

Abstract
Among naturally-occurring polymorphisms in the 5’ flanking region of the human angiotensinogen (AGT) gene, the −20 and −217 polymorphisms have the strongest effects on AGT regulation in AGT-expressing cells derived from liver, kidney, brain, and fat. These polymorphisms may affect allele-specific transcription factor binding, and the high-expressing alleles are both relatively common. We show herein that the −20C allele has higher transcriptional activity than −20A, and the −20A allele confers no additional transactivation potential beyond that of a mutated vector. Gel shift assays show that USF1 and USF2 preferentially bind the −20C allele whereas the −20A allele retains a low affinity USF binding site. Plasmid immunoprecipitation assays confirmed preferential association of USF1 with the −20C allele in transfected HepG2 cells. Chromatin immunoprecipitation confirmed that USF1 binds to the endogenous AGT −20C allele in CCF cells, the only cell line tested that carries the −20C allele, and to the human AGT promoter in liver and adipose tissue from transgenic mice. Transduction of AGT-expressing cells with shRNAs specifically targeting USF1 or USF2, resulted in cell- and allele-specific attenuation of AGT promoter activity. In vivo, knockdown of USF expression in the liver of transgenic mice expressing the −20C allele of AGT resulted in lower AGT expression, a decrease in circulating human AGT protein, but no change in expression of GAPDH or HNF-4α. We conclude that USF1 functionally and differentially regulates AGT expression via the −20 polymorphism and that the differential expression exhibited by −20 can be accounted for by differential association with USF1.
Keywords: Transcription, Genetics, Hypertension, Chromatin
Introduction
The renin-angiotensin system (RAS) is an important regulator of salt, water, and blood pressure homeostasis. It is commonly accepted that the rate-limiting step in the production of angiotensin-II, and other angiotensin peptides, is the enzymatic cleavage of angiotensinogen (AGT) by renin. Since the level of AGT is close to the Km for renin, any variation in the level of either can cause differences in angiotensin production. While the generation of blood-borne angiotensin is recognized as the classical pathway, there is considerable evidence that RAS expression and angiotensin generation in tissues can also play an important role in blood pressure regulation. Indeed, transgenic studies show that local production of RAS products in the kidney and brain can have profound effects on systemic arterial pressure without affecting circulating levels of angiotensin peptides 1, 2.
While it is known that expression of AGT directly correlates with blood pressure 3, the etiology of inter-individual variability in its expression is incompletely understood. Single nucleotide polymorphisms (SNPs) in the AGT gene were reported to correlate with altered tissue AGT expression patterns and/or hypertension in humans (recently reviewed in 4). The most frequently studied AGT SNPs are at nucleotide −6 and codon 235 5. The 235 polymorphism does not effect the kinetics of the renin-AGT enzymatic reaction 6, and the only transcription factor known to bind the −6 position does so in an allele-independent fashion 6. On the contrary, the −20 and −217 SNPs are thought to elicit differential transcription factor binding in the AGT promoter 7, 8, are found in varying degrees of linkage disequilibrium with other AGT SNPs, including −6 and 235 9, and may be related to differences in plasma AGT 10. Because of a historical bias toward a small subset of AGT polymorphisms (−6 and 235), it has been difficult to determine which SNPs are mechanistically linked to differences in AGT promoter activity, AGT expression and hypertension. Moreover, most molecular studies have focused entirely on AGT expression in hepatic cell lines, ignoring AGT expression in cells derived from other physiologically relevant tissues.
We recently examined the effects of 14 SNPs in the AGT promoter assembled into 8 haplotypes accounting for 90% of the haplotype diversity previously reported in whites, Japanese, and Africans, using AGT-expressing cell lines from four tissues known to express AGT in vivo 11. We showed that the −20 and −217 polymorphisms play dominant roles in differential AGT regulation in cells derived from the liver, kidney, brain and adipose tissue, respectively. In the current study we tested the hypothesis that the A-20C polymorphism plays an important functional role in vitro and in vivo and that these effects are due to direct, preferential activity of the leucine zipper transcription factors upstream stimulatory factor 1 (USF1) and 2 (USF2) on the −20C allele of AGT. Functional ablation of USF1 and USF2 with shRNAs eliminated the preferential transactivation from the −20C allele in AGT-expressing cells, significantly lowered expression of human AGT in liver, and lowered circulating human AGT in transgenic mice carrying the −20C allele of the gene. These results along with DNA and chromatin immunoprecipitation studies define USF1 as the transcription factor that modulates differential AGT expression by the A-20C polymorphism.
Methods
Cell Culture Studies
Culture conditions, AGT promoter reporter vectors (haplotypes 4 and 8), transfections, and transcriptional reporter assays were as previously described 11. These naturally-occurring haplotypes extend from −1219 to +125 bp, and their sequences differ only at position −20 (haplotype: 4=A, 8=C). Mutant vector generation, adfections and luciferase assays are detailed in Supplemental Methods.
Western and Gel Shift Analysis
Western blotting employed antibodies against USF1 (1:2000 sc-229X, Santa Cruz Biotechnology), USF2 (1:2000 sc-861X, Santa Cruz Biotechnology), hAGT (1:10000, gift from Duane Tewksbury, Marshfield Medical Research Foundation, Marshfield, WI), GFP (1:4000 sc-9996, Santa Cruz Biotechnology), HNF-4α (1:300 sc-8987, Santa Cruz Biotechnology), GAPDH (1:6000 sc-32233 or 1:5000 sc-25778, Santa Cruz Biotechnology), α-tubulin (1:800000 T5168, Sigma), or β-actin (1:5000 ab8227, Abcam). Quantitative Western analysis utilized ImageJ software (National Institutes of Health). Gel shift experiments were performed as described in Supplemental Methods.
Adenovirus Design
Four 21nt core sequences for hUSF1 (5’-GACAGCTGCTGAGACGCACTA-3’, 5’-GGAGTACAGCTGCTGTTGTTA-3’, 5’-GTGCAGCTCTCCAAGATAATC-3’, 5’- GGTGGGATTCTATCCAAAGCT-3’) and hUSF2 (5’-GTCCAGGTGACTGATGGTCAG-3’, 5’-GCCAGTTCTACGTCATGATGA-3’, 5’-GGATCGTCCAGCTTTCGAAAA-3’, 5’-GCCTGCGATTACATCCGGGAG-3’) were chosen based on high predicted secondary structure formation as candidate sequences for shRNA development. These sequences in the form of hairpins separated by a 6 nucleotide loop (TATCGC) were cloned into the PmeI and EcoR1 sites in an adenoviral shuttle vector (pacAd5K-NpA, University of Iowa Vector Core) modified to contain the U6 promoter (pacAd5pU6, gift of Dr. Robin L. Davisson, Cornell University) as described 12. Plasmids were transfected into HEK-293 cells and tested for their ability to knockdown FLAG-tagged USF expression by real-time RT-PCR using SYBR green and primers listed in Supplemental Methods. Effective knockdown was observed with the second and third sequences for hUSF1 (1.2, 1.3), and the third sequence for hUSF2 (2.3). Two shRNA sequences (1.3 and 2.3) also caused effective knockdown of FLAG-tagged mouse USF expression. These three shRNA vectors were converted into type-5 adenoviruses by recombination with a helper vector which expressed a CMV-eGFP reporter gene (Gene Transfer Vector Core at the University of Iowa). The effectiveness of the adenoviruses on USF1 and USF2 was then retested using primers listed in Supplemental Methods. The viruses designated LacZ and eGFP are type-5 adenoviruses encoding shRNAs targeting either E. coli LacZ or eGFP (gift from Beverly Davidson, Vector Core, University of Iowa) 13. HepG2, HK-2, and CCF cells had optimum MOIs of 200, 200, and 500, respectively. We could not effectively infect differentiated 3T3-L1 cells with adenovirus.
Plasmid (PIP) and Chromatin (ChIP) Immunoprecipitation
PIP assays were performed in transfected HepG2 cells. Chromatin was sonicated to 200−1000 bp. IP was performed with the EZ-ChIP Kit (Millipore) using 10 μg antibody against USF1 or USF2, or non-specific IgG. Real-time PCR reactions utilized the TaqMan genotyping master mix (Applied Biosystems), and probes conferring at least 1000-fold allele-specificity. Some experiments utilized haplotype reporter vectors that were independently transfected into separate batches of HepG2 cells while others utilized co-transfections in the same cells. Results were similar between the two methods, so summary data represent a mixture of the two protocols. ChIP was performed on chromatin from CCF cells and tissues from mice using an analogous protocol. Primers and probes used for these experiments are listed in Supplemental Methods.
Intravenous Adenoviral Delivery
The experimental protocol was approved by our institution's animal care and use committee. All breeding and genotyping was performed in the Transgenic Animal Facility. All studies were performed in transgenic mice expressing hAGT 14. Intravenous jugular cannulation, adenoviral delivery, and extraction of RNA and proteins from tissues were performed as previously described 15, 16. We injected 2×1010 pfu of AdshGFP or 1×1010 pfu each of AdshUSF1.3 and AdshUSF2.3. Tissues were harvested 5 days after virus injection. Real-time PCR was performed using primers listed in Supplemental Methods.
Statistical Analysis
All statistical analyses were performed using SigmaStat (Systat Software, Inc.). The normality assumption of data distribution was tested using the Shapiro-Wilk's test. When the data was not normally distributed, a normal log transformation was applied or a nonparametric statistical analysis ensued using ANOVA on ranks followed by a post-hoc examination using the Tukey adjustment. If the data distribution was normally distributed, then results were analyzed using either unpaired, two-tailed Student's t-test or an ANOVA followed by a post-hoc examination with the Tukey adjustment. P<0.05 was considered significant.
Results
Compared with the consensus binding site for USF1, a C at position −20 of hAGT has much greater relative entropy than the corresponding A (Figure 1A). We generated mutant reporter vectors to verify the importance of the −20 hAGT polymorphism and to determine the amount of residual transactivation offered by factors interacting with the −20A allele (Figure 1B). Reporter activities using these vectors were assayed in transfected HepG2, HK-2, and differentiated 3T3-L1 cells (Figure 1C-E). The −20C allele (haplotype 8) gave 2- to 3-fold higher expression than the −20A allele (haplotype 4) in each cell type. Mutating the USF binding sequence attenuated expression of the −20C (labeled 8U) allele to the level of the −20A allele, but the same alteration (labeled 4U) had no effect on the −20A allele, suggesting that the −20A allele has little transactivation capacity conferred by any factor that binds to the sequence surrounding −20.
Figure 1. Transfection of Mutant AGT reporter vectors.

A. Hidden Markov model for hUSF1 32. The −20 polymorphism occurs at the nucleotide with the highest relative entropy for binding. B. hAGT sequence from −33 to +1 showing the sequence of the mutant reporter (U) eliminating the USF1/2 binding site and oligonucleotide sequences used in EMSA. C-E. Dual-Luciferase activities using AGT promoter reporter vectors and normalizing results to expression from haplotype 4 (−20A allele). Each graph is data acquired using multiple DNA preparations generated on multiple days. Graphs show mean±SEM with the indicated N. *, P<0.05 vs haplotype 4; **, P<0.05 vs haplotype 8.
Western blotting confirmed the expression of USF1 and USF2 in each cell line (Figure S1). Electrophoretic mobility shift assays (EMSAs) were next used to examine USF1/2 binding to the AGT promoter region surrounding −20 (Figure 2). Double-stranded oligonucleotides used in this study are shown in Figure 1B and include variants at positions −20 and −6. Consistent with previous reports 17, there were no differences observed in EMSAs due to variation at the −6 position in any cell line tested. In HK-2 cells, a stronger shift, abolished by self competitor but not with mutant competitor was observed on the −20C allele (Figure 2A). Altering the −6 position (while holding the −20 allele constant) had no reproducible effect in any cell line. Unlabeled oligonucleotides containing −20C were consistently more effective competitors than −20A (Figure 2B) or a competitor containing a mutant USF1 binding site (Figure 2D) when −20C was used as a probe. Supershift using antibodies specific for USF1 and USF2 resulted in distinct products indicating that both proteins can bind to the −20 region of the AGT promoter (Figure 2C). Interestingly, the gel shift resolved into two separate products upon use of individual USF antibody, allowing the potential identification of residual USF1/1 and USF2/2 homodimers (indicated by the arrows in Figure 2C). This is because USF1 antibody should supershift USF1/1 homodimers and USF1/2 heterodimers without effecting USF2/2 homodimers. The reverse is true of USF2 antibody. The gel shift was completely eliminated when both antibodies were employed together (Figure 2D). Antibodies against β-actin or the transcription factor FOS-related antigen 1 (FRA1) had no effect on the gel shift thus demonstrating specificity (Figure 2D). Similar results were obtained using extracts from HepG2 cells (Figure S2), although the difference in affinity of USF for −20A as compared to −20C was more pronounced in HK-2 nuclear extracts than in HepG2 nuclear extracts.
Figure 2. Electrophoretic Gel Mobility Shift Assays.

EMSA utilizing nuclear extracts from HK-2 cells and the oligonucleotides detailed in Figure 1B. A. EMSA using cold competitor (50-fold) derived from a mismatched sequence (M) or the same sequence as the radiolabeled probe (labeled as C). * indicates free probe using CA oligo without nuclear extract. B. EMSA using radiolabeled CA probe with increasing amounts (10-, 25- and 50-fold molar excess) of the indicated cold or mismatched (M) competitor. C. EMSA using the indicated probes and antibody against USF1 (1) or USF2 (2). Predicted USF1/1 and USF2/2 homodimers are indicated. D. EMSA using the radiolabeled CA probe and indicated competitors (10-, 25- and 50-fold excess). Supershifts were performed using the CA probe and antibodies directed against both USF1 and USF2 (1,2), FRA1 (F) or β-Actin (A). Arrow, specific gel shift band; Open arrowhead, supershift bands. NS, non-specific shift products.
We next sought to confirm the gel shift findings using an in vivo assay. Because none of the AGT-expressing cell lines we studied were heterozygous for −20A and −20C, we first performed plasmid immunoprecipitation (PIP) rather than chromatin immunoprecipitation (ChIP). Our results indicate that USF1 preferentially associates with the −20C allele in HepG2 cells (Figure 3A). The USF1 result was confirmed in co-transfection experiments where the recovery after PIP was measured with allele-specific real-time PCR (Figure 3B). There was no apparent increase in association of USF2 with either allele. It is important to note that although equal amounts of −20A and −20C were recovered using non-specific IgG, the recovery was about 20-fold less than for USF1 antisera. HepG2 and HK-2 carry the −20A allele endogenously, whereas the astrocyte cell line CCF, which also expresses AGT, carries the −20C allele endogenously. We first confirmed that the EMSA pattern observed for HepG2 and HK-2 cells was also observed in CCF cells (Figure S3). Genomic ChIP suggests an interaction between USF1 and the −20C hAGT allele in CCF cells (Figure 3C). We therefore included CCF cells in the assays to follow.
Figure 3. Plasmid and Chromatin Immunoprecipitation.

A. HepG2 cells were mock transfected (first in each set of three lanes) or transfected with AGT reporter vectors containing −20A (haplotype 4) or −20C (haplotype 8). IP was carried out using the indicated antibodies followed by PCR using primers specific for the plasmid DNA. Results from two independent experiments are shown. B. Summary of HepG2 PIP experiments quantified by a real-time PCR SNP assay. The relative recovery of −20A (gray) and −20C (filled) DNA IP by the indicated antisera is shown. ΔCT values were corrected for input, converted to a fraction of total (USF1/USF1+USF2 or USF2/USF1+USF2), and are presented as mean±SEM. *, P<0.05 for −20C vs −20A (n=5). The dotted line represents equal recovery of −20A vs −20C. C. ChIP using input, USF1, USF2 or IgG antibody control from CCF cell chromatin amplified with primers specific for hAGT. The IgG was cut from the same gel as the other lanes.
We generated adenoviruses expressing shRNAs directed against USF1 (AdshUSF1) or USF2 (AdshUSF2) and tested the specificity of each shRNA to knockdown expression of USF1 and USF2. Two shRNA viruses directed against USF1 (1.2 and 1.3) and one shRNA virus directed against USF2 (2.3) were shown to effectively and specifically knockdown USF1 or USF2 mRNA (Figure 4A-B) and protein (Figure 4C) as compared to treatment with a control shRNA virus targeting LacZ. We also demonstrated that nuclear extracts derived from AdshUSF1- and AdshUSF2-treated cells exhibited a marked decrease in the amount of the USF1 and USF2 gel shift product (Figure 4D). Like the supershift studies described above, the use of shRNAs allowed the resolution of two distinct gel shift products, one by USF1/1 homodimers and the other by USF2/2 homodimers (Figure 4D and Figure S4).
Figure 4. USF1 and USF2 Expression After AdshUSF1 or AdshUSF2 Transduction.

A-B. Quantitative real-time RT-PCR for USF1 (A) and USF2 (B) mRNA in the indicated cell lines transduced with the indicated adenoviruses. shRNAs 1.2 and 1.3 target USF1 whereas shRNA 2.3 targets USF2. Data are mean±SEM normalized to cells treated with AdshLacZ. C. Western analyses for USF1 and USF2 performed using total cellular protein from the indicated cells treated with the indicated adenoviruses. α-tubulin (TUBA) is the internal control. Z, AdshLacZ. D. Cells were transduced with the indicated adenoviruses. One of two independent preparations of nuclear extracts from these cells used for the gel shift analyses is shown. The CA radiolabeled probe (−20C, −6A) was used. Predicted USF1/1 and USF2/2 homodimers are indicated. *, free probe using CA oligo without nuclear extract. Arrow, specific gel shift band. Z, AdshLacZ.
We next performed co-transfection/transduction studies using AdshUSF1 and AdshUSF2 viruses along with luciferase reporter vectors driven by the −20A or −20C human AGT promoter (Figure 5). Viruses with shRNA targeting LacZ (AdshLacZ) served as controls. In HepG2 cells transduced with the control shRNA virus, there was a 3.3±0.6 fold (mean±SEM) increase in human AGT promoter activity comparing the −20C with the −20A allele (Figure 5A). Addition of AdshUSF1 or AdshUSF2 selectively attenuated reporter activity from the −20C allele to the levels of the −20A allele. Considering all shUSF viruses either together or independently, there was no significant difference in the level of −20A promoter activity in cells transduced with AdshUSF1, AdshUSF2, or AdshLacZ in HepG2 cells. This data confirmed that in HepG2 cells there is little residual USF-mediated transactivation activity of the −20A site, whereas the difference in transactivation between −20C and −20A is USF-dependent. Baseline activity of the −20C promoter was higher than the −20A promoter in HK-2 (Figure 5B) and CCF cells (Figure 5C). −20C promoter activity was attenuated by AdshUSF1 or AdshUSF2 in both cell types. However, unlike HepG2, activity of the −20A promoter was also attenuated by the AdshUSF1/2 viruses, suggesting that in these cell types the −20A allele may support some transactivation by USF, perhaps through the binding of USF to other sites in the hAGT promoter.
Figure 5. Role of USF1 and USF2 in hAGT Promoter Activity.

Dual-Luciferase activity assays of HepG2 (A), HK-2 (B), or CCF (C) cells transduced with the indicated adenovirus and transfected with the indicated reporter vector. Results are mean±SEM normalized to expression from haplotype 4 transduced with AdshLacZ (labeled Z). Each graph represents data acquired using multiple DNA preparations and transfected on multiple days. NS=Non-significant. *, p<0.05 vs. haplotype 4 LacZ; **, P<0.05 AdshUSF vs AdshLacZ from the same haplotype. Statistics utilized two-way ANOVA comparing AdshUSF vs AdshLacZ.
We then tested whether we could achieve knockdown of mouse USF expression in vivo following intravenous adenoviral delivery and determined the effect on AGT expression. We co-injected AdshUSF1 and AdshUSF2 simultaneously, and as a control injected shRNA-containing adenovirus targeting GFP (AdshGFP). The AdshGFP was used since the same amount of the AdshLacZ virus was lethal. Switching from human cell lines to mouse tissues preserved effective knockdown of USF1 and USF2 in the liver, although the subtype specificity was diminished somewhat (data not shown).
To assess the effects of USF ablation on human AGT expression, experiments were performed in transgenic mice expressing the human AGT gene driven by its own endogenous promoter 14. This transgene carries the high-expressing −20C allele which binds both USF1 and USF2 in chromatin from white adipose tissue and liver (Figure 6). Relative to AdshGFP, the AdshUSF viruses resulted in significant knockdown of USF1 and USF2 mRNA in the liver (Figure 7). Consistent with effective infection of the liver, but poor infection of other tissues, there was no effect on USF1 or USF2 mRNA in kidney or brain (data not shown). The levels of hepatic USF1 (0.35±0.1 AdshUSF vs 1.04±0.1 AdshGFP, 34% of baseline, P<0.001) and USF2 (0.4±0.1 AdshUSF vs 0.9±0.1 AdshGFP, 47% of baseline, P<0.001) proteins were also decreased in mice receiving AdshUSF (Figure 8A-B). Expression of eGFP, which is encoded on the AdshUSF viruses, was detected in the liver (Figure 8C). Importantly, the ablation of USF1 and USF2 expression in liver was associated with a significant decrease in both mouse and human AGT mRNA (Figure 7), and a decrease in hepatic human AGT protein (0.14±0.02 AdshUSF vs 2.2±0.1 AdshGFP, 7±0.8% of baseline, P<0.001, Figure 8D). Consistent with the liver being the primary site of AGT secretion, there was significant attenuation of hAGT protein levels in the plasma (0.9±0.05 AdshUSF vs 2.1±0.1 AdshGFP, 44±2.5% of baseline, P<0.001, Figure 8E). Note that multiple glycosylation forms of hAGT exist (Figure 8D and E). In an independent experiment, 10 of 12 transgenic mice receiving AdshUSF, but 0 of 13 transgenic mice receiving the AdshGFP, exhibited a decrease in plasma hAGT (data not shown). As an indication of specificity, there was no change in the hepatic expression of GAPDH (Figure 8B,D,F) or HNF-4α (0.43±0.06 AdshUSF vs 0.36±0.12 AdshGFP, P=0.26) in the liver of AdshUSF vs AdshGFP treated mice (Figure 8F). HNF-4α is a liver-specific transcription factor implicated to regulate several genes including AGT 18. Finally, there was no change in the level of human or mouse AGT mRNA in the kidney or brain, consistent with the absence of USF1 and USF2 knockdown (data not shown).
Figure 6. ChIP for USF1 and USF2 in Tissues from hAGT Transgenic Mice.
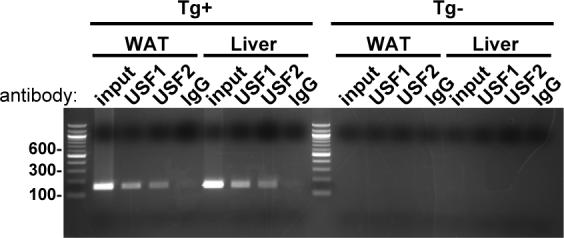
ChIP was performed using the indicated antibody and from chromatin isolated from white adipose tissue (WAT) or liver. Tissues were harvested from transgenic mouse (Tg+) containing hAGT and from a non-transgenic littermate control (Tg-).
Figure 7. Effect of AdshUSF on Gene Expression in Liver In Vivo.

Age-matched female hAGT transgenic mice were treated with AdshGFP (N=4) or both AdshUSF1 and AdshUSF2 (N=5), and liver was harvested for RNA 5 days later. Real time PCR data show relative expression of the indicated genes. CT values were corrected for expression of β-actin and GAPDH (ΔCT), and then compared between groups using the 2-ΔΔCT method.
Figure 8. Effect of IV AdshUSF on USF1, USF2, and hAGT Protein In Vivo.

Age-matched female hAGT transgenic mice were treated with AdshGFP (N=4) or both AdshUSF1 and AdshUSF2 (N=5). Western blots from liver tissue or plasma were probed with the indicated antibody. GFP signal indicates presence of adenoviruses targeting USF since the viruses also express eGFP. The blot for hAGT and GAPDH was stripped and re-probed for USF1 (note a small residual hAGT signal above the USF1 band). Each lane reflects samples from independent mice. The brackets in panels D and E refer to the multiple glycosylation forms of hAGT protein in liver and plasma.
Discussion
Previous work on the functional effects of hAGT polymorphisms suggests that the −20 and −217 promoter variants play dominant roles in differential regulation of hAGT haplotypes in human and mouse cell lines derived from important sites of AGT expression 11. These studies are in concordance with studies showing that these are the only two AGT polymorphisms known to differentially affect transcription factor binding in cultured hepatocytes 7, 8. In the present study, we confirmed the functionally important role of the −20 polymorphism in cells derived from human hepatocytes, renal proximal tubule cells, astrocytes, and mouse adipocytes, by showing that the −20C allele correlates with higher expression in these cell lines.
Our data show that the low-activity −20A allele has similar transactivation potential to an allele with a mutant USF binding site indicating the −20A allele harbors minimal ability to be functionally transactivated via USF (or other factors binding a similar sequence). Gel shift experiments confirm expression of USF1 and USF2 and further show that they preferentially bind −20C despite some residual binding capacity for −20A. The gel shift and transfection data leads us to suggest that USF has some capacity to bind the −20A allele but that −20A does not functionally support transcriptional activation by USF. Prior studies show that native human USF1 activates, whereas a dominant-negative splice variant of USF1 attenuates hAGT promoter reporter expression in HepG2 cells, although the importance of the −20 polymorphism was not examined 19. Although USF1 and USF2 can homodimerize, they preferentially heterodimerize in vivo and have similar transactivating capacity 20. USF1 or USF2 null mutants survive, whereas the double null is lethal 21, suggesting the possibility of some functional redundancy in USF activity.
The mechanism of transcriptional activation by USF at the AGT locus is not known. Two general hypotheses include either recruitment of coactivators or modification of surrounding chromatin 22. Recent evidence from the chicken β-globin locus favors a mechanism whereby USF1 promotes opening of local chromatin while simultaneously preventing spreading of any nearby heterochromatin 23. Whichever mechanism occurs, it is interesting to note that there are only 6 nucleotides separating the −20 polymorphism from the TATA box in hAGT. This is noteworthy since it has been suggested that genetic alterations are more likely to affect transcription when located close to a TATA box, and USF is thought to more strongly affect transcription when its binding site is close to a TATA box 24, 25.
When we attempted to confirm USF1 and USF2 preferential association with the −20C allele of AGT using plasmid immunoprecipitation in HepG2 cells, we found that USF1, but not USF2, preferentially associated with −20C. We generated shRNA-expressing adenoviruses specifically targeting both factors, and confirmed their effectiveness at the mRNA, protein and functional levels. These viruses offer allele-specific attenuation of AGT haplotype reporter activity in cultured hepatocytes. Expression from the −20C allele was diminished upon either USF1 or USF2 knockdown to the point where it mimicked expression from the −20A allele. The data support a functional role of USF in differentially transactivating the −20C vs. −20A alleles of AGT. Knockdown of either USF1 or USF2 within cultured astrocytes or renal proximal tubule cells attenuated promoter activity for both the −20C and −20A alleles. We suggest two possible explanations for this observation. First, we have noted additional predicted USF binding sites beginning at −1145 and −717 in the hAGT promoter that do not overlie any known polymorphisms. Both sites have the sequence CATGTG as opposed to [A/C]TCGTG at the −20 position. These additional sites may still support USF-dependent transactivation even when the −20 site is mutated but not when USF1 and USF2 are ablated. Alternatively, USF proteins may regulate other genes in a cell type-specific manner that could, in turn, alter expression of AGT. Neither of these possibilities rule out that USF functionally regulates via the −20 polymorphism even in astrocytes and renal proximal tubule cells.
Intravenous co-administration of AdshUSF1 and AdshUSF2 resulted in knockdown of USF1 and USF2 in the liver of transgenic mice and, concomitantly, a marked attenuation of AGT mRNA and protein in the liver. The observation that there was a decrease in plasma AGT is consistent with the observation that most circulating AGT is derived from the liver 15. Of course we cannot rule out that the residual circulating hAGT may be derived from an extra-hepatic site, such as adipose tissue. Indeed, both USF1 and USF2 bind to the hAGT promoter in chromatin from adipose tissue. Consequently, our data clearly demonstrate the dependence on USF for hepatic expression of the AGT gene, and further suggest that USF binding to the −20 position in the hAGT promoter is important for regulating the levels of hepatic and plasma AGT. Therefore we can conclude that differential binding of USF1 can account for the differences in transcriptional activity caused by genetic variation at −20. Further studies directly targeting renal proximal tubules, adipocytes, astrocytes and neurons in the brain will be required to test the USF-dependence in these cell types.
Our data are interesting when one considers that USF1 polymorphisms correlate with hyperlipidemia phenotypes in families ascertained for hypertension and premature stroke, especially in males 26, and USF1 polymorphisms associate with AGT expression in human fat biopsies 27. Although a genetic link between USF1 and type 2 diabetes remains controversial 28, it has been reported that USF expression and transactivation of target genes varies in proportion with glucose levels in vitro and in vivo. For example, hyperglycemic conditions correlated with increased USF expression in cultured mesangial cells and USF expression increased in diabetic rat kidneys and in kidneys of mice subjected to 24 hours of fasting followed by 18 hours of refeeding a high-carbohydrate diet 29. Moreover, USF knockout mice had attenuated USF target gene expression after a similar fasting-refeeding diet 30. Finally, a genetic link between USF1 and familial combined hyperlipidemia is well recognized 31, which in turn is closely associated with the metabolic syndrome. It is thus possible that USF expression and its allele-specific effects on AGT expression may contribute to blood pressure control in humans, especially in the presence of diabetes or obesity. Testing whether our findings are directly relevant to human diseases will require further basic studies as well as clinical trials paying careful attention to not only the AGT genotype but also the USF genotype and measurement of patient characteristics such as body mass index, presence of diabetes/pre-diabetes, blood pressure, plasma lipids, and perhaps other factors.
Supplementary Material
Acknowledgements
We gratefully acknowledge the generous research support of the Roy J. Carver Trust.
Sources of Funding Supported by NIH grants HL61446, HL48058, and HL84207 (CDS) and by AHA Predoctoral Fellowship 0710043Z (MED).
Footnotes
Publisher's Disclaimer: This is an un-copyedited author manuscript accepted for publication in Circulation Research, copyright The American Heart Association. This may not be duplicated or reproduced, other than for personal use or within the “Fair Use of Copyrighted Materials” (section 107, title 17, U.S. Code) without prior permission of the copyright owner, The American Heart Association. The final copyedited article, which is the version of record, can be found at http://circres.ahajournals.org/. The American Heart Association disclaims any responsibility or liability for errors or omissions in this version of the manuscript or in any version derived from it by the National Institutes of Health or other parties.
Conflict of Interest None.
Disclosures NIH funding to Sigmund AHA Funding to Dickson
References
- 1.Davisson RL, Ding Y, Stec DE, Catterall JF, Sigmund CD. Novel mechanism of hypertension revealed by cell-specific targeting of human angiotensinogen in transgenic mice. Physiol Genomics. 1999;1:3–9. doi: 10.1152/physiolgenomics.1999.1.1.3. [DOI] [PubMed] [Google Scholar]
- 2.Morimoto S, Cassell MD, Sigmund CD. Glial- and neuronal-specific expression of the renin-angiotensin system in brain alters blood pressure, water intake, and salt preference. J Biol Chem. 2002;277:33235–33241. doi: 10.1074/jbc.M204309200. [DOI] [PubMed] [Google Scholar]
- 3.Kim HS, Krege JH, Kluckman KD, Hagaman JR, Hodgin JB, Best CF, Jennette JC, Coffman TM, Maeda N, Smithies O. Genetic control of blood pressure and the angiotensinogen locus. Proc Natl Acad Sci USA. 1995;92:2735–2739. doi: 10.1073/pnas.92.7.2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dickson ME, Sigmund CD. Genetic basis of hypertension: revisiting angiotensinogen. Hypertension. 2006;48:14–20. doi: 10.1161/01.HYP.0000227932.13687.60. [DOI] [PubMed] [Google Scholar]
- 5.Jeunemaitre X, Soubrier F, Kotelevtsev YV, Lifton RP, Williams CS, Charru A, Hunt SC, Hopkins PN, Williams RR, Lalouel J-M, Corvol P. Molecular Basis of Human Hypertension: Role of Angiotensinogen. Cell. 1992;71:169–180. doi: 10.1016/0092-8674(92)90275-h. [DOI] [PubMed] [Google Scholar]
- 6.Inoue I, Nakajima T, Williams CS, Quackenbush J, Puryear R, Powers M, Cheng T, Ludwig EH, Sharma AM, Hata A, Jeunemaitre X, Lalouel JM. A nucleotide substitution in the promoter of human angiotensinogen is associated with essential hypertension and affects basal transcription in vitro. J Clin Invest. 1997;99:1786–1797. doi: 10.1172/JCI119343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jain S, Li Y, Patil S, Kumar A. A single-nucleotide polymorphism in human angiotensinogen gene is associated with essential hypertension and affects glucocorticoid induced promoter activity. J Mol Med. 2005;83:121–131. doi: 10.1007/s00109-004-0621-5. [DOI] [PubMed] [Google Scholar]
- 8.Zhao YY, Zhou J, Narayanan CS, Cui Y, Kumar A. Role of C/A polymorphism at −20 on the expression of human angiotensinogen gene. Hypertension. 1999;33:108–115. doi: 10.1161/01.hyp.33.1.108. [DOI] [PubMed] [Google Scholar]
- 9.Nakajima T, Jorde LB, Ishigami T, Umemura S, Emi M, Lalouel JM, Inoue I. Nucleotide diversity and haplotype structure of the human angiotensinogen gene in two populations. Am J Hum Genet. 2002;70:108–123. doi: 10.1086/338454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ishigami T, Umemura S, Tamura K, Hibi K, Nyui N, Kihara M, Yabana M, Watanabe Y, Sumida Y, Nagahara T, Ochiai H, Ishii M. Essential hypertension and 5' upstream core promoter region of human angiotensinogen gene. Hypertension. 1997;30:1325–1330. doi: 10.1161/01.hyp.30.6.1325. [DOI] [PubMed] [Google Scholar]
- 11.Dickson ME, Zimmerman MBRK, Sigmund CD. The −20 and −217 promoter variants dominate differential angiotensinogen haplotype regulation in angiotensinogen-expressing cells. Hypertension. 2007;49:631–639. doi: 10.1161/01.HYP.0000254350.62876.b1. [DOI] [PubMed] [Google Scholar]
- 12.Gou D, Zhang H, Baviskar PS, Liu L. Primer extension-based method for the generation of a siRNA/miRNA expression vector. Physiol Genomics. 2007;31:554–562. doi: 10.1152/physiolgenomics.00005.2007. [DOI] [PubMed] [Google Scholar]
- 13.Harper SQ, Davidson BL. Plasmid-based RNA interference: construction of small-hairpin RNA expression vectors. Methods Mol Biol. 2005;309:219–235. doi: 10.1385/1-59259-935-4:219. [DOI] [PubMed] [Google Scholar]
- 14.Yang G, Merrill DC, Thompson MW, Robillard JE, Sigmund CD. Functional expression of the human angiotensinogen gene in transgenic mice. J Biol Chem. 1994;269:32497–32502. [PubMed] [Google Scholar]
- 15.Stec DE, Davisson RL, Haskell RE, Davidson BL, Sigmund CD. Efficient liver-specific deletion of a floxed human angiotensinogen transgene by adenoviral delivery of cre-recombinase in vivo. J Biol Chem. 1999;274:21285–21290. doi: 10.1074/jbc.274.30.21285. [DOI] [PubMed] [Google Scholar]
- 16.Stec DE, Keen HL, Sigmund CD. Lower blood pressure in floxed angiotensinogen mice after adenoviral delivery of cre-recombinase. Hypertension. 2002;39:629–633. doi: 10.1161/hy0202.103418. [DOI] [PubMed] [Google Scholar]
- 17.Nakajima T, Inoue I, Cheng T, Lalouel JM. Molecular cloning and functional analysis of a factor that binds to the proximal promoter of human angiotensinogen. J Hum Genet. 2002;47:7–13. doi: 10.1007/s10038-002-8649-2. [DOI] [PubMed] [Google Scholar]
- 18.Shimamoto Y, Ishida J, Yamagata K, Saito T, Kato H, Matsuoka T, Hirota K, Daitoku H, Nangaku M, Yamagata K, Fujii H, Takeda J, Fukamizu A. Inhibitory effect of the small heterodimer partner on hepatocyte nuclear factor-4 mediates bile acid-induced repression of the human angiotensinogen gene. J Biol Chem. 2004;279:7770–7776. doi: 10.1074/jbc.M310577200. [DOI] [PubMed] [Google Scholar]
- 19.Saito T, Oishi T, Yanai K, Shimamoto Y, Fukamizu A. Cloning and characterization of a novel splicing isoform of USF1. Int J Mol Med. 2003;12:161–167. [PubMed] [Google Scholar]
- 20.Viollet B, Lefrancois-Martinez AM, Henrion A, Kahn A, Raymondjean M, Martinez A. Immunochemical characterization and transacting properties of upstream stimulatory factor isoforms. J Biol Chem. 1996;271:1405–1415. doi: 10.1074/jbc.271.3.1405. [DOI] [PubMed] [Google Scholar]
- 21.Sirito M, Lin Q, Deng JM, Behringer RR, Sawadogo M. Overlapping roles and asymmetrical cross-regulation of the USF proteins in mice. Proc Natl Acad Sci U S A. 1998;95:3758–3763. doi: 10.1073/pnas.95.7.3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.West AG, Huang S, Gaszner M, Litt MD, Felsenfeld G. Recruitment of histone modifications by USF proteins at a vertebrate barrier element. Mol Cell. 2004;16:453–463. doi: 10.1016/j.molcel.2004.10.005. [DOI] [PubMed] [Google Scholar]
- 23.Huang S, Li X, Yusufzai TM, Qiu Y, Felsenfeld G. USF1 recruits histone modification complexes and is critical for maintenance of a chromatin barrier. Mol Cell Biol. 2007;27:7991–8002. doi: 10.1128/MCB.01326-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Landry CR, Lemos B, Rifkin SA, Dickinson WJ, Hartl DL. Genetic properties influencing the evolvability of gene expression. Science. 2007 July 6;317:118–121. doi: 10.1126/science.1140247. [DOI] [PubMed] [Google Scholar]
- 25.Griffin MJ, Wong RH, Pandya N, Sul HS. Direct interaction between USF and SREBP-1c mediates synergistic activation of the fatty-acid synthase promoter. J Biol Chem. 2007;282:5453–5467. doi: 10.1074/jbc.M610566200. [DOI] [PubMed] [Google Scholar]
- 26.Coon H, Xin Y, Hopkins PN, Cawthon RM, Hasstedt SJ, Hunt SC. Upstream stimulatory factor 1 associated with familial combined hyperlipidemia, LDL cholesterol, and triglycerides. Hum Genet. 2005;117:444–451. doi: 10.1007/s00439-005-1340-x. [DOI] [PubMed] [Google Scholar]
- 27.Naukkarinen J, Gentile M, Soro-Paavonen A, Saarela J, Koistinen HA, Pajukanta P, Taskinen MR, Peltonen L. USF1 and dyslipidemias: converging evidence for a functional intronic variant. Hum Mol Genet. 2005;14:2595–2605. doi: 10.1093/hmg/ddi294. [DOI] [PubMed] [Google Scholar]
- 28.Ng MC, Miyake K, So WY, Poon EW, Lam VK, Li JK, Cox NJ, Bell GI, Chan JC. The linkage and association of the gene encoding upstream stimulatory factor 1 with type 2 diabetes and metabolic syndrome in the Chinese population. Diabetologia. 2005 October;48:2018–2024. doi: 10.1007/s00125-005-1914-0. [DOI] [PubMed] [Google Scholar]
- 29.Zhu Y, Casado M, Vaulont S, Sharma K. Role of upstream stimulatory factors in regulation of renal transforming growth factor-beta1. Diabetes. 2005;54:1976–1984. doi: 10.2337/diabetes.54.7.1976. [DOI] [PubMed] [Google Scholar]
- 30.Vallet VS, Casado M, Henrion AA, Bucchini D, Raymondjean M, Kahn A, Vaulont S. Differential roles of upstream stimulatory factors 1 and 2 in the transcriptional response of liver genes to glucose. J Biol Chem. 1998;273:20175–20179. doi: 10.1074/jbc.273.32.20175. [DOI] [PubMed] [Google Scholar]
- 31.Hopkins PN, Heiss G, Ellison RC, Province MA, Pankow JS, Eckfeldt JH, Hunt SC. Coronary artery disease risk in familial combined hyperlipidemia and familial hypertriglyceridemia: a case-control comparison from the National Heart, Lung, and Blood Institute Family Heart Study. Circulation. 2003;108:519–523. doi: 10.1161/01.CIR.0000081777.17879.85. [DOI] [PubMed] [Google Scholar]
- 32.Marinescu VD, Kohane IS, Riva A. MAPPER: a search engine for the computational identification of putative transcription factor binding sites in multiple genomes. BMC Bioinformatics. 2005;6:79. doi: 10.1186/1471-2105-6-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.