

Abstract
We studied 613 genes which regulate immunity and, utilizing predictive algorithms, identified 285 genes as microRNA (miRNA or miR) targets. Of these, ~250 are newly predicted gene-miR interactions. The frequency of predicted miRNA binding sites in immune gene 3′ UTRs indicated preferential targeting of immune genes compared to the genome. Major targets include transcription factors, cofactors and chromatin modifiers whereas upstream factors, such as ligands and receptors (cytokines, chemokines and TLRs), were, in general, non-targets. About 10% of the immune genes were ‘hubs’ with eight or more different miRNAs predicted to target their 3′UTRs. Hubs were focused on certain key immune genes, such as BCL6, SMAD7, BLIMP1, NFAT5, EP300 and others. NF-κB and p53 do not themselves have binding sites for miRNAs but rather these pathways are targeted by miRNAs at downstream sites. MHC class II genes lacked miRNA targets but binding sites were identified in the CIITA gene and were shown experimentally to repress IFN-γ induced MHC class II activation. Unexpectedly, factors involved in regulating message stability via AU-rich elements (ARE) were heavily targeted. Moreover, multiple components involved in the generation and effector functions of miRNAs (Dicer and Argonautes) were themselves miRNA targets suggesting that a subset of miRNAs may indirectly control their own production as well as other miRNAs.
Keywords: immune genes, microRNA, AU-rich element, Dicer, chromatin factors, RNAi
1. Introduction
MicroRNAs are small, highly conserved, 20–24nt non-coding RNAs which function in cell differentiation and development. Currently 475 human miRNA sequences are listed in the miRNA registry (miRBase 9.2, May 2007) (Griffith-Jones, 2004) and it is estimated that there may be 1000 or more human miRNAs (Bentwich et al., 2005) which target ~30% of the genome and thereby regulate ~8,000 genes. Each miRNA may suppress multiple targets (average ~200) and one mRNA can be targeted by many miRNAs thereby controlling a broad spectrum of cellular process (Bartel 2007). MiRNA regulate gene expression by sequence-specific targeting of mRNA for translational repression and/or degradation. Computational methods have been designed to identify the gene targets of miRNA (Chaudhuri and Chatterjee 2007) and nearly a dozen algorithms designed to identify specific miRNA targets based on the location and extent of 3′ UTR-miRNA complementarity, free-energy parameters and evolutionary conservation. Each algorithm has somewhat different but overlapping requirements for gene target selection and their predictive capacity and use in biology and medicine has been reviewed (Chaudhari and Chatterjee 2007; Mazière et al., 2007). Integration of data from multiple algorithms as employed here has been shown to enhance the reliability of predictions (to over 90%) by reduction of the rate of false positive predictions. However, this is accompanied by increases in the false negative rate (Sethupathy et al., 2006). Experimental validations of the in silico predictions are essential for establishing the validity of the algorithm selection and understanding the biological functions of miRNA.
In animals, miRNAs repress by inhibiting translational initiation or, less commonly, by degrading mRNA. MiRNAs can also inhibit transcription although it is unclear whether they bind directly to DNA or to nascent nuclear transcripts (Wassenegger, 2005). In either case, these miR-gene interactions facilitate recruitment of chromatin modifying proteins to nuclear sites mediating heterochromatic silencing (Grewal and Moazed, 2003). MiRNAs are known to regulate gene expression during differentiation and development, but may also be involved in acute processes that require dynamic responses to specific environmental toxins, nutritional factors such as amino acid depletion, osmotic stress, nerve cell synaptic stimulation and stress-dependent cardiac hypertrophy (Bhattacharyya et al., 2006; Chiou, 2007; Marsit et al., 2006; van Rooji et al., 2007). An unresolved issue is whether miRNA expression is altered acutely by growth factors and cytokines and data addressing this is discussed below. A recent report (Taganov et al., 2006) demonstrating that miRNAs respond to bacterial components, such as LPS, which activate Toll-like receptors (TLRs) supports the view that miRNAs do respond to acute stresses including stimuli known to alter immune responses. The involvement of miRNAs in immunity is also suggested by studies of Dicer knockdown mice showing defects in T cell differentiation (Muljo et al., 2005) and T-regulatory cell development (Cobb et al., 2005). Recently, single miRs have been shown to modulate germinal center responses (Thai et al., 2007; Rodriguez et al., 2007), B cell maturation (Zhou et al., 2007) and also thymic T cell selection and the sensitivity of T cells to antigens (Li et al., 2007).
The present study demonstrates that numerous immune genes are predicted targets of miRNAs and identifies specific pathways, and locations within pathways, where miRNA control is likely to be focused. For example, although MHC genes were not predicted targets of miRNAs using multiple algorithms, the CIITA gene is shown to have 3′UTR binding sites for two miRNAs and experimental evidence is presented for miRNA regulation of the IFN-γ response. Our findings also suggest potential mechanisms by which miRNAs may activate genes, albeit indirectly, via repression of inhibitors, such as transcriptional co-factors and chromatin factors. Finally and unanticipated, was the observation of high probability miRNA targeting of the genes involved in regulating message stability via AU-rich elements (AREs) as well as the machinery components (Dicer/Argonaute) involved in miRNA silencing.
2. Materials and Methods
To examine miRNA targeting of immune genes, we first assembled a list of 613 immune genes which include certain genes known to indirectly regulate immune gene expression via general pathways and chromatin. These genes were selected from databases described below and from the immunological literature. This list of genes was then submitted to publicly available websites designed to identify potential miRNA target sites in the 3′UTR of the genes. Each of the algorithms (listed below) uses somewhat different criteria for target for target predictions and examination of predictions from two or three independent algorithms for consensus miRNA enhances the functional correlation. A UNIX script was written to sort the predictions and identify those selected by multiple algorithms. Supplemental Table 1 shows our entire gene list, the predictions of each of the three algorithms and indicates those miRs selected by 2 or 3 algorithms. The data in this table thus provides the basis for experimental studies to validate the predictions and examine their biological significance. Validation of predicted results is critical and the methods used to validate the miRNA selected to target CIITA and their impact on MHC class II expression is detailed below.
2.1 Data sets and miRNA target prediction
The list of 613 immune genes was compiled based on their known involvement in immune processes, data mining tools and the gene ontology (GO) databases which classifies genes into functional categories (http://www.geneontology.org/GO.database.shtml). We employed the GO immune category (http://david.abcc.ncifcrf.gov/) to establish our set of immune genes but also added certain genes from other existing databases of immune genes. We also compared our selected set with other immune databases. For example seventy-three percent of our selected genes are present in the ImmPort (https://www.immport.org/) and Gene Ontology Wiki Immune Gene databases (http://wiki.geneontology.org/index.php/Immunology). Importantly some genes were added which are not present in immune databases including transcription factors, cofactors, selected signaling pathway components, apoptotic genes and chromatin factors which although not classical immune genes were found in the literature to be involved in immune processes. The 27% of our genes not listed in ImmPort were in these categories.
Three algorithms TargetscanS, (http://genes.mit.edu/tscan/targetscanS2005.html) (Lewis et al., 2003), MiRanda (http://www.microrna.org/) (John et al., 2004), and PicTar (http://pictar.bio.nyu.edu/) (Krek et al., 2005) were employed for the prediction of miRNA targets. Integration of miRNA target predictions from three algorithms substantially increases the functional correlations compared to single algorithms as reported by Sethupathy (Sethupathy et al., 2006). For most of the data discussed in this study, except when noted in the text, we required consensus between two algorithms as it increases the stringency of the predictions and reduces the rate of false positive target predictions although it may exclude detection of some important functional target genes. From the databases of the three algorithms used in this study, after exclusion of any duplication, we assembled a set of 20232 unique genes that formed the basis for genome-wide analyses and is the overall population size used for computing p-values. There are currently 475 identified miRNAs in the human genome (miRBase9.2). Enrichment analyses were performed by comparing the number of miRNA target genes in the 613 immune genes to that in the 20232 genomic set. The degree of enrichment (p-values) of immune gene targets relative to the genome was assessed by Fisher’s exact test.
2.2 Cells and Reagents
The human cervical carcinoma cell line HeLa was from American Type Culture Collection (ATCC) (Manassas, VA) and cultured according to ATCC instructions. Recombinant human IFN-γ (R&D systems, Minneapolis, MN) was used at concentrations and time periods indicated.
2.3 CIITA 3′UTR reporters
RT-PCR primers were designed to amplify the proximal 1kb of the CIITA 3′UTR. The primers were designed with a 5′ extension encoding a SpeI restriction endonuclease recognition site on the sense primer and an extension encoding a HindIII restriction site on the anti-sense primer (CIITA 3′ UTR sense [3535SpeI] 5′-GACTAGTCAGCTGTGCTCTGGACAG-3′, CIITA 3′ UTR antisense [4549 HindIII] 5′-GCTAAGCTTAGGACCTGCCTGTGACATT-3′). IFN-γ treated HeLa RNA was used in RT-PCR, products of the predicted size were amplified, purified and subcloned into the SpeI and HindIII sites of the pMIR-REPORT luciferase vector (Ambion). The sequence of reporter plasmids carrying the 1000bp CIITA 3′UTR fragment downstream of the luciferase coding region were verified and termed pMIR-Luc-CIITA.
2.4 Reporter assays
Luciferase reporters (vector or 2C) were co-transfected (10:1) with pMIR-REPORT-βgal. Whole cell lysates were prepared in Passive Lysis Buffer (Promega, Madison, Wisconsin) and assayed for protein concentration (BCA, Pierce, Rockford, IL), β-galactosidase activity (Promega) and luciferase activity (Promega). Luciferase activity was normalized to β-gal activity and expressed as RLU/mU. For presentation, the normalized activity of the CIITA reporter is presented relative to the luciferase control vector.
2.5 Pre-miR transfections
HeLa cells were seeded at 40% confluency in 6 well plates and transfections were carried out using siIMPORTER™ (Millipore, Temecula, CA) according to the manufacturer’s instructions. Pre-miR-145 and -198 (Ambion) were transfected at 5–100nM followed by IFN-γ treatment for 24hrs and RNA isolation.
2.6 MiRNA isolation and RT-PCR for miRNA quantitation
Total RNA was isolated (miRVana™, Ambion) and used to perform real-time RT-PCR according to the manufacturer’s protocol (Applied Biosystems, Foster City, CA). The RT-PCR was performed in triplicate in at least three independent experiments.
2.7 RT-PCR for detection of CIITA mRNA
TaqMan real-time RT-PCR was performed to detect the CIITA transcript levels after pre-miR-145 and 198 transfections. The RNA was isolated as described above and 2μg was used for reverse transcription with the Superscript™ II enzyme (Invitrogen, Carlsbad, CA). Real-Time PCR was performed as previously described (Magner et al., 2000). The experiments were performed in triplicate in three independent experiments. Error was reported as + SEM, while values were reported as relative fold change compared to untransfected samples.
3. Results
Using computational analyses, we identified potential miRNA targets among 613 genes selected on the basis of their known involvement in immune processes (see Methods and Supplemental Table 1). Comparison of our dataset with experimentally validated functional miRNA targets (Sethupathy et al., 2006) identified 31 miR:gene interactions and of these we were able to predict 28 using three algorithms. This represents ~90% probability of miRNA predictions in accord with a previous report using a similar multi-algorithm approach (Sethupathy et al., 2006). Although agreement among three algorithms is highly predictive of binding, its stringency may lead to the exclusion of some genes with functional miRNA targets. Therefore, we present data that represents consensus of two algorithms and also includes single algorithm predictions (Supplemental Table 1).
Analysis of our gene list (see Methods), although selecting immune genes as the major targets, also identified certain general pathways including transcriptional factors and cofactors, signal transduction, chromatin remodeling, apoptosis and inflammation. These pathways, although not classified by GO as immune, are intimately involved in immune processes. Additionally, we added genes in several pathways (cell adhesion, tumor antigens, TGF-β, SNARE/vesicular transport) known to be important in immunity (see also Methods). We call miRNAs that bind to well-established immune targets ‘immunomiRs’ in analogy with the term oncomiRs for miRNAs predicted to be involved in cancer (Calin and Croce, 2006). The Supplemental Table 2 indicates the top 20 miRNAs on the basis of frequency of predicted targets in the immune genes. Alternatively, we also define immunomiRs as the miRs that preferentially target (p<0.005) immune genes relative to other genes (Supplemental Table 2). Some immunomiRs may be oncomiRs and both types of miRNAs often target genes in multiple general cellular pathways.
3.1 Immune genes are differentially regulated and enriched for miRNA targets compared to the genome
Analyses of the distribution of miRNA targets revealed three categories, depending on the number of miRNAs targeting each gene (Supplemental Figure 1). The majority (54%) of the 613 immune genes had no targets, many (~36%) had one or a few targets while ~10% are ‘hubs’ with multiple (≥8) miRNA binding sites selected by at least two algorithms. Representative genes in these categories are shown in Figure 1A. In our dataset, genes that have short 3′UTR (<2000) are seldom targets (30%) while genes with long 3′UTRs (>2000nt) are common miRNA targets (80%). The number of 3′UTR binding sites is an important determinant of the level of inhibition achieved by miRs, with substantially enhanced repression upon binding of multiple miRNAs (see Discussion).
Figure 1. Examples of miRNA targeting of immune genes.

A, Genes were placed in three categories according to the number of miRNA binding sites in the 3′UTR. B, MiRNA regulation of major cellular and immune pathways showing the contrast between genes which are targets versus non-targets in the same general pathway. (* indicates genes in the pathway that do not have miRNA target sites)
As shown in Table 1, immune genes as a group are significantly enriched for miRNA targets compared to the genome as a whole. The enrichment analyses indicated that specific pathways were preferentially targeted by miRNAs. For example, genes related to transcription, signal transduction and chromatin factors were highly enriched for targets of miRNA (Table 1). Similarly, the TGF-β, JAK/STAT pathways and SNAREs were frequent targets. In contrast, chemokines, cytokines, TLRs, complement components, costimulatory factors, adhesion molecules and tumor antigens were generally poor or non-targets and there were no hubs in these groups. T cell, B cell and apoptotic pathways were targeted by miRNAs but were not enriched compared to the genome. However, it is important to note that the analyses employed in Table 1 were based on the number of genes in the pathway having miRNA targets and did not consider hubs. For example, in the T cell, B cell and apoptotic pathways there are multiple hubs and specific components may be heavily targeted. It is emphasized, however, that single miRs selected by one algorithm can have important biological properties (see below). Forty-three of the immune genes are high probability targets of single miRNAs (for example, FAS, CTLA4, RAG1, IL12B (Supplemental Table 1)). An analysis of immune gene targets in relation to their cellular location, similar to that suggested by Cui et al (Cui et al., 2006) for neural genes, is shown in Figure 2. Although the methods of analyses were somewhat different, overall the immune and neural genes have similar patterns of miRNA targeting with ligands and receptors being poor targets compared to the signaling and nuclear transcriptional and chromatin pathways.
Figure 2. Cellular distribution of miRNA targets in immune pathways.

The protein targets are classified based on their cellular localization in four categories. The percentages are based on the number of genes in each location predicted to be targeted by miRNAs by at least 2 algorithms. A number of important pathways (RNA stability, apoptosis and RNAi pathway) could not be distributed into these spatial categories and were not included in the analysis. Figure patterned after the neuronal pathways reported by Cui, Q. et al., 2006.
Analysis of pathways that are rich in miRNA binding sites often revealed unexpected selectivity among structurally and functionally related genes. Examples of immune genes, chosen to illustrate the contrast observed within the same general pathway between genes predicted to be targets or non-targets, are shown in Figure 1B. For example, STAT1, 2 are non-targets whereas STAT3, 4, 5 and 6 are predicted to each bind a distinct set of miRNAs. Numerous other examples were identified (Supplemental Table 1). For instance, IL12 is targeted by a single miRNA (miR-153) but only the IL-12 Beta-chain has a miR-153 binding site. Elucidation of the mechanisms governing the differential selection of miRNA targets by pathway components may lead to enhanced insight into the regulation of immune responses.
3.2 MiRNAs in innate immunity
Of importance in understanding how miRNAs may function in the immune system is the observation that all ten toll-like receptors (TLRs) examined have no predicted target sites. Similarly, the genes in the complement pathway, which are important constituents of innate as well as adaptive immunity, had no predicted miRNA targets (Supplemental Table 1). Moreover, only 23% of the 43 chemokine & chemokine receptor genes and 29% of the 34 cytokine genes examined were miRNA targets (Supplemental Table 3). The relative paucity of miRNA binding sites in TLRs, chemokines and most cytokines compared to the genome (Table 1), suggests that other elements may regulate message stability in these genes. It is well known that AU-rich elements (AREs) located in the 3′UTRs mediate mRNA stability (Bakheet et al., 2006). Examination of the mRNAs of TLRs and the majority of chemokines revealed no AREs (Supplemental Table 1), however, 16 (~49%) of the cytokines studied have AREs (Figure 3). Thus, overall 63% of the cytokines examined are potentially regulated by message stability via miRNAs and/or AREs. Furthermore regulation of ARE-binding proteins by miRNAs may add another layer of control (discussed below).
Figure 3. MiRNAs and MAP Kinase may indirectly regulate multiple inflammatory genes by regulating the components of the ARE machinery.

The expression of multiple inflammatory molecules and immune genes is known to be regulated by mRNA stability mediated by AU-rich elements located in their 3′UTRs. The predictions suggest that the expression of the machinery components for ARE are regulated by miRNA. The specific miRNAs targeting the ARE machinery are listed in Supplemental Table 7.
3.3 MiRNA in adaptive immunity
Multiple miRNAs are predicted to be involved in Th1, Th2, Th17 and Treg differentiation and are localized to at least eight specific steps in these pathways (Figure 4 and Supplemental Table 4). Undoubtedly this minimalistic representation of T cell differentiation is oversimplified. For example, we predict no miRNA targets in FOXP3, but high probability sites in IL-10 and CTLA4 genes that play a role in the development of Treg (Li et al., 2006; Rudensky et al., 2006 and Read et al., 2006). As mentioned above single miRNAs can dramatically effect gene expression and function and an important recent study (Li et al., 2007) has shown that miR-181a is critical in T cell development and regulates the quantitative levels of the T cell response to antigens. These effects were attributed to repression of several phosphatases by miR-181a but definitive targets were not identified. We find that miR-181a targets ~600 genes (single algorithm prediction -PicTar) which include 10 phosphatases but also other factors that could influence T cell function, including CD4, BCL6, MECP2, and TGFBR1 (Supplemental Table 1).
Figure 4. Potential miRNA regulation of T cell differentiation.

The schematic diagram describes the primary effectors of Th1, Th2, Th17 and Treg differentiation pathways and indicates the predicted miRNA targeting of these pathways. The specific miRNAs predicted to target each of the effector genes can be found in Supplemental Table 3.
The genes for TGF-β1 and its receptors, TGF-βRII, Endoglin and Cripto-1, have no miRNA binding sites. However, all 7 SMADs examined are predicted miRNA targets and also TGIF, a SMAD pathway co-repressor, has multiple high probability miRNA binding sites. MiRNAs which repress these TGF-β inhibitors would be expected to enhance TGF-β signaling. Thus, the TGF-β pathway is a rich source of miRNA binding sites; of 24 components analyzed, 17 were predicted targets and of those nine were hubs for a total of 64 different miRNAs (Supplemental Table 5).
B cell differentiation and activity are also potentially regulated by multiple miRNAs. For example, the miRNA hub BCL6 binds several co-repressors (BCOR, NCOR, SMRT), as well as the remodeling complex NURD/Mi2 (Fugita et al., 2004), which are all likely sites of miRNA regulation (Supplemental Table 6). A recent publication demonstrated that miR-127 translationally inhibits Bcl6 protein levels in cancer cells (Saito et al., 2006). One algorithm, miRanda, identified a binding site for miR-127 again emphasizing the necessity of examining the results from individual algorithms (Supplemental Table 1). The Bcl6 repressor also binds and recruits HDAC4 which is a miRNA hub with 14 target sites (Lemercier et al., 2002) (Supplemental Table 1). The Blimp1 transcription factor which has been implicated in chromatin mediated repression during B cell to plasma cell differentiation and specifically in silencing the CIITA gene (Gyorgy et al., 2004) is targeted by 3 miRNAs (Supplemental Table 1). Thus, induction of plasma cell differentiation might be expected to be associated with down-regulation of miRNAs targeting Blimp, a testable thesis using RT-PCR detection of miRNA levels and anti-miRs. The unique patterns of miRNA targets, illustrated by the above data are likely important clues to some of the underlying regulatory mechanisms in T and B cell differentiation and function.
3.4 MiRNA involved in general pathways (transcription factors, cofactors and chromatin modifiers) that also regulate immunity
3.4.1 MiRNA targeting of transcription factors
We surveyed 96 transcription factors thought to be important in immune pathways (Supplemental Tables 1 and 7). Even in this relatively small group interesting relationships emerge. For example, the NFAT family consists of 5 members (NFAT1-5) and all, except NFAT3, are expressed in cells of the immune system. Although they are required for T cell activation, NFAT1 and NFAT2 do not have miRNA binding sites, but rather NFAT4 and NFAT5 are likely targets. All of the NFATs are regulated by calmodulin (4 miRNA sites) and calcineurin (15 miRNA sites) and thus, the calcium pathway appears to be heavily targeted by miRNAs (Supplemental Table 8).
The NF-κB family of transcription factors regulates the expression of a large number of genes, many of which are involved in apoptosis, inflammatory processes and stress response, as well as in T and B cell responses (Karin and Greten, 2005). LPS signaling through TLR-4 activates NF-κB and induces miR-146. The molecular targets for miR-146 include the downstream factors IL-1 receptor associated kinase (IRAK-1) and the TNF receptor associated factor 6 (TRAF6) (Taganov et al., 2006). We also predict these targets of miR-146 and, in addition, NFKB1 itself, but not other NF-κB species, has a high probability binding site for miR-9. The transcription factor p53 is a central player in cell proliferation and apoptosis and has extensive cross-talk with NF-κB (Huang et al., 2007). The paucity of miRNA binding sites in TP53 3′UTRs (Supplemental Table 1) was therefore surprising. Perhaps, like NF-κB, miRNAs also affect p53 pathways via downstream targets such as the cell cycle inhibitors p21 and p27. Indeed, our algorithms predict that both the p21 and p27 repressive factors but not p53 are regulated by miRNAs (Supplemental Table 9). Moreover, the miR-34 family (miR-34a, b, & c) has recently been found to target multiple downstream genes involved in the p53 tumor suppressor pathways and transfection of miR-34 leads to cell cycle arrest and senescence (He et al., 2007). Thus, it appears from the above data that the regulation by miRNAs of both the NF-κB and p53 pathways may occur at downstream targets.
3.4.2. MiRNA targets of transcription cofactors and chromatin modifier
In addition to regulating transcription factors, miRNAs may alter immunity by targeting cofactors, as well as chromatin modifiers, which frequently function as cofactors. MiRNAs target both coactivators and corepressors and thus can potentially regulate gene expression both positively and negatively. Chromatin regulators are known to be involved in diverse immune pathways (Tomasi et al., 2006; Smale and Fisher, 2002) and are frequent components of cofactor complexes. The chromatin pathways and their specific miRNAs are illustrated in Figure 5 and specific miRNA targets are listed in Supplemental Table 10. It is notable that both HATs and HDACs are targets and therefore miRNAs may also mediate activation or silencing via chromatin effects. The closely related HATs, CBP and p300, have binding sites for 19 and 31 individual miRNAs respectively and of these ten are common to both genes (Supplemental Table 10). Striking selectivity was found in HDAC targets with miRNA sites in class II (HDAC4, 5, 9), especially the hub HDAC4, but not class I HDACs. SIRT1, a sirtuin class III HDAC, is a predicted target of 29 individual miRNAs (Supplemental Table 10). Selective targeting by miRNAs is also shown for the enzymes that regulate histone methylation (Supplemental Table 10), such as SUV39H1 and EZH2, which methylate H3K9 and H3K27 respectively. MiRNAs also target Jmjc domain histone demethylases (Supplemental Table 10). For example, miR-23 is predicted to bind JMJD2A which removes trimethyl groups from H3K9me3 and H3K36me3. The effect of miR-23 targeting of JMJD2A is unknown but would be expected to enhance histone methylation. Although Jmjc proteins are likely involved in immunity there are, to our knowledge, no reports of this to date.
Figure 5. Role of miRNA in chromatin regulation.
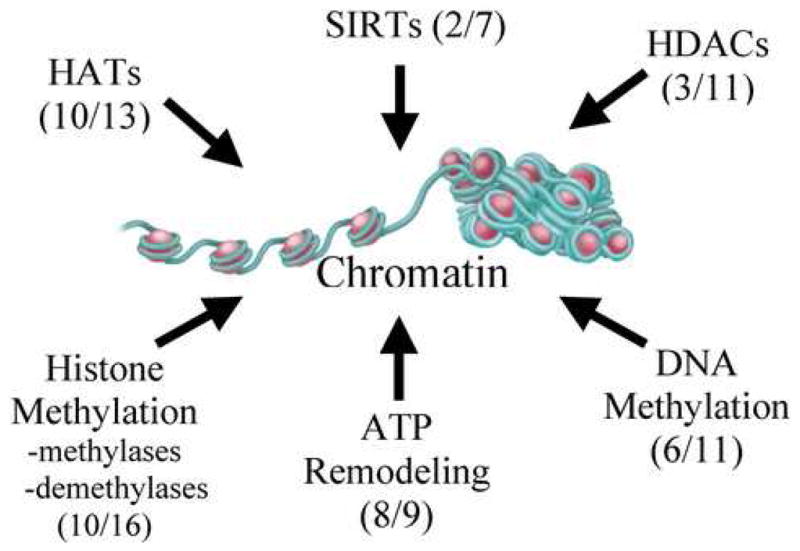
Multiple pathways contribute to the regulation of chromatin structure and thereby gene expression. For each category of chromatin regulator, the number of genes found to have miRNA targets (consensus of two algorithms) and the total number of genes analyzed in that category are shown in parentheses. Our predictions suggest significant contributions to chromatin regulation in multiple pathways from miRNA. See Supplemental Table 11 for specific miRNA targets.
3.4.3. MiRNAs targeting DNA methylation
DNA methylation is initiated and maintained in mammalian cells by three major enzymes; the de novo DNA methyltransferases DNMT3A/B and the maintenance enzyme DNMT1 (Baylin, 2005) each of which has predicted miRNA targets, and DNMT3A is a hub (Supplemental Table 11). Both methyltransferases are targeted by a common miR (miR-29) and a recent report (Fabbri et al., 2007) demonstrates that in lung cancers having low miR-29 and high Dnmts, the enforced expression of miR-29 restores normal methylation patterns. DNA methylation is also associated with gene repression mediated by methyl-CpG binding domain (MBD) proteins. We find that MECP2 mRNA is predicted to bind 76 individual miRNAs and is the most highly targeted gene in our data set.
3.5 The machinery components involved in the generation of ARE mediated message stability are targeted by miRNAs
Since both miRNAs and AREs regulate message stability, the question arises as to the relation between these two post-transcriptional mechanisms. At least one miRNA (miR-16) binds to an ARE and degrades mRNA by an Argonaute 2 dependent mechanism (Jing et al., 2005). MiR-16 has 801 gene targets (single algorithm prediction-PicTar) of which 217 potentially contain AU-rich sequences in their 3′UTR (ARED registry) (http://rc.kfshrc.edu.sa/ared). MiR-16 is ubiquitous, present at high levels in cells and is therefore potentially a ‘master miR’ involved in determining mRNA stability via AREs. However, only three of the genes in our data set with miR-16 target sites have AREs (SOCS2, AGO1, RARB). Interestingly, examination of the SOCS2 miR-16 target (miRanda) (Supplemental Figure 2) indicates that the ARE binding site is at position 11–15 and therefore outside the 5′ seed region of miR-16 (see Discussion). Message stability is influenced by several protein factors (ARE binding proteins - ARE-BP) which bind directly or indirectly with other protein factors at or near the ARE and either stabilize or destabilize the message (Barreau et al., 2005). These proteins regulate distinct types of AREs and are thought to function by recruitment and/or affecting the activity of deadenylation and exosome enzymes (Hau et al., 2007; Mukerjee et al., 2002). Importantly, the AU binding proteins TTP, AUF1 and the ELAV family members (HuA/HuR, HuB, HuC and HuD) are miRNA targets (Figure 3 and Supplemental Table 12). MiRNA regulation of the components of the ARE machinery factors potentially has broad implications (see discussion).
3.6 Regulation of Dicer and Argonaute by miRNAs
We recently found that Dicer levels of human cells vary substantially between cell types (data not shown) and several reports demonstrate that Dicer may be up or down-regulated in different cancer types (Chiosea et al., 2006; Karube et al., 2005). Unexpectedly, we found that key components of the miRNA machinery were themselves predicted targets of specific sets of miRNAs (Supplemental Table 13). The RNase III enzyme, Drosha, and its RNA binding partner, Pasha/DGCR8, were not targets of miRNAs. However, Exportin-5, required for pre-miRNA transport from the nucleus to the cytoplasm, was the target of two miRNAs. Strikingly, the DICER1 3′UTR was found to have 25 individual miRNA binding sites of which 6 were selected by all three algorithms. Notably, an intense focus of miRNAs occurred at AGO1, with 62 potential miRNA binding sites in its 5682 bp 3′UTR. Of these, 20 high probability sites were selected by three algorithms. AGO 2, 3 and 4 had 4, 1 and 12 miRNA binding sites respectively.
The let-7 miRNA targeted all four Argonaute family members and one algorithm (PicTar) also indicated a let-7d site in DICER1. Other common miRNAs between DICER1 and AGO1 include miR-122, -103, -107, -124, -130, and -29. These findings suggest feedback regulation of miRNA synthesis and have broad implications. Additional experiments, currently in progress, are obviously required to clarify the functional significance of the miRNA targets we identified in the RNAi machinery components.
3.7 The MHC class II response to IFN-γ is regulated by miRNAs; functional validation
The large menu of immune genes potentially targeted by miRNAs, discussed above, represents a rich source of gene-miRNA interactions which have important implications in immunity. However, the computational approach represents but a first step in elucidating the scope of immune regulation by miRNAs and establishing the function of miRNA targets defined computationally is essential. We have selected the MHC class II genes for further investigation since class II is a critical factor in the initiation of antigen presentation and this pathway is a major determinant of immune surveillance to infectious agents and tumors (Dunn et al., 2004).
MiRNA target predictions failed to detect sites in the 3′UTR of 20 MHC class I and II genes (Supplemental Table 1), although several sites were found in the MHC co-activator CIITA (Figure 6). DeLerma Barbaro et al. characterized a segment of the CIITA 3′UTR that regulates mRNA stability during phorbol ester induced differentiation of U937 cells (DeLerma Barbaro et al., 2005) and contains two non-canonical AREs. The target sites for miR-145 and -198, although in this distal segment (nucleotides 3893–4543) of the CIITA 3′UTR, were distinct from the putative AREs. A second miR-198 site was identified proximal to the translation terminator (Figure 6). At each of the CIITA binding sites, the miR-145 and -198 5′ seeds showed perfect complementarity with their targets. To confirm these observations, we engineered luciferase reporter constructs which included the predicted miRNA target sites. HeLa cells co-transfected with these reporters and pre-miR-145 or -198 displayed decreases in luciferase activity relative to cells carrying the reporter without a miRNA target site (Figure 6). Transfection of combinations of miR-145 and -198 did not, however, enhance repression over that observed for the individual miRNAs (data not shown).
Figure 6. MiRNA regulation of CIITA and MHC class II induction.

A, Schematic representation of the CIITA transcript illustrating the predicted miRNA binding sites and AREs in the 3′UTR. The black bar indicates the fragment present in the pMir-Luc-CIITA reporter construct. B, miR-145 inhibits the IFN- γ induction of HLA-DR in HeLa cells. HeLa cultures were transfected with pre-miR-145, treated with IFN- γ and, after 24hrs, total RNA (mirVana, Ambion) was analyzed for CIITA and HLA-DR α expression by real time RT-PCR. The negative control, NC1, non-targeting pre-miR, and untransfected cells are shown as controls. C, miR-145 and 198 inhibit a heterologous reporter when the CIITA 3′UTR is present. HeLa cells were transfected with reporter constructs carrying 1kb of the CIITA 3′UTR (pMir-Luc-CIITA) or no insert (pMir-Luc) along with 100nM pre-miR-145 or 198 and, 24hr later, whole cell lysates were analyzed for luciferase activity. Co-transfection with pMIR- βgal allowed normalization to β-galactosidase activity.
To determine whether miR-145 and -198 were capable of functionally regulating CIITA, we transfected pre-miRs-145 and 198 into HeLa cells and analyzed MHC class II expression in response to IFN-γ. Since active CIITA protein is required for the IFN-γ induction of MHC class II genes, inhibition of CIITA translation by miR-145 transfection can be assessed by monitoring HLA-DRα mRNA. As shown in Figure 6, miR-145 transfection of HeLa cells inhibited the induction of MHC class II mRNA by IFN-γ with little change in CIITA mRNA. This data also suggests that miR-145 is primarily repressing translation. Since active CIITA protein is required for the IFN-γ induction of MHC class II genes, the inhibition of HLA-DRα mRNA by miR-145 transfection demonstrated the functional repression of CIITA. MiR-198 transfection also inhibited the induction of HLA-DRα, though less efficiently than miR-145, without affecting CIITA mRNA levels (data not shown).
None of the predictive algorithms used here identified miR-145 targets in five other interferon responsive genes (GBP1, ICAM1, IRF1, ISG54, 2′5′OAS). Interestingly however, one algorithm (TargetScanS) predicts that miR-198 targets the CIITA, GBP, ICAM, ISG54 and 2′5′OAS but not IRF1 genes. This suggests that miR-198, but not -145, could have broader specificity for the IFN-γ regulated genes and further studies are needed to clarify this issue.
It is noteworthy that, in addition to CIITA, miRNAs may regulate other components of the MHC class II enhanceosome. A count of all miRNAs predicted in our study that focus on the components of the class II enhanceosome (Supplemental Table 14) indicated the potential for regulation by ~50 different miRNAs.
4. Discussion
Since little information is currently available on immune gene regulation by miRNAs, we initially utilized a bioinformatics approach to predict immune gene targets of miRNAs. Forty-six percent (285) of the 613 immune genes studied were targets of one or several miRNAs. Of the 285 immune genes targeted by miRNAs in our study, 26 are currently in TARBase (Sethupathy et al., 2006) and several others have been separately reported (Jing et al., 2005; Taganov et al., 2006; Si et al., 2007). Our data therefore identifies ~250 immune genes which are newly predicted miRNA targets. Additionally, in this study we provide functional data and verification of miRNA regulation of CIITA and thereby the MHC class II genes. It should be emphasized however that our dataset does not encompass all immune genes and, moreover, the multi-algorithm approach employed, although highly predictive, may well miss some functionally relevant miR-gene interactions.
Of the 285 immune genes we predict as miRNA targets, 65 are hubs with eight or more 3′UTR binding sites. The potential significance of the number of binding sites is suggested by previous reports indicating that the level of gene repression is enhanced by multiple 3′UTR binding sites for the same miRNAs (Pillai et al., 2005; Mayr et al., 2007) as well as the observation of exponential enhancement of gene repression when different miRNAs target different sites on the same UTR (Doench and Sharp, 2004). In this study, we did not determine the number of binding sites for a given miRNA except for the CIITA gene. The data obtained indicates that immune genes are targeted by miRNAs statistically more frequently than the genome as a whole (Table 1). The analyses also show that miRNAs generally tend to avoid upstream factors in that ligands and receptors are poor or non-targets of miRNAs. It is emphasized that the data in Table 1, which presents an overview of miRNA gene targets, does not necessarily indicate a functional activity of miRNAs in regulating each pathway but rather the potential for regulation.
MiRNAs are known to regulate many important cellular pathways but in many the identity of the direct miRNA targets is elusive. For example, miR-155 has been identified as a requirement for normal immune function and regulation of a spectrum of genes in germinal centers, including cytokines and chemokines. However, its exact gene targets have not been defined (Rodriguez et al., 2007; Thai et al 2007). We find that miR-155 likely does not directly target cytokines, or their receptors, and have identified several potential targets, including the hub BCL11A. The BCL11A repressor is an attractive target since it interacts with Bcl6, is expressed abundantly in germinal centers and is required for germinal center differentiation of T and B cells (Liu et al., 2003).
Transcriptional and chromatin cofactors are common miRNA targets and can act as sensors which integrate numerous signals from environmental stimuli to generate appropriate tissue specific responses (Rosenfeld et al., 2006). While miRNAs probably function solely by repression, the various cofactors they target can repress or activate gene expression. Therefore miRNAs, by regulating cofactors, may either activate or inhibit gene expression albeit indirectly. Surprisingly, class I HDACs, often components of immune pathways, are non-targets of miRs, while HDAC4 and SIRT1 are hubs. HDAC4 is involved in muscle differentiation but also in MHC class II expression (Wang et al., 2005) while SIRT1 serves as a sensor for the metabolic state of a cell and has been implicated in stress, aging and cancer (Longo and Kennedy, 2006). Both are excellent candidates for miRNA regulation of important immune and other pathways. SIRT1 synergizes with other HDACs, including HDAC4, and preferentially deacetylates H3K9, H4K16 and H4K20 in mammalian cells. It has widespread affects on cellular processes through modulation of NF-κB and FOXO transcription factors. Despite the involvement of SIRT1 in many important pathways (Anastasiou and Krek, 2006) its immune function remains largely unexplored. Moreover, since little is known of the pathway that regulates sirtuins, the finding of multiple high probability miRNA binding sites is of substantial interest.
Highly selective targeting by miRNAs was also suggested for the enzymes that regulate histone methylation (Supplemental Table 11) including SUV39H1 and EZH2 which methylate H3K9 and H3K27 respectively. H3K9me3 is a repressive modification whereas H3K27me3, a polycomb binding site, has been characterized as a memory mark (Bernstein et al., 2006) and also may be an epigenetic signature of cancer stem cells (Widschwendter et al., 2007).
In cancer cells DNMTs mediate DNA methylations which repress tumor suppressor and immune genes, via methyl binding proteins and HDAC, and these factors are frequently targeted by miRNAs. The miR-29 family members (29a and 29b) which target DNMTs are deficient in lung cancer and this has been related to enhanced DNA methylation and gene silencing of tumor suppressor genes. The levels of DnmT3A/B in lung cancer are inversely correlated with miR-29 and overexpression of miR-29 in lung cancer lines induces re-expression of silenced tumor suppressor genes and inhibits tumors in vivo (Fabbri et al., 2007). The Let-7 miRNA is also reported to be deficient in poor prognosis lung cancer (Karube et al., 2005; Harris et al., 2006). Since the RAS oncogene is a target of Let-7 (Johnson et al., 2005) it has been suggested that let-7 is a tumor suppressor (Takamizawa et al., 2004) which functions by repressing RAS. In sum, most pathways of chromatin regulation examined, including HAT, HDAC, histone methyltransferases and demethylases, DNA methylases, methyl binding proteins and chromatin remodeling factors, are heavily targeted by miRNAs (Supplemental Tables 10 and 11). It appears likely therefore, that a major focus of miRNA targeting of immune gene expression occurs via chromatin and that both gene activating and repressing chromatin factors are regulated.
There are numerous examples in this data in which the rationale for the cell’s choice of miRNA:gene targets is not obvious (Figure 1B). These may be ‘clues’ and future in-depth studies of these miRNA:gene interactions may enhance our understanding of important regulatory pathways. For example, the differential targeting of the methyl binding proteins MBD2 and MBD3 by miRNAs led us to identify five other pairs of duplicated genes in which only one gene of the pair has a miRNA target site (data not shown). Speculatively, miRNAs could have evolved to regulate the overlapping functions of closely related duplicated genes and studies of selected duplicated genes are ongoing to evaluate this thesis. However, this phenomenon is not restricted to duplicated genes, and RAG1/2 and HRAS/KRAS are also examples where only one member of the gene pair is a miRNA target (Supplemental Table 1). As mentioned, structurally and functionally closely related genes are most often targeted by quite different sets of miRNAs. However, as illustrated by DnmT3a/b, CBP/p300 and all four argonaute genes, certain genes with related functions are targeted by one or more common miRNAs.
Recent reports of miRNA regulation by LPS (Taganov et al., 2006), amino acid depletion (Bhattacharyya et al., 2006), folate deficiency and sodium arsenite (Marsit et al., 2006) suggest the existence of stress miRNAs. Importantly, these data also indicate that stress may induce transcriptional factors, such as NF-κB and p53 which bind to promoter sequences in both coding and non-coding (miRNA) genes. This could lead to gene activation on the one hand and miRNA repression of a different set of downstream genes on the other. Since miRNAs often target hundreds of genes, miRNA induction may be a rapid and efficient means (without making protein) of silencing a bank of genes following various stresses. Additionally, miR-127 and let-7 have promoter CpG islands and are upregulated by inhibitors of DNA methylation (Saito et al., 2006; Brueckner et al., 2007). HDACi have also been shown to alter miRNA levels in a breast cancer cell line (Scott et al., 2006). Our recent studies show a rapid enhancement of miR-127, -122a and several other miRNA following histone deacetylase inhibitor treatment but this is highly contextual and dependent on the cell type (unpublished data). Additional studies are required to elucidate this mechanisms and pathways by which acute stresses regulate miRs and genes.
This study also establishes a cytokine mediated pathway in which miRNAs directly modulate MHC expression. Both miR-145 and -198 inhibit the function of CIITA, but do not affect mRNA levels, suggesting translational inhibition. MiR-145 is not predicted to target other components of the IFN-γ pathway. However, miR-198 has homology sites in multiple IFN-γ regulated genes suggesting the possibility of two types of control mechanisms by miRNAs; a broader pathway (miR-198) encompassing several components and gene specific (miR-145) regulation. These results suggest a novel mechanism of MHC class II regulation not previously recognized. MiR-145 is a tumor suppressor whose levels are reduced in certain colon cancers (Calin and Croce, 2006) and thus, miR-145 may be both an oncomiR and an immunomiR.
An unanticipated finding was the identification of numerous high probability miRNA targets in several of the machinery components of the ARE binding proteins regulating message stability. As the ARE sequences are present in the 3′ UTR of many cytokines and interferons, we propose that message levels of these inflammatory genes can be indirectly controlled by miRNAs via their effects on the ARE binding proteins. Since, about 8% of human mRNA contain ARE, these sequences may affect many important biological processes including cell growth, DNA binding, transcription, signal transduction, host defense (inflammation and immune response), cell cycle, apoptosis and energy metabolism (Khabar et al., 2005). Thus miRNA regulation of the ARE binding proteins may indirectly affect diverse biological processes and cellular stresses. Additionally, many of the ARE-BPs are regulated by phosphorylation which is linked to stress pathways; for example, MAPK phosphorylation of TTP inactivates this destabilizing protein and enhances mRNA expression levels for TNF-α and other inflammatory cytokines (Deleault et al., 2008; Hitti et al., 2006; Dean et al., 2004). Considering that the miRNAs themselves may be indirectly activated by various stresses suggests that complex circuitries with potential feedback mechanisms may regulate ARE function (Leung et al., 2007). Defects in these ARE-mediated processes, as illustrated by the autoimmune syndrome following TTP knockdown, could be involved in inflammatory disorders (Taylor et al., 1996; Carrick et al., 2007). The finding of a potential ‘collaboration’ between the miR-16 seed region and an ARE at position 11–15 of the target site in the SOCS2 gene is also of interest. These additional interactions may be necessary for the functional activity of miR-16: SOCS2 interactions, similar to that described for miR-16: TNF-α (Jing et al., 2005). Our findings are consistent with the previous report of Jing et al (Jing et al., 2005) showing that TTP does not by itself bind miR-16 but does associate with the RISC complex and assists in ARE mediated degradation. Speculatively, this type of cooperative activity could have arisen to extend the functional activity of miRNAs and AREs.
The Dicer/Argonaute pathway is currently under intensive investigation and the data presented here suggests a level of regulation not previously appreciated. These results, if verified by functional studies currently in progress, introduce the possibility of feedback loops whereby activation or repression of specific miRNAs, by a set of ‘master miRNAs’ targeting exportin-5/Dicer-1/Ago components, could alter overall levels of miRNA and potentially affect gene expression levels of ~30% of the genome estimated to be regulated by miRNAs.
The findings in this study implicate miRNAs in the regulation of a broad landscape of immune genes. The data suggests a novel explanation (i.e. repression via miRNA) for the failure of certain immune genes to respond to external stimuli and, helps explain how a general signal via a receptor and its pathway, that activates multiple genes (e.g. IFN-γ), can, in certain cells and conditions, mediate a restricted response in one or a small subset of its regulated genes. Such a system, mediated by miRNAs, would add substantial plasticity to regulatory pathways. The study also outlines additional mechanisms by which chromatin, inflammation and perhaps immune memory may be regulated and therefore potentially provides data helpful in the design of agents that target miRNAs as treatment modalities in immune and other diseases including cancer.
Supplementary Material
Acknowledgments
This work was supported by an NIH grant HD17013 and utilized core facilities supported in part by an NCI Cancer Support Grant CA16056.
The authors wish to thank A. Latif Kazim for reviewing the manuscript and helpful advice and members of the Molecular Immunology laboratory for their assistance and comments.
Abbreviations
- miRNA
microRNA
- miR
microRNA
- TLRs
toll-like receptors
- CIITA
MHC class II transactivator
- UTR
untranslated region
- ARE
AU-rich element
- GO
gene ontology
- HAT
histone acetyltransferase
- HDAC
histone deacetylase
- Luc
luciferase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anastasiou D, Krek W. SIRT1: Linking Adaptive Cellular Responses to Aging-Associated Changes in Organismal Physiology. Physiology. 2006;21:404–410. doi: 10.1152/physiol.00031.2006. [DOI] [PubMed] [Google Scholar]
- Bakheet T, Williams BR, Khabar KS. ARED 3.0: the large and diverse AU-rich transcriptome. Nucleic Acids Res. 2006;34:D111–114. doi: 10.1093/nar/gkj052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barreau C, Paillard L, Osborne HB. AU-rich elements and associated factors: are there unifying principles? Nucleic Acids Res. 2005;33:7138–7150. doi: 10.1093/nar/gki1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- Baylin SB. DNA methylation and gene silencing in cancer. Nat Clin Pract Oncol. 2005;2:S4–11. doi: 10.1038/ncponc0354. [DOI] [PubMed] [Google Scholar]
- Bennett D, Alphey L. PP1 binds Sara and negatively regulates Dpp signaling in Drosophila melanogaster. Nat Genet. 2002;31:419–423. doi: 10.1038/ng938. [DOI] [PubMed] [Google Scholar]
- Bentwich I, Avniel A, Karov Y, Aharonov R, Gilad S, Barad O, Barzilai A, Einat P, Einav U, Meiri E, Sharon E, Spector Y, Bentwich Z. Identification of hundreds of conserved and nonconserved human microRNAs. Nat Genet. 2005;37:766–770. doi: 10.1038/ng1590. [DOI] [PubMed] [Google Scholar]
- Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, Fry B, Meissner A, Wernig M, Plath K, Jaenisch R, Wagschal A, Feil R, Schreiber SL, Landers ES. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006;125:315–326. doi: 10.1016/j.cell.2006.02.041. [DOI] [PubMed] [Google Scholar]
- Bhattacharyya SN, Habermacher R, Martine U, Closs EI, Filipowicz W. Relief of microRNA-mediated translational repression in human cells subjected to stress. Cell. 2006;125:1111–11124. doi: 10.1016/j.cell.2006.04.031. [DOI] [PubMed] [Google Scholar]
- Brueckner B, Stresemann C, Kuner R, Mund C, Musch T, Meister M, Sultman H, Lyko F. The Human let-7a-3 Locus Contains an Epigenetically Regulated MicroRNA Gene with Oncogenic Function. Cancer Res. 2007;67:1419–1423. doi: 10.1158/0008-5472.CAN-06-4074. [DOI] [PubMed] [Google Scholar]
- Calin GA, Croce CM. MicroRNA-cancer connection: the beginning of a new tale. Cancer Res. 2006;66:7390–7394. doi: 10.1158/0008-5472.CAN-06-0800. [DOI] [PubMed] [Google Scholar]
- Calin GA, Croce CM. MicroRNA signatures in human cancers. Nat Rev Cancer. 2006;6:857–866. doi: 10.1038/nrc1997. [DOI] [PubMed] [Google Scholar]
- Carrick DM, Chulada P, Donn R, Fabris M, McNicholl J, Whitworth W, Blackshear PJ. Genetic variations in ZFP36 and their possible relationship to autoimmune diseases. J Autoimmun. 2006;26:182–196. doi: 10.1016/j.jaut.2006.01.004. [DOI] [PubMed] [Google Scholar]
- Chan JA, Krichevsky AM, Kosik KS. MicroRNA-21 is an antiapoptotic factor in human glioblastoma cells. Cancer Res. 2005;65:6029–6033. doi: 10.1158/0008-5472.CAN-05-0137. [DOI] [PubMed] [Google Scholar]
- Chaudhary K, Chatterjee T. MicroRNA detection and target prediction: integration of computational and experimental approaches. DNA Cell Biol. 2007;26:321–337. doi: 10.1089/dna.2006.0549. [DOI] [PubMed] [Google Scholar]
- Chiosea S, Jelezcova E, Chandran U, Acquafondata M, McHale T, Sobol RW, Dhir R. Up-regulation of dicer, a component of the MicroRNA machinery, in prostate adenocarcinoma. Am J Pathol. 2006;169:1812–1820. doi: 10.2353/ajpath.2006.060480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiou TJ. The role of microRNAs in sensing nutrient stress. Plant Cell Environ. 2007;30:323–332. doi: 10.1111/j.1365-3040.2007.01643.x. [DOI] [PubMed] [Google Scholar]
- Cimmino A, Calin GA, Fabbri M, Iorio MV, Ferracin M, Shimizu M, Wojcik SE, Aqeilan RI, Zupo S, Dono M, Rassenti L, Alder H, Volinia S, Liu CG, Kipps TJ, Neqrini M, Croce CM. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc Natl Acad Sci U S A. 2005;102:13944–13949. doi: 10.1073/pnas.0506654102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cismasiu VB, Ghanta S, Duque J, Albu DI, Chen HM, Kasturi R, Avram D. BCL11B participates in the activation of IL2 gene expression in CD4+ T lymphocytes. Blood. 2006;108:2695–2702. doi: 10.1182/blood-2006-05-021790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobb BS, Nesterova TB, Thompson E, Hertweck A, O’Connor E, Godwin J, Wilson CB, Brockdorff N, Fisher AG, Smale ST, Merkenschlager M. T cell lineage choice and differentiation in the absence of the RNase III enzyme Dicer. J Exp Med. 2005;201:1367–1373. doi: 10.1084/jem.20050572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Q, Yu Z, Purisima EO, Wang E. Principles of microRNA regulation of a human cellular signaling network. Mol Syst Biol. 2006;2:46–52. doi: 10.1038/msb4100089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean JL, Sully G, Clark AR, Saklatvala J. The involvement of AU-rich element-binding proteins in p38 mitogen-activated protein kinase pathway-mediated mRNA stabilisation. Cell Signal. 2004;10:1113–1121. doi: 10.1016/j.cellsig.2004.04.006. [DOI] [PubMed] [Google Scholar]
- De Lerma Barbaro A, Procopio FA, Mortara L, Tosi G, Accolla RS. The MHC class II transactivator (CIITA) mRNA stability is critical for the HLA class II gene expression in myelomonocytic cells. Eur J Immunol. 2005;35:603–611. doi: 10.1002/eji.200425378. [DOI] [PubMed] [Google Scholar]
- Deleault KM, Skinner SJ, Brooks SA. Tristetraprolin regulates TNF TNF-alpha mRNA stability via a proteasome dependent mechanism involving the combined action of the ERK and p38 pathways. Mol Immunol. 2008;45:13–24. doi: 10.1016/j.molimm.2007.05.017. [DOI] [PubMed] [Google Scholar]
- Doench JG, Sharp PA. Specificity of microRNA target selection in translational repression. Genes Dev. 2004;18:504–511. doi: 10.1101/gad.1184404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn GP, Old LJ, Schreiber RD. The three Es of cancer immunoediting. Ann Rev Immunol. 2004;22:329–360. doi: 10.1146/annurev.immunol.22.012703.104803. [DOI] [PubMed] [Google Scholar]
- Fabbri M, Garzon R, Cimmino A, Liu Z, Zanesi N, Callegari E, Liu S, Alder H, Costinean S, Fernandez-Cymering C, Volinia S, Guler G, Morrison CD, Chan KK, Marcucci G, Calin GA, Huebner K, Croce CM. MicroRNA-29 family reverts aberrant methylation in lung cancer by targeting DNA methyltransferases 3A and 3B. Proc Natl Acad Sci U S A. 2007;104:15805–15810. doi: 10.1073/pnas.0707628104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita N, Jaye DL, Geigerman C, Akyildiz A, Mooney MR, Boss JM, Wade PA. MTA3 and the Mi-2/NuRD complex regulate cell fate during B lymphocyte differentiation. Cell. 2004;119:75–86. doi: 10.1016/j.cell.2004.09.014. [DOI] [PubMed] [Google Scholar]
- Goriely S, Vincart B, Stordeur P, Vekemans J, Willems F, Goldman M, De Witt D. Deficient IL-12 (p35) gene expression by dendritic cells derived from neonatal monocytes. J Immunol. 2001;166:2141–2146. doi: 10.4049/jimmunol.166.3.2141. [DOI] [PubMed] [Google Scholar]
- Grewal SI, Moazed D. Heterochromatin and epigenetic control of gene expression. Science. 2005;301:798–802. doi: 10.1126/science.1086887. [DOI] [PubMed] [Google Scholar]
- Griffiths-Jones S. The microRNA Registry. Nucleic Acids Res. 2004;32:D109–D111. doi: 10.1093/nar/gkh023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyory I, Wu J, Fejér G, Seto E, Wright KL. PRDI-BF1 recruits the histone H3 methyltransferase G9a in transcriptional silencing. Nat Immunol. 2007;5:299–308. doi: 10.1038/ni1046. [DOI] [PubMed] [Google Scholar]
- Harris KS, Zhang Z, McManus MT, Harfe BD, Sun X. Dicer function is essential for lung epithelium morphogenesis. Proc Natl Acad Sci U S A. 2006;103:2208–2213. doi: 10.1073/pnas.0510839103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hau HH, Walsh RJ, Ogilvie RL, Williams DA, Reilly CS, Bohjanen PR. Tristetraprolin recruits functional mRNA decay complexes to ARE sequences. J Cell Biochem. 2007;100:1477–1492. doi: 10.1002/jcb.21130. [DOI] [PubMed] [Google Scholar]
- He L, He X, Lim LP, de Stanchina E, Xuan Z, Liang Y, Xue W, Zender L, Magnus J, Ridzon D, Jackson AL, Linsley PS, Chen C, Lowe SW, Cleary MA, Hannon GJ. A microRNA component of the p53 tumour suppressor network. Nature. 2007;447:1130–1134. doi: 10.1038/nature05939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrich B, Bird A. Identification and characterization of a family of mammalian methyl-CpG binding proteins. Mol Cell Biol. 1998;18:6538–6547. doi: 10.1128/mcb.18.11.6538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hitti E, Iakovleva T, Brook M, Deppenmeier S, Gruber AD, Radzioch D, Clark AR, Blackshear PJ, Kotlyarov A, Gaestel M. Mitogen-activated protein kinase-activated protein kinase 2 regulates tumor necrosis factor mRNA stability and translation mainly by altering tristetraprolin expression, stability, and binding to adenine/uridine-rich element. Mol Cell Biol. 2006;6:2399–2407. doi: 10.1128/MCB.26.6.2399-2407.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang WC, Ju TK, Hung MC, Chen CC. Phosphorylation of CBP by IKKalpha promotes cell growth by switching the binding preference of CBP from p53 to NF-kappaB. Mol Cell. 2007;26:75–87. doi: 10.1016/j.molcel.2007.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jing Q, Huang S, Guth S, Zarubin T, Motoyama A, Chen J, Di Padova F, Lin SC, Gram H, Han J. Involvement of microRNA in AU-rich element-mediated mRNA instability. Cell. 2005;120:623–634. doi: 10.1016/j.cell.2004.12.038. [DOI] [PubMed] [Google Scholar]
- John B, Enright AJ, Aravin A, Tuschl T, Sander C, Marks DS. Human micro-RNA targets. Plos Biol. 2004;2:e363–e381. doi: 10.1371/journal.pbio.0020363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SM, Grosshans H, Shingara J, Byrom M, Jarvis R, Cheng A, Labourier E, Reinert KL, Brown D, Slack FJ. RAS is regulated by the let-7 microRNA family. Cell. 2005;120:635–647. doi: 10.1016/j.cell.2005.01.014. [DOI] [PubMed] [Google Scholar]
- Karin M, Greten FR. NF-kappaB: linking inflammation and immunity to cancer development and progression. Nat Rev Immunol. 2005;5:749–759. doi: 10.1038/nri1703. [DOI] [PubMed] [Google Scholar]
- Karube Y, Tanaka H, Osada H, Tomida S, Tatematsu Y, Yanagisawa K, Yatabe Y, Takamizawa J, Miyoshi S, Mitsudomi T, Takahashi T. Reduced expression of Dicer associated with poor prognosis in lung cancer patients. Cancer Sci. 2005;96:111–115. doi: 10.1111/j.1349-7006.2005.00015.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khabar KS. The AU-rich transcriptome: more than interferons and cytokines, and its role in disease. J Interferon Cytokine Res. 2005;25:1–10. doi: 10.1089/jir.2005.25.1. [DOI] [PubMed] [Google Scholar]
- Krek A, Grun D, Poy MN, Wolf R, Rosenberg L, Epstein EJ, MacMenamin P, da Piedade I, Gunsalus KC, Stoffel M, Rajewsky N. Combinatorial microRNA target predictions. Nat Genet. 2005;37:495–500. doi: 10.1038/ng1536. [DOI] [PubMed] [Google Scholar]
- Kumar MS, Lu J, Mercer KL, Golub TR, Jacks T. Impaired microRNA processing enhances cellular transformation and tumorigenesis. Nat Genet. 2007;39:673–677. doi: 10.1038/ng2003. [DOI] [PubMed] [Google Scholar]
- Leung AK, Sharp PA. microRNAs: A Safeguard against Turmoil? Cell. 2007;130:581–585. doi: 10.1016/j.cell.2007.08.010. [DOI] [PubMed] [Google Scholar]
- Lewis BP, Shih IH, Jones-Rhoades MW, Bartel DP, Burge CB. Prediction of mammalian micro-RNA targets. Cell. 2003;115:787–798. doi: 10.1016/s0092-8674(03)01018-3. [DOI] [PubMed] [Google Scholar]
- Lemercier C, Brocard MP, Puvion-Dutilleul F, Kao HY, Albagli O, Khochbin S. Class II histone deacetylases are directly recruited by BCL6 transcriptional repressor. J Biol Chem. 2002;277:22045–22052. doi: 10.1074/jbc.M201736200. [DOI] [PubMed] [Google Scholar]
- Li MO, Sanjabi S, Flavell RA. Transforming growth factor-beta controls development, homeostasis, and tolerance of T cells by regulatory T cell-dependent and -independent mechanisms. Immunity. 2006;25:455–471. doi: 10.1016/j.immuni.2006.07.011. [DOI] [PubMed] [Google Scholar]
- Li QJ, Chau J, Ebert PJ, Sylvester G, Min H, Liu G, Braich R, Manoharan M, Soutschek J, Skare P, Klein LO, Davis MM, Chen CZ. MiR-181a is an intrinsic modulator of T cell sensitivity and selection. Cell. 2007;129:147–161. doi: 10.1016/j.cell.2007.03.008. [DOI] [PubMed] [Google Scholar]
- Liu P, Keller JR, Ortiz M, Tessarollo L, Rachel RA, Nakamura T, Jenkins NA, Copeland NG. Bcl11a is essential for normal lymphoid development. Nat Immunol. 2003;4:525–532. doi: 10.1038/ni925. [DOI] [PubMed] [Google Scholar]
- Longo VD, Kennedy BK. Sirtuins in aging and age-related disease. Cell. 2006;126:257–268. doi: 10.1016/j.cell.2006.07.002. [DOI] [PubMed] [Google Scholar]
- Magner WJ, Kazim AL, Stewart C, Romano MA, Catalano G, Grande C, Keiser N, Santaniello F, Tomasi TB. Activation of MHC class I, II, and CD40 gene expression by histone deacetylase inhibitors. J Immunol. 2000;165:7017–7024. doi: 10.4049/jimmunol.165.12.7017. [DOI] [PubMed] [Google Scholar]
- Marsit CJ, Eddy K, Kelsey KT. MicroRNA responses to cellular stress. Cancer Res. 2006;22:843–848. doi: 10.1158/0008-5472.CAN-06-1894. [DOI] [PubMed] [Google Scholar]
- Mayr C, Hemann MT, Bartel DP. Disrupting the pairing between let-7 and Hmga2 enhances oncogenic transformation. Science. 2007;315:1576–1579. doi: 10.1126/science.1137999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazière P, Enright AJ. Prediction of microRNA targets. Drug Discov Today. 2007;12:452–458. doi: 10.1016/j.drudis.2007.04.002. [DOI] [PubMed] [Google Scholar]
- Muljo SA, Ansel KM, Kanellopoulou C, Livingston DM, Rao A, Rajewsky K. Aberrant T cell differentiation in the absence of Dicer. J Exp Med. 2005;202:261–269. doi: 10.1084/jem.20050678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee D, Gao M, O’Connor JP, Raijmakers R, Pruijn G, Lutz CS, Wilusz J. The mammalian exosome mediates the efficient degradation of mRNAs that contain AU-rich elements. EMBO J. 2002;21:165–174. doi: 10.1093/emboj/21.1.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillai RS, Bhattacharyya SN, Artus CG, Zoller T, Cougot N, Basyuk E, Bertrand E, Filipowicz W. Inhibition of translational initiation by Let-7 MicroRNA in human cells. Science. 2005;309:1573–1576. doi: 10.1126/science.1115079. [DOI] [PubMed] [Google Scholar]
- Read S, Greenwald R, Izcue A, Robinson N, Mandelbrot D, Francisco L, Sharpe AH, Powrie F. Blockade of CTLA-4 on CD4+CD25+ Regulatory T Cells Abrogates Their Function In Vivo. J Immunol. 2006;177:4376–4383. doi: 10.4049/jimmunol.177.7.4376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez A, Vigorito E, Clare S, Warren MV, Couttet P, Soond DR, van Dongen S, Grocock RJ, Das PP, Miska EA, Vetrie D, Okkenhaug K, Enright AJ, Douqan G, Turner M, Bradley A. Requirement of bic/microRNA-155 for normal immune function. Science. 2007;316:608–611. doi: 10.1126/science.1139253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenfeld MG, Lunyak VV, Glass CK. Sensors and signals: a coactivator/corepressor/epigenetic code for integrating signal-dependent programs of transcriptional response. Genes Dev. 2006;20:1405–1428. doi: 10.1101/gad.1424806. [DOI] [PubMed] [Google Scholar]
- Rudensky AY, Campbell DJ. In vivo sites and cellular mechanisms of T reg cell-mediated suppression. J Exp Med. 2006;203:489–492. doi: 10.1084/jem.20060214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito Y, Liang G, Egger G, Friedman JM, Chuang JC, Coetzee GA, Jones PA. Specific activation of microRNA-127 with downregulation of the proto-oncogene BCL6 by chromatin-modifying drugs in human cancer cells. Cancer Cell. 2006;9:435–443. doi: 10.1016/j.ccr.2006.04.020. [DOI] [PubMed] [Google Scholar]
- Scott GK, Mattie MD, Berger CE, Benz SC, Benz CC. Rapid Alteration of MicroRNA Levels by Histone Deacetylase Inhibition. Cancer Res. 2006;66:1277–1281. doi: 10.1158/0008-5472.CAN-05-3632. [DOI] [PubMed] [Google Scholar]
- Sethupathy P, Corda B, Hatzigeorgiou AG. TarBase: A comprehensive database of experimentally supported animal microRNA targets. RNA. 2006;12:192–197. doi: 10.1261/rna.2239606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sethupathy P, Megraw M, Hatzigeorgiou AG. A guide through present computational approaches for the identification of mammalian microRNA targets. Nat Methods. 2006;3:881–886. doi: 10.1038/nmeth954. [DOI] [PubMed] [Google Scholar]
- Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, Casero RA, Shi Y. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell. 2004;119:941–953. doi: 10.1016/j.cell.2004.12.012. [DOI] [PubMed] [Google Scholar]
- Si ML, Zhu S, Wu H, Lu Z, Wu F, Mo YY. MiR-21-mediated tumor growth. Oncogene. 2007;26:2799–2803. doi: 10.1038/sj.onc.1210083. [DOI] [PubMed] [Google Scholar]
- Smale ST, Fisher AG. Chromatin structure and gene regulation in the immune system. Ann Rev Immunol. 2002;20:427–462. doi: 10.1146/annurev.immunol.20.100301.064739. [DOI] [PubMed] [Google Scholar]
- Taganov KD, Boldin MP, Chang KJ, Baltimore D. NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc Natl Acad Sci U S A. 2006;103:12481–12486. doi: 10.1073/pnas.0605298103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takamizawa J, Konishi H, Yanagisawa K, Tomida S, Osada H, Endoh H, Harano T, Yatabe Y, Nagino M, Nimura Y, Mitsudomi T, Takahashi T. Reduced expression of the let-7 microRNAs in human lung cancers in association with shortened postoperative survival. Cancer Res. 2004;64:3753–3756. doi: 10.1158/0008-5472.CAN-04-0637. [DOI] [PubMed] [Google Scholar]
- Taylor GA, Carballo E, Lee DM, Lai WS, Thompson MJ, Patel DD, Schenkman DI, Gilkeson GS, Broxmeyer HE, Haynes BF, Blackshear PJ. A pathogenetic role for TNF alpha in the syndrome of cachexia, arthritis, and autoimmunity resulting from tristetraprolin (TTP) deficiency. Immunity. 1996;4:445–454. doi: 10.1016/s1074-7613(00)80411-2. [DOI] [PubMed] [Google Scholar]
- Thai TH, Calado DP, Casola S, Ansel KM, Xiao C, Xue Y, Murphy A, Frendewey D, Valenzuela D, Kutok JL, Schmidt-Supprian M, Rajewsky N, Yancopoulos G, Rao A, Rajewsky K. Regulation of the germinal centre response by microRNA-155. Science. 2007;316:604–608. doi: 10.1126/science.1141229. [DOI] [PubMed] [Google Scholar]
- Tomasi TB, Magner WJ, Khan AN. Epigenetic regulation of immune escape genes in cancer. Cancer Immunol Immunother. 2006;55:1159–1184. doi: 10.1007/s00262-006-0164-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Rooji E, Sutherland LB, Qi X, Richardson JA, Hill J, Olson EN. Control of Stress-Dependent cardiac growth and gene expression by a micro-RNA. Science. 2007;316:575–579. doi: 10.1126/science.1139089. [DOI] [PubMed] [Google Scholar]
- Vasudevan S, Steitz JA. AU-rich-element-mediated upregulation of translation by FXR1 and Argonaute 2. Cell. 2007;128:1105–1118. doi: 10.1016/j.cell.2007.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakabayashi Y, Watanabe H, Inoue J, Takeda N, Sakata J, Mishima Y, Hitomi J, Yamamoto T, Utsuyama M, Niwa O, Aizawa S, Kominami R. Bcl11b is required for differentiation and survival of αβT lymphocytes. Nat Immunol. 2003;4:533–539. doi: 10.1038/ni927. [DOI] [PubMed] [Google Scholar]
- Wang AH, Gregoire S, Zika E, Xiao L, Li CS, Li H, Wright KL, Ting JP, Yang XJ. Identification of the ankyrin repeat proteins ANKRA and RFXANK as novel partners of class IIa histone deacetylases. J Biol Chem. 2005;280:29117–29127. doi: 10.1074/jbc.M500295200. [DOI] [PubMed] [Google Scholar]
- Wassenegger M. The role of the RNAi machinery in heterochromatin formation. Cell. 2005;122:13–16. doi: 10.1016/j.cell.2005.06.034. [DOI] [PubMed] [Google Scholar]
- Widschwendter M, Fiegl H, Egle D, Mueller-Holzner E, Spizzo G, Marth C, Weisenberger DJ, Campan M, Young J, Jacobs I, Laird PW. Epigenetic stem cell signature in cancer. Nat Genet. 2007;39:157–158. doi: 10.1038/ng1941. [DOI] [PubMed] [Google Scholar]
- Zhou B, Wang S, Mayr C, Bartel DP, Lodish FH. MiR-150, a microRNA expressed in mature B and T cells, blocks early B cell development when expressed prematurely. Proc Natl Acad Sci U S A. 2007;104:7080–7085. doi: 10.1073/pnas.0702409104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.