. Nonenzymatic cleavages of peptide chains at the cysteine and serine residues through their conversion to dehydroalanine (DHAL). II The specific chemical cleavage of cysteinyl peptides. J Am Chem Soc. 1964;86:1212–1217. [Google Scholar]
- Srivastava A, Poonkuzhali B, Shaji RV, George B, Mathews V, Chandy M, Krishnamoorthy R. Glutathione S-transferase M1 polymorphism: a risk factor for hepatic venoocclusive disease in bone marrow transplantation. Blood. 2004;104:1574–1577. doi: 10.1182/blood-2003-11-3778. [DOI] [PubMed] [Google Scholar]
- Stone RM. Optimizing treatment of chronic myeloid leukemia: a rational approach. Oncologist. 2004;9:259–270. doi: 10.1634/theoncologist.9-3-259. [DOI] [PubMed] [Google Scholar]
- Wadleigh M, Ho V, Momtaz P, Richardson P. Hepatic veno-occlusive disease: pathogenesis, diagnosis and treatment. Curr Opin Hematol. 2003;10:451–462. doi: 10.1097/00062752-200311000-00010. [DOI] [PubMed] [Google Scholar]
- Wang J, Schiller SM, Schultz PG. A biosynthetic route to dehydroalanine-containing proteins. Angew Chem Int Ed Engl. 2007;46:6849–6851. doi: 10.1002/anie.200702305. [DOI] [PubMed] [Google Scholar]
- Willey JM, van der Donk WA. Lantibiotics: peptides of diverse structure and function. Annu Rev Microbiol. 2007;61:477–501. doi: 10.1146/annurev.micro.61.080706.093501. [DOI] [PubMed] [Google Scholar]