

Abstract
Sialyltransferases transfer sialic acid from CMP-NeuAc to an acceptor molecule. Trans-sialidases of parasites transfer α2,3 linked sialic acid from one molecule to another without the involvement of CMP-NeuAc. Here, we report another type of sialylation termed reverse sialylation catalyzed by mammalian sialyltransferase ST3Gal-II. This enzyme synthesizes CMP-NeuAc by transferring NeuAc from the NeuAcα2,3Galβ1,3GalNAcα-unit of O-glycans, 3-sialyl globo unit of glycolipids and sialylated macromolecules to 5′-CMP. CMP-NeuAc produced in situ is utilized by the same enzyme to sialylate other O-glycans and by other sialyltransferases such as ST6Gal-I and ST6GalNAc-I forming α2,6 sialylated compounds. ST3Gal-II also catalyzed the conversion of 5′-UMP to UMP-NeuAc, which was found to be an inactive sialyl donor. Reverse sialylation proceeded without the need for free sialic acid, divalent metal ions or energy. The direct sialylation using CMP-NeuAc as well as the formation of CMP-NeuAc from 5′-CMP had a wide optimum range (pH 5.2–7.2 and 4.8–6.4 respectively) whereas the entire reaction comprising in situ production of CMP-NeuAc and sialylation of acceptor had a sharp optimum at pH 5.6 (the activity level 50% at pH 5.2 & 6.8 and 25% at pH 4.8 & 7.2). Several properties distinguish forward/conventional vs. reverse sialylation: i. Sodium citrate inhibited forward sialylation but not reverse sialylation. ii. 5′-CDP, a potent forward sialyltransferase inhibitor, did not inhibit the conversion of 5′-CMP to CMP-NeuAc. iii. The mucin core 2 compound 3-O-sulfoα2,3Galβ1,4GlcNAcβ1,6(Galβ1,3)GalNAcα-O-Bn, an efficient acceptor for ST3Gal-II, inhibited the conversion of 5′-CMP to CMP-NeuAc. A significant level of reverse sialylation activity is noted in human prostate cancer cell lines LNCaP and PC3. Overall, the study demonstrates that the sialyltransferase reaction is readily reversible in the case of ST3Gal-II and can be exploited for the enzymatic synthesis of diverse sialyl products.
The sialylation of carbohydrates is catalyzed by sialyltransferases (sialylTs) and trans-sialidases. During the biosynthesis of glycoconjugates, enzymes belonging to the glycosyltransferase family (including sialylT) catalyze the transfer of a monosaccharide unit from an activated glycosyl donor to an appropriate acceptor molecule. In the case of sialic acid (NeuAc), the activated donor consists of NeuAc that is β-glycosidically linked to the aglycan cytidine 5′-monophosphate (CMP) to form CMP-NeuAc (1). In contrast to other activated nucleotide sugars, CMP-NeuAc contains a monophosphate. This activated sugar is also unique since it exclusive forms α–glycosidically linked sialic acid residues in nature (2).
Besides sialylTs, trans-sialidases (TS) exist in parasites and these acquire sialic acids from the host for incorporation into the parasite’s surface (3). For example, Trypanosomas cruzi, an agent of Chagas’ disease, is unable to synthesize sialic acid de novo (4). This parasite employs TS to transfer terminal α2,3-linked sialic acid residues to terminal β-linked Gal in glycoproteins and glycolipids (5,6). In the absence of a suitable acceptor, TS transfers sialic acid to water molecules, thus functioning as a sialidase similar to viral, mammalian and bacterial sialidases (7). Some members of the TS family lack enzyme activity but still retain their Gal-binding properties. Such molecules function as lectins during parasite-host interaction (8). Recently, a multifunctional sialyltransferase has also been identified from Pasteurella multocida that exhibits multiple functions including α2,3 and α2,6 sialylation, and trans-sialidase activity (9).
Besides sialylT and TS activity, in the current paper, we report yet another action of sialylTs. This activity which we term “reversible sialylation” involves the enzymatic transfer of NeuAc by mammalian sialylT ST3Gal-II, from linear carbohydrate, branched core-2 mucin and fetuin glycoprotein based sialylated donors to acceptor CMP. This results in the formation of CMP-NeuAc. This newly synthesized CMP-NeuAc is then available for transfer to another acceptor using the same sialylT (ST3Gal-II). It is also available to other sialylTs like ST6Gal-I and ST6GalNAc-I for sialylation of other macromolecules. Thus, using reversible sialylation, diverse chemical entities can be readily synthesized using an array of sialyl donors without the need for CMP-NeuAc.
Indeed, reversible biochemical reactions are known to occur in metabolic pathways. For example, the hydrolysis of maltose by maltase is reversible provided high glucose concentrations (40%w/v) are present in the reaction mixture (10). Plant sucrose synthase can also reversibly catalyze the synthesis of UDP-Glc from sucrose and UDP (11, 12).Recently, Zhang et al. (13) also showed that four different glycosyltransferases that participate in natural product biosynthetic pathways related to calicheamicin and vancomycin, readily catalyze reversible reactions allowing sugars and aglycons to be exchanged with ease. Our studies now show that sialylation is also a reversible process. Of the three enzymes studied (ST3Gal-II, ST3Gal-III and ST6Gal-I), reversible sialylation was observed to occur readily in the case of ST3Gal-II. Forward and reverse sialylation could also be independently regulated using citrate ions, 5′-CDP and selected synthetic molecules, and this suggests that two distinct catalytic activities of the enzyme may regulate the forward and reverse sialylation reactions. Overall, our data support the concept that reverse sialylT activity can be exploited for the chemical synthesis of diverse sialylated glycoconjugates. The physiological significance of this novel enzymatic activity remains to be established.
MATERIALS AND METHODS
Materials
Rat recombinant ST3Gal-II (α2,3(O)ST), ST3Gal-III (α2,3(N)ST) and ST6Gal-I (α2,6(N)ST) were purchased from Calbiochem (14). Several different lots of ST3Gal-II have been used for this study and all yielded similar findings. Cloned ST6GalNAc-I (chicken) was kindly provided by Dr. James C. Paulson (Scripps Research Institute, La Jolla, CA). All nucleotide phosphates and CMP-NeuAc were from Sigma. The synthesis strategy of most acceptors/compounds used in the present study is described elsewhere (15, 16). Acrylamide copolymer of Galβ1,3GalNAcα-O-Al, asialo Cowper’s gland mucin (CGM), Anti-Freeze glycoproteins, Fetuin-O-glycosidic asialo glycopeptide (asialo FOG) and Fetuin Triantennary asialo Glycopeptide (asialo FTG) were available from earlier studies (17–19). The culturing of human cancer cell lines and the preparation of cell extracts is reported elsewhere (20). All cell extracts were frozen at −20ºC prior to use. Asialo bovine submaxillary mucin (asialo BSM) was made by heating BSM (Sigma) (5mg/ml) at 80ºC in 0.1N HCl for 1h, neutralizing with 1.0N NaOH, dialyzing against distilled deionized water in the cold room for 24h with four changes and then lyophilizing the product.
Enzymology studies
All enzymatic sialylation reactions were typically carried out in 100mM NaCacodylate buffer pH6.0 in the presence of enzyme, synthetic acceptor (at 7.5mM or as indicated in each experiment) and 0.2μCi CMP-[9-3H]NeuAc (NEN-Dupont, 29mCi/μmol). The concentration of total CMP-NeuAc was adjusted in individual reactions by supplementing with additional cold CMP-NeuAc. Reaction volume was 20μl. Products formed were separated using four different chromatography procedures (below). In all cases, the radioactive content of isolated products was determined by using 3a70 scintillation cocktail (Research Products International, Mount Prospect, IL), and a Beckman LS6500 scintillation counter.
a) Biogel P2 chromatography
A Biogel P2 column (Fine Mesh; 1.0×116.0 cm) was used with 0.1 M pyridine acetate (pH5.4) as the eluent at room temperature. In cases where radiolabeled donor compounds were prepared using this column, the peak fraction containing radioactivity was collected, lyophilized to dryness, dissolved in small volume of water and stored frozen at −20ºC for further experimentation.
b) Lectin- agarose affinity chromatography
A column of 5ml bed volume of WGA-agarose or PNA-agarose (Vector Lab, Burlingame, CA) was employed using 10mM Hepes pH7.5 containing 0.1mM CaCl2, 0.01mM MnCl2 and 0.1% NaN3 as the running buffer. Fractions of 1.0ml were collected. The bound material was then eluted with 0.5M GlcNAc or 0.2M Gal in the same buffer. SNA-agarose (Vector Lab) affinity chromatography was also carried out as above except that fractions of 2.0ml were collected and the bound material was eluted with 0.5M lactose.
c) Hydrophobic chromatography
This was done using Sep-Pak C18 cartridge (Waters, Milford, MA) and eluting the product with 3.0ml methanol.
d) Dowex-1-Formate column
Radioactive products from neutral allyl and methyl glycosides were measured by fractionation on Dowex-1-Formate (Bio-Rad:AG-1X8; 200–400 mesh; formate form) as described previously (14).
Calculation of equilibrium constant, Keq
Equilibrium constant was calculated for selected reversible bimolecular reactions that are denoted by:
Here, [9-3H]D* denotes the radiolabeled donor and D’ is the donor after removal of sialic acid. Similarly, A and [9-3H]A* denote the acceptor before and after incorporation of NeuAc. In each run, unreacted [9-3H]D* and product [9-3H] A* are measured using radioactivity measurements. The equilibrium constant (Keq=kreverse/kforward, dimensionless units) are then determined using the following equation, where terms in square brackets denote concentrations:
[D0] and [A0] are initial donor and acceptor concentrations. A plot of [D*]/([D0]-[D*]) versus [A*]/([A0]-[A*]) yields Keq from slope data. Keq<1 implies that the forward reaction is favored and the reaction proceeds to the right side to form [9-3H]A* efficiently.
Liquid chromatography coupled with tandem mass spectrometry (LC/MS/MS)
The LC separation was performed using a C18 reverse phase column at a flow rate of 220μL/min. Two buffers, 0.1% formic acid in acetonitrile and 0.1% formic acid in water were used, with a linear gradient of 8%/min increase of the organic buffer starting from 20%. The sample injection volume was 20 μL. Negative-ion ESI was used for the detection of sialic acid derivatives due to its superior sensitivity to positive-ion ESI (14). The identification was accomplished in precursor ion scan mode at unit resolution (FWHM 0.6–0.8 amu) by selectively detecting the parent ions in the third quadrupole (Q3) of a triple quadrupole instrument that give rise to the diagnostic fragment ion (sialic acid ion [M-H]− at m/z 290) created by collisions in Q2.
RESULTS
ST3Gal-II reverse sialylates 5′-CMP
The cloned and purified rat sialyltransferase α2,3(O)ST (ST3Gal-II) (14) is used in many experiments presented in this manuscript. This enzyme has been shown to mediate α2,3 sialylation of terminal Gal residues in the O-glycan core-2 trisaccharide unit Galβ1,3(GlcNAcβ1,6)GalNAcα (14, 21). We examined if this sialylation reaction is reversible (Fig. 1). For this, [9-3H]NeuAcα2,3Galβ,13(GlcNAcβ1,6)GalNAcα-O-Al [9-3H[1]] was prepared using ST3Gal-II in the presence of CMP-[9-3H]NeuAc and the trisaccharide acceptor Galβ1,3(GlcNAcβ1,6)GalNAcα-O-Al as previously described (14), and the radiolabeled product was isolated using Biogel P2 chromatography. During this isolation, radiolabeled [9-3H[1]] appeared prior to unreacted trisaccharide (data not shown). Two different reaction mixtures were then prepared with: i) [9-3H[1]] and ST3Gal-II but without 5′-CMP and ii) [9-3H[1]] and 5′-CMP along with ST3Gal-II. When the products formed were subjected to WGA-agarose affinity chromatography, a majority of the radioactive component from i) but not ii) bound the column (Fig. 1). The results of the first run indicate that the enzyme ST3Gal-II does not exhibit any sialidase activity. Further, the efficient (>90%) transfer of radioactive [9-3H]NeuAc from 150μM [9-3H[1]] to 1.0mM 5′-CMP in the presence of ST3Gal-II in the second panel suggests that the reverse reaction in scheme-I proceeds at an appreciable rate.
Fig. 1.
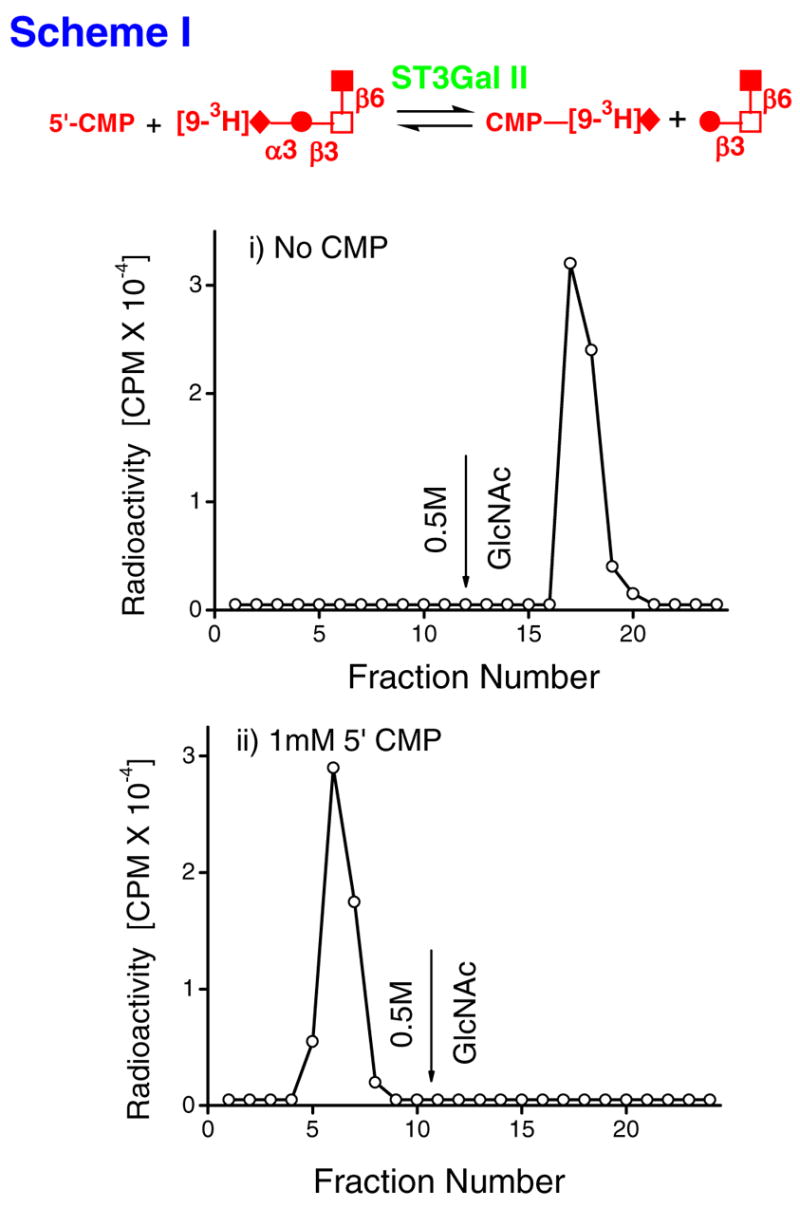
Nucleotide phosphates as acceptors of radioactivity from [9-3H]NeuAcα2,3Galβ,13 (GlcNAcβ1,6)GalNAcα-O-Al (Al=Allyl) [[9-3H][1]] in the presence of ST3Gal-II (scheme-I). Reaction mixtures (RM) with the following composition were incubated at 37ºC for 21h in 100mM NaCacodylate buffer, pH6.0: i) RM containing 150μM (0.4μCi) [[9-3H][1]] (donor) along with 0.8mU ST3Gal-II but no 5′-CMP. ii) RM containing [[9-3H][1]] and 1.0mM 5′-CMP, along with ST3Gal-II. Products formed were fractionated on a WGA-agarose column. Bound products were released using 0.5M GlcNAc at fraction 12. The radioactive product formed in ii) does not bind WGA. Thus, the forward reaction of scheme-I occurs at an appreciable rate. The feasibility of the reverse reaction is well established in literature. Schematic symbols: ◆:sialic acid, ●:Gal, □:GalNAc, ■:GlcNAc
Similar results were observed upon increasing concentrations of 5′-CMP when [9-3H]NeuAcα2,3Galβ1,3(4-FGlcNAcβ1,6)GalNAcα-O-Bn [9-3H[2]] was donor (Fig. 2a, b). The concentration of 5′-CMP that mediated half-maximal transfer of [9-3H]NeuAc was 80μM. The bimolecular equilibrium constant (Keq) for this reaction was 0.35 (dimensionless units, Table 1). Indeed, the Keq varied depending on the donor and it was 98.9 when the donor was based on the globo glycolipid, [9-3H]NeuAcα2,3Galβ1,3GalNAcβ1,3 Galα-O-Me [[9-3H][3]] (Fig. 2c, d). Upon comparing [[9-3H][2]] and [[9-3H][3]], it is apparent that reverse sialylation proceeds more effectively in the case of [[9-3H][2]]. To further confirm the above estimates of Keq for core-2 based structures, we also calculated this parameter for our previously published data (Fig. 3C, (14)) where [9-3H]CMP-NeuAc was donor and mucin core-2 tetrasaccharide Galβ1,4GlcNAcβ1,6(Galβ1,3)GalNAcα-O-Bn was acceptor. In this case, Keq measured with respect to the rate of mucin core-2 sialylation was 5.55 (data not shown), which translates to a reverse sialylation Keq of 0.18. Table 1 provides a summary of Keq values for reverse sialylation.
Fig. 2.

Equilibrium constant. a. Reversible sialyltransferase activity at increasing concentration of 5′-CMP. 0.15mM of donor [9-3H]NeuAcα2,3Galβ1,3(4-FGlcNAcβ1,6)GalNAcα-O-Bn [9-3H[2]] was incubated with varying concentrations of 5′-CMP and 0.15mU ST3Gal-II under conditions described in Fig. 1 for 2 h. The consumption of the [9-3H]NeuAc benzyl glycoside donor was measured by subjecting the incubation mixture to Sep-Pak C18 fractionation, which binds the donor and not CMP-[9-3H]NeuAc. The reaction proceeded to ½ the maximum extent at 80μM 5′-CMP. b. Equilibrium constant Keq (=0.35) calculated for data in panel a. c. Varying concentrations of 5′-CMP was sialylated with ST3Gal-II (2.0 mU) using either 0.15mM (panel c) or 1.5mM (not shown) sialyl donor [9-3H]NeuAcα2,3Galβ1,3GalNAcβ1,3Galα-O-Me [9-3H[3]] for 4h. The product CMP-[9-3H]NeuAc and the unused [9-3H[3]] were separated and quantitated by Dowex-1-Formate method. d. Equilibrium constant, Keq =98.9. Diamonds denote data from 1.5mM runs while data from 0.15mM runs are depicted by squares.
Table 1.
Reverse sialylation equilibrium constant
| Donor | Acceptor |
Keq (dimensionless units) |
|---|---|---|
|
| ||
| [9-3H]NeuAcα2,3Galβ1,3(4-FGlcNAcβ1,6) GalNAcα-O-Bn, [[9-3H]2] | 5′-CMP | 0.35 |
| [9-3H]NeuAcα2,3Galβ1,3GalNAcβ1,3Galα-O-Me, [[9-3H]3] | " | 98.9 |
| [9-3H]NeuAcα2,3Galβ1,3(Galβ1,4 GlcNAcβ1,6)GalNAcα-O-Bn | " | 0.18 |
Fig. 3.

Serial transfer of sialic acid using scheme-II was followed at increasing concentrations of acceptor D-Fucβ1,3GalNAcα-O-Bn [5]. The donor used was either: i) 0.15mM [9-3H]CMP-NeuAc, ii) 0.15mM [9-3H]NeuAcα2,3Galβ1,3(4-FGlcNAcβ1,6)GalNAcα-O-Bn [9-3H [2]] in the presence of 2.0mM 5′-CMP, or iii) [9-3H]NeuAcα2,3Galβ1,3(6-O-SulfoGlcNAcβ1,6)GalNAcα-O-Al [[9-3H][4]] in the presence of 2.0mM 5′-CMP. In runs where [9-3H [2]] was donor since both [5] and [2] bind C18 cartridges, control runs were performed in the absence of D-Fucβ1,3GalNAcα-O-Bn to estimate the amount of [9-3H]CMP-NeuAc formed under the experimental conditions. 80–85% of the radioactivity was transferred to CMP-NeuAc in these runs. The amount of CMP-NeuAc remaining in runs with [5] was then subtracted from CMP-NeuAc radioactivity in the control run to determine the amount of sialylated [5]. To complement this run, studies were also performed with [[9-3H][4]], a molecule with Allyl at the anomeric position, which does not bind C18. In these runs, the amount of sialylated [5] was determined by measuring radioactivity retained in the C18-cartridge. a. Equilibrium constant was 5.55 when CMP-[9-3H]NeuAc was donor b. ~10-fold lower acceptor concentration was required for similar extents of reaction when CMP-[9-3H]NeuAc was donor, compared to core-2 based donors [2] and [4].
When other nucleotide phosphates were substituted for 5′-CMP, their effectiveness varied. 5′-UMP, 5′-CDP and 2′deoxy 5′-CMP were 55.9%, 28.4% and 26.3% effective in comparison with 5′-CMP. Dosage studies further confirm the formation of UMP-NeuAc via the reverse sialylation mechanism (Supplemental Fig. 1). In these studies where [9-3H[3]] was donor and 5′-CMP or 5′-UMP was acceptor, CMP-[9-3H]NeuAc was observed to form more efficiently than UMP-[9-3H]NeuAc. Other nucleotide phosphates (3′-CMP, 5′-CTP, 5′-GMP, 5′-AMP, 5′-TMP and 5′-IMP) had lower activity (< 3.0%). 5′-CMP in the presence of cold sialic acid did not show any decrease in accepting [9-3H]NeuAc from donor (100.3%), indicating that reverse sialylation did not involve the formation of free sialic acid as intermediate. Such free sialic acid could be formed following hydrolysis of donor. Overall, the data support the reverse sialylation mechanism shown in scheme-I.
ST3Gal-II utilizes CMP-NeuAc formed in reverse reaction to sialylate O-glycans
We tested the possibility that CMP-NeuAc formed above may be available for transfer to other acceptors using ST3Gal-II (scheme-II, Fig. 3). For this, three radiolabeled donors were prepared using methods described above: two were based on the mucin Core-2 structure ([[9-3H][1]] and [9-3H]NeuAcα2,3Galβ1,3(6-O-SulfoGlcNAcβ1,6)GalNAcα-O-Al [[9-3H][4]]) while the third was based on the Globo glycolipid ([9-3H][3]). The transfer of [9-3H]NeuAc from these sialylated donors to various T-hapten (Galβ1,3GalNAcα) and mucin core2-based glycoside acceptors in the presence of 5′-CMP and ST3Gal-II was assessed (Table 2). All three donors of ST3Gal-II ([9-3H] siayl [1], [3] and [4]) allowed the formation of CMP-[9-3H]NeuAc, and a diverse array of products. In contrast to this, two other molecules ([9-3H] NeuAcα2,3Galβ1,4GlcNAcβ-O-Al and [9-3H]NeuAcα2,6Galβ1,4GlcNAcβ-O-Al), which were formed by sialylation of a poor acceptor of ST3Gal-II (Galβ1,4GlcNAcβ-O-Al) by enzymes ST3Gal-III and ST6Gal-I, did not act as donors in the reverse sialylation reaction. Thus, while the reversible function of ST3Gal-II is not unique to a given donor-acceptor pair, the α2,3 sialic acid linkage on different donors have vastly different Keq values (see Table 1).
Table 2.
| Acceptor (2.5mM) | [9-H]NeuAcα2,3Galβ1,3(GlcNAcβ1,6)GalNAcα-O-Al [[9-3H][1]] | [9-3H]NeuAcα2,3Galβ1,3(6-O-sulfoGlcNAcβ1,6) GalNAc-O-Al [[9-3H][4] | [9-3H]NeuAcα2,3Galβ1,3GalNAcβ1,3Galα-O-Me[[9-3H][3]] | |
|---|---|---|---|---|
|
| ||||
| D-Fucβ1,3GalNAcα-O-Bn | [5] | 100.0 (34569 CPM) | 100.0 (97675 CPM) | 100.0 (6224 CPM)*** |
| D-Fucβ1,3(GlcNAcβ1,6)GalNAcα-O-Bn | [6] | ND | 96.9 | 106.5 |
| 4-FGalβ1,3GalNAcα-O-Bn | [7] | 60.8 | 33.3 | 4.1 |
| Galβ1,3(6-O-Me)GalNAcα-O-Bn | [8] | 43.0 | 21.5 | 3.3 |
| 3-O-MeGalβ1,4GlcNAcβ1,6(Galβ1,3) | [9] | ND | ||
| GalNAcα-O-Bn | 22.4 | 10.0 | ||
| Galβ1,4GlcNAcβ1,6(Galβ1,3) | [10] | |||
| GalNAc3-O-Bn | 20.5 | 8.4 | ND | |
| 4-O-MeGalβ1,3GalNAcα-O-Bn | [11] | 8.9 | 2.7 | 0.2 |
| Galβ1,4GlcNAcβ-O-Bn | [12] | ND | 1.9 | 0.5 |
| Galβ1,4GlcNAcβ1,6(4-O-MeGalβ1,3) | [13] | |||
| GalNAcα-O-Bn | 1.3 | 0 | ND | |
ND: Not Determined
150μM [9-3H] labeled donor was incubated with 1mM 5′-CMP and 2.5mM either T-hapten or mucin core-2 based acceptor in the presence of 0.2mU ST3Gal-II for 4h. under reaction conditions identical to Fig. 1. Acceptors were separated from donor using C-18 cartridge due to hydrophobicity of Bn (Benzyl) group. Radioactivity of eluate was quantified. The blank containing no acceptor had CPM<100.
Incorporation of [9-3H]NeuAc expressed as percent of the CPM incorporated into [5]
CPM for this donor is low since 10 fold cold CMP-NeuAc (i.e., lower specific radioactivity of CMP-[9-3H]NeuAc) was used to synthesize it as compared to other two donors.
The transfer of [9-3H]NeuAc from [[9-3H][1]] to D-Fucβ1,3GalNAcα-O-Bn [5] in the presence of 5′-CMP and ST3Gal-II increased linearly in the first two hours and it reached saturation at 4h (Supplemental Fig. 2), and thus the data in Table 3 (4h time point) represent equilibrium conditions. In Table 3, [5] and D-Fucβ1,3(GlcNAcβ1,6)GalNAcα-O-Bn [6] were observed to serve as good acceptors followed by 4-FGalβ1,3GalNAcα-O-Bn [7] and Galβ1,3(6-O-Me)GalNAcα-O-Bn [8], while 4-O-methylation of β1,3-linked Gal [11] reduced acceptor efficiency. As anticipated, Galβ1,4GlcNAcβ-O-Bn [12] was an inactive acceptor since ST3Gal-II does not act on it. Since CMP-NeuAc is formed via the same first reaction of scheme-II for a given donor, the data in Table 3 suggest that acceptor specificity for ST3Gal-II may govern the extent of reaction. Further, diverse sialylated products can be formed using the same synthetic sialylated donor.
TABLE 3.
* CMP-NeuAc but not UMP-NeuAc formed by reverse sialylation is an efficient sialyl donor for forward sialyltransferase reaction
| Reverse sialylation | ||
|---|---|---|
| 5′CMP→CMP-[9-3H]NeuAc | 5′UMP→UMP-[9-3H] NeuAc | |
| Incorporation of [9-3H]NeuAc into the acceptor | ||
| CPM | CPM | |
| a) ST3Gal-II activity
[Acceptor: D-Fucβ1,3GalNAcα-O-Bn] |
134122 | 1243 |
| b) ST3Gal-III activity
[Acceptor: 4-O-MeGalβ1,4GlcNAcβ-O-Bn] |
59531 | 3804 |
| c) ST6Gal-I activity
[Acceptor: Galβ1,4GlcNAcβ-O-Bn] |
15066 | 182 |
Incubation mixtures run in duplicate contained the sialyl donor [[9-3H] [3]] (0.0625mM), 8mM 5′-CMP (first column) or 5′-UMP (second column) and benzyl disaccharide glycoside acceptor (2.5mM). In addition: a) contained 3.0mU of ST3Gal-II, b) contained 3.0mU each of ST3Gal-II and ST3Gal-III, and c) contained 3.0mU of ST3Gal-II and 1.0mU of ST6GalI. Blank samples did not contain 5′-CMP or 5′UMP.
The reaction mixtures described above were incubated for 4h at 37°C and then processed using Sep-Pak C18 method, which binds benzyl glycosides. The amount of [9-3H] NeuAc transferred from donor to individual acceptors was obtained after subtracting blank values (<400 CPM). The variation of duplicate values was less than 5% in all cases.
In order to compare the efficiency of individual reactions of scheme II, independent runs were performed where either 0.15mM [9-3H]CMP-NeuAc, 0.15mM [9-3H][2] or 0.15mM [9-3H][4] was the donor, and [5] was the acceptor (Fig. 3). 2mM 5′-CMP was added in runs with core-2 donors [2] and [4], and thus reverse sialylation of these molecules was feasible. Keq for transfer of sialic acid from CMP-NeuAc to [5] was 5.55 (Fig. 3a). As seen in Fig. 3b, 10-fold lower amounts of acceptor (0.6mM [5]) was required for comparable transfer when CMP-NeuAc was donor versus the case where the donor was either [9-3H [2]] (~6mM) or [9-3H[4]] (~6mM). Thus, while the two step mechanism results in lower conversion than CMP-NeuAc alone, the transfer of NeuAc still takes place at an appreciable rate.
Reverse sialylation occurs more readily with ST3Gal-II compared to ST3Gal-III and ST6Gal-I
We determined if reverse sialylation was more pronounced for ST3Gal-II in comparison to other sialyltrasferases. Thus two other cloned rat sialyltransferases ST3Gal-III (or α2,3(N)ST) and ST6Gal-I (or α2,6(N)ST) (14) were examined. For these studies, we generated two molecules: i) [9-3H]NeuAcα2,6Galβ1,4GlcNAcβ-O-Al [14] was made by the reaction of Galβ1,4GlcNAcβ-O-Al and CMP-[9-3H]NeuAc in the presence of α2,6sialyltransferase, ST6Gal-I. The radioactive product was separated using Biogel P2 column using a protocol similar to that described above; ii) [9-3H]NeuAcα2,3Galβ1,4 GlcNAcβ-O-Al [15] was similarly produced by reacting Galβ1,4GlcNAcβ-O-Al and CMP-[9-3H]NeuAc in the presence of α2,3sialyltransferase ST3Gal-III. We note that the acceptor used in the above runs, Galβ1,4GlcNAcβ-O-Al, does not undergo significant sialylation in the presence of CMP-NeuAc and the sialyltransferase that is the focus of our paper, ST3Gal-II (14). However, it is efficiently sialylated by both ST3Gal-III and ST6Gal-I (14). The ability of these two radiolabeled molecules ([14] and [15]) to act as [9-3H]NeuAc donors was assayed by using reaction mixtures where these compounds were mixed with excess 5′-CMP, enzyme and acceptors with benzyl aglycan group. C18 cartridges were then used to measure the extend of [9-3H]NeuAc transferred to acceptor. The formation of CMP-NeuAc was negligible when either [9-3H] NeuAcα2,6Galβ1,4GlcNAcβ-O-Al or [9-3H]NeuAcα2,3Galβ1,4GlcNAcβ-O-Al was the donor in the presence of a series of acceptors and sialyltransferases ST6Gal-I and ST3Gal-III respectively. Thus, reverse sialylation takes place more readily with ST3Gal-II. When these two radiolabeled donors were used in the presence of ST3Gal-II, also, we observed the formation of negligible amounts of product suggesting that CMP-NeuAc was not formed via reverse sialylation from these two donors. While the possibility that ST3Gal-III and ST6Gal-I can mediate reverse sialylation under different conditions cannot be ruled out, the data do suggest that ST3Gal -II may have unique structural properties that confer the reverse sialylation activity.
pH dependence of reverse sialylation
We examined if the pH range in which ST3Gal-II catalyzes the formation of CMP-NeuAc from sialylated donor and the total reaction ie the incorporation of NeuAc from CMP-NeuAc produced in situ into acceptor are distinct. First, in studies that measured the transfer of [93H]NeuAc from donor [9-3H[2]] to 5′-CMP, we observed optimum transfer at pH range 4.8–6.4 (Supplemental Fig. 3a). In contrast, the transfer of [9-3H]NeuAc from CMP-[9-3H]NeuAc to Galβ1,3GalNAcα-O-Bn [16] was maximum at pH 5.2–7.2(Supplemental Fig. 3b). Even though the pH curves for the direct sialylation using CMP-[9-3H] NeuAc and for the formation of CMP-[9-3H] NeuAc by reverse sialylation have wide range (almost two pH units) and appear very much overlapping, it is really significant to note that when the entire reaction was measured by monitoring the transfer of [9-3H]NeuAc to [5] from the donor [9-3H[3]] via 5′-CMP a sharp optimum at pH 5.6 (Supplemental Fig. 3c) was observed. The possibility that reversible sialylation may have additional unique properties was explored as described below.
Distinct effects of citrate on forward and reverse sialyltransferase activities
Besides pH other distinctions were also observed between forward and reverse sialylation. In this regard, citrate ions tended to inhibit forward sialylation but not reverse sialylation activity. For these studies, the effect of citrate on the forward sialylation activity of ST3Gal-I, ST3Gal-II, ST3Gal-III and ST6Gal-I was examined. Reverse sialylation activity of ST3Gal-II was also measured in terms of: i) the formation of CMP-NeuAc from 5′-CMP and ii) the transfer of NeuAc from this newly synthesized CMP-NeuAc to another O-glycan. Citrate inhibited 48%–98% the direct sialylation activity of all the enzymes mentioned above. However, both the synthesis of CMP-[9-3H]NeuAc from 5′-CMP as well as the synthesis of [9-3H]NeuAcα2,3D-Fucβ1,3 GalNAcα-O-Bn using the newly formed CMP-[9-3H]NeuAc through reverse sialylation of ST3Gal-II was not inhibited at all by citrate. The effect of citrate at high concentration (40 mM ) may be due to complexing with cytidine but it is interesting to note that citrate was present in excess (twice the concentration) of 5′-CMP in the reverse sialylation incubation mixtures.
Effect of 5′-nucleotides on reverse sialylation
The effect of 5′-nucleotides on forward sialylation (Fig. 4a) and reverse sialylation (Fig. 4b) were examined. As seen in Fig. 5a, both 5′-CDP and 5′-CMP inhibited the forward sialylation activities of ST3Gal-II, ST3Gal-III, and ST6Gal-I using the sialyl donor CMP-[9-3H]NeuAc and specific acceptors for the respective enzymes. 5′-CDP was a more potent inhibitor in all cases. In studies of reverse sialylation (Fig. 4b), 5′-CDP did not inhibit the first step in the reverse sialylation reaction of ST3Gal-II namely the formation of CMP-[9-3H]NeuAc from 5′-CMP. The overall formation of [9-3H]NeuAcα2,3D-Fucβ1,3GalNAcα-O-Bn by the two-step reverse sialylation process, however, was inhibited by 5′-CDP and this is consistent with the finding that 5′-CDP inhibits forward sialylation in the second step. 5′-UMP and 2-deoxy 5′-CMP served as controls in these experiments since they did not alter the extent of product formation (Fig. 5b). Overall, our studies show that 5′-CDP, an analog of 5′-CMP inhibits the forward sialylation reaction without altering the reverse reaction.
Fig. 4.
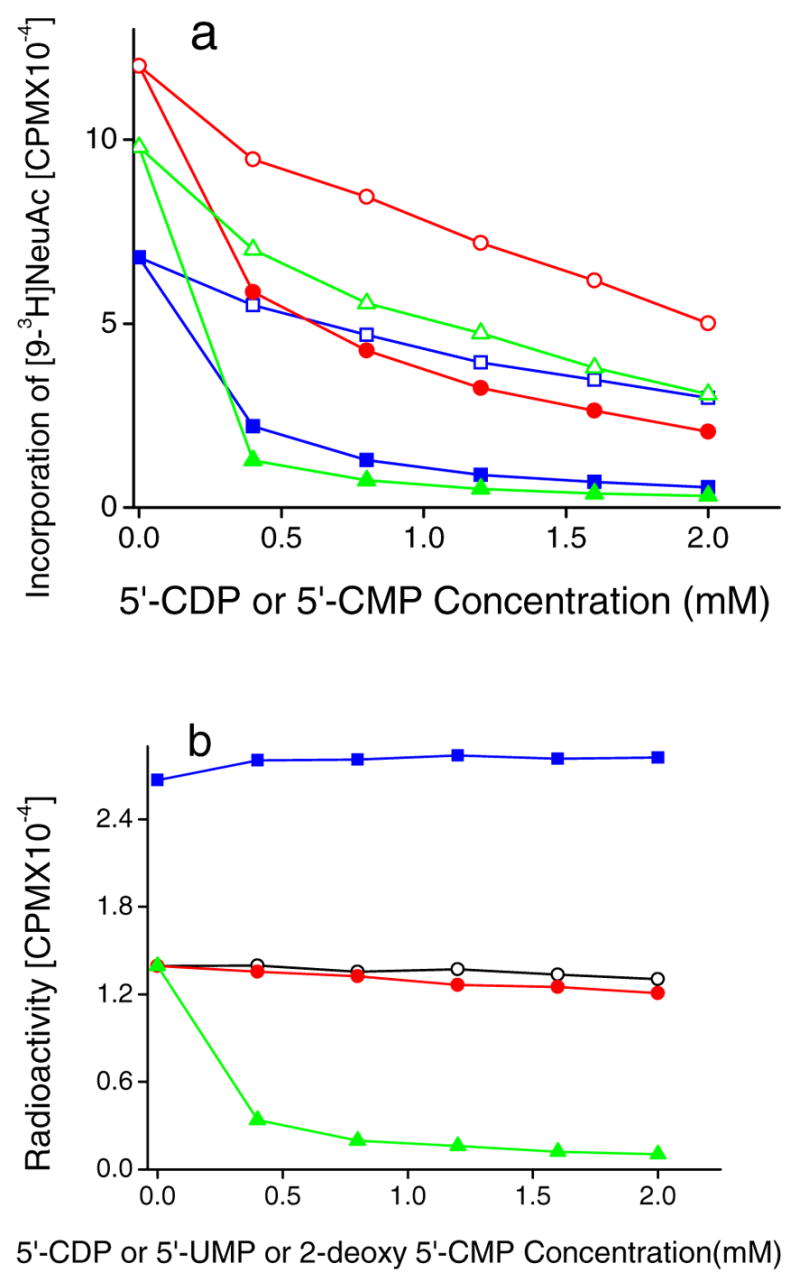
Effect of 5′-nucleotides on direct sialyltransferase activity (panel a) and reverse sialylation (panel b). a. Varying concentrations of 5′-CDP or 5′-CMP were added to 100 mM acceptor (specified below), 0.15mM CMP-[9-3H]NeuAc and either ST3Gal-II (0.2mU), ST3Gal -III (0.5mU) or ST6Gal-I(0.2mU) for 4h at 37ºC. Products were separated using Sep-Pak C18 method. ST3Gal-III activity was measured using acceptor 4-O-MeGalβ1,4GlcNAcβ-O-Bn in the presence of 5′-CDP (●) and 5′-CMP (○). ST6Gal-I activity was measured using Gal β1,4GlcNAcβ-O-Bn in the presence of 5′-CDP (▲) and 5′-CMP (Δ). ST3Gal-II using D-Fucβ1,3GalNAcα-O-Bn [5] in the presence of 5′-CDP (■) and 5′-CMP (□). b) Reverse sialylation by ST3Gal-II was measured in reaction mixtures containing 0.2 mM [9-3H][3], 7mM 5′-CMP, ST3Gal-II (2mU), D-Fucβ1,3GalNAcα-OBn (3.0mM) and varying doses of either 5′-CDP (▲), 5′-UMP (○) or 2-deoxy 5′-CMP (●) for 4h at 37ºC. Reaction product ([9-3H]NeuAcα2,3DFucβ1,3 GalNAcα-O-Bn) was isolated using Sep-Pak C18 method. In some runs, where [5] was absent, the amount of CMP-NeuAc produced was quantified using the Dowex-1 Formate method in the presence of varying doses of 5′-CDP (■)
Fig. 5.
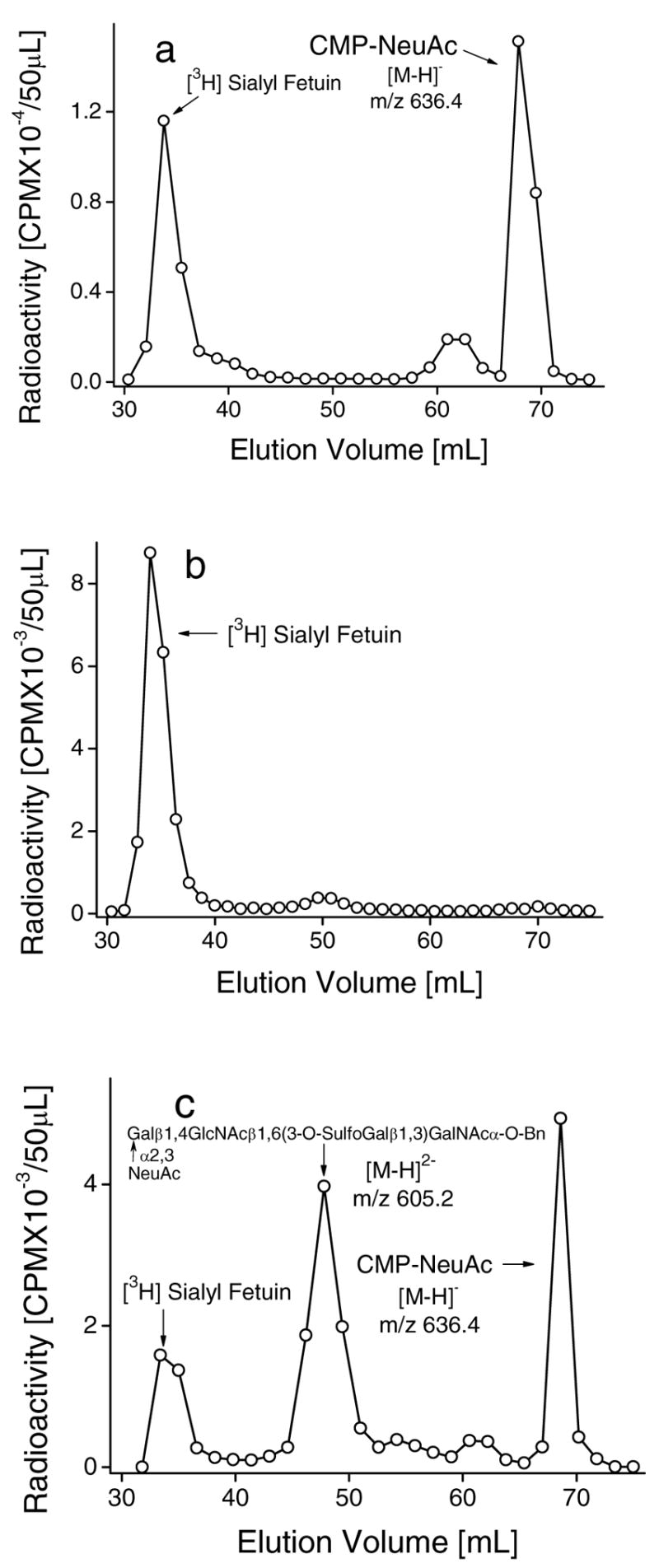
Effect of sialyl or sulfo substituents in O-glycan chain on the reverse sialylation by ST3Gal-II. Incubation mixtures (600μl) contained [9-3H]Sialyl Fetuin (5mg), 200mM NaCacodylate pH 6.0, 20mM 5′-CMP, 50mM ST3Gal-II and the following: a. 6.0mM NeuAcα2,3Galβ1,4GlcNAcβ1,6 (Galβ1,3)GalNAcα-O-Bn, b. 6.0mM 3-O-SulfoGalβ1,4GlcNAcβ1,6(Galβ1,3)GalNAcα-O-Bn or c. 6.0mM Galα1,6GlcNAcβ1,6(3-O-Sulphoβ1,3Gal)GalNAcα-O-Bn along with 50mU ST3Gal-III. Products were fractionated using Biogel P2 column after 20h at 37ºC. Unused [9-3H]Sialyl Fetuin (peak 1) appears prior to [9-3H]Sialyl product from acceptor (peak 2) and CMP-[9-3H]NeuAc (peak 3). Product identities were verified using LC-MS as indicated by molecular weights noted in the panels.
Effect of O-glycans with sialyl or sulfo substituents on reverse sialyltransferase activity
Another distinction between forward and reverse sialylation was observed in studies where the effect of three compounds, NeuAcα2,3Galβ1,4 GlcNAcβ1,6(Galβ1,3)GalNAcα-O-Bn (Fig. 5a), 3-O-SulfoGalβ1,4GlcNAcβ1,6(Galβ1,3)GalNAcα-O-Bn (Fig. 5b) and Galβ1,4GlcNAcβ1,6(3-O-SulfoGalβ1,3)GalNAcα-O-Bn (Fig. 5c), on the sialylation reaction was measured. For these studies, [9-3H] sialyl fetuin was prepared by the action of ST3Gal-II on fetuin in the presence of CMP-[9-3H]NeuAc. Radioactivity incorporation into the O-glycan chain of the glycoprotein was confirmed by treating the labeled molecule with alkaline borohyrdride (1MNaBH4 in 0.1MNaOH at 45ºC for 24 hours) and detecting the released radioactivity. During these studies, [9-3H] sialyl fetuin was incubated with 5′-CMP, one of the above mentioned compounds, and sialyltransferase for 20h., and the product formed was seperated using a Biogel P2 column. As seen in Fig. 5, the radioactivity associated with fetuin can be resolved from that associated with CMP-NeuAc and the synthetic acceptor. The radioactive fractions under the peaks corresponding to the product (the sialylated compound) and CMP-NeuAc were pooled separately, concentrated to dryness by lyophilization and then identified by mass spectrometry as shown in Fig 5. Here, it is observed that 3-O-SulfoGalβ1,4GlcNAcβ1,6(Galβ1,3)GalNAcα-O-Bn (Fig. 5b) inhibits formation of the intermediate CMP-NeuAc. Consequuently no radioactivity was associated with the acceptor. NeuAcα2,3Galβ1,4GlcNAcβ1,6(Galβ1,3)GalNAcα-O-Bn (Fig. 5a), on the other hand, prevents product formation without inhibiting CMP-NeuAc formation. Thus the sulfated compound 3-O-SulfoGalβ1,4GlcNAcβ1,6(Galβ1,3)GalNAcα-O-Bn in contrast to its sialylated analog NeuAcα2,3Galβ1,4GlcNAcβ1,6(Galβ1,3)GalNAcα-O-Bn inhibits the formation of CMP-NeuAc from 5′-CMP. Finally, Galβ1,4GlcNAcβ1,6(3-O-SulfoGalβ1,3)GalNAcα-O-Bn permits both the formation of CMP-NeuAc from 5′-CMP, and the transfer of [9-3H]NeuAc from the newly synthesized CMP-[9-3H]NeuAc into Galβ1,4 GlcNAcβ1,6(3-O-SulfoGalβ1,3)GalNAcα-O-Bn in the presence of ST3Gal-III (Fig. 5c). The use of distinct compounds to differentially alter reverse and forward sialylation function further supports the concept that ST3Gal-II may have more than one catalytic function.
Reverse sialylation activity of ST3Gal-II allows formation of sialylated globo backbone structure from sialyl fetuin
As shown in Fig. 6a, [9-3H]sialyl fetuin served as a good donor that allowed the formation of CMP-[9-3H]NeuAc by ST3Gal-II through the reverse sialylation mechanism. Fig. 6b further shows that ST3Gal-II can synthesize NeuAcα2,3Galβ1,3GalNAcβ1,3Galα-O-Me from [9-3H] sialyl fetuin using reaction scheme-II. Thus sialyl fetuin may be a useful compound for the inexpensive, enzymatic synthesis of an array of α2,3sialylated compounds including globo backbone based analogs.
Fig. 6.
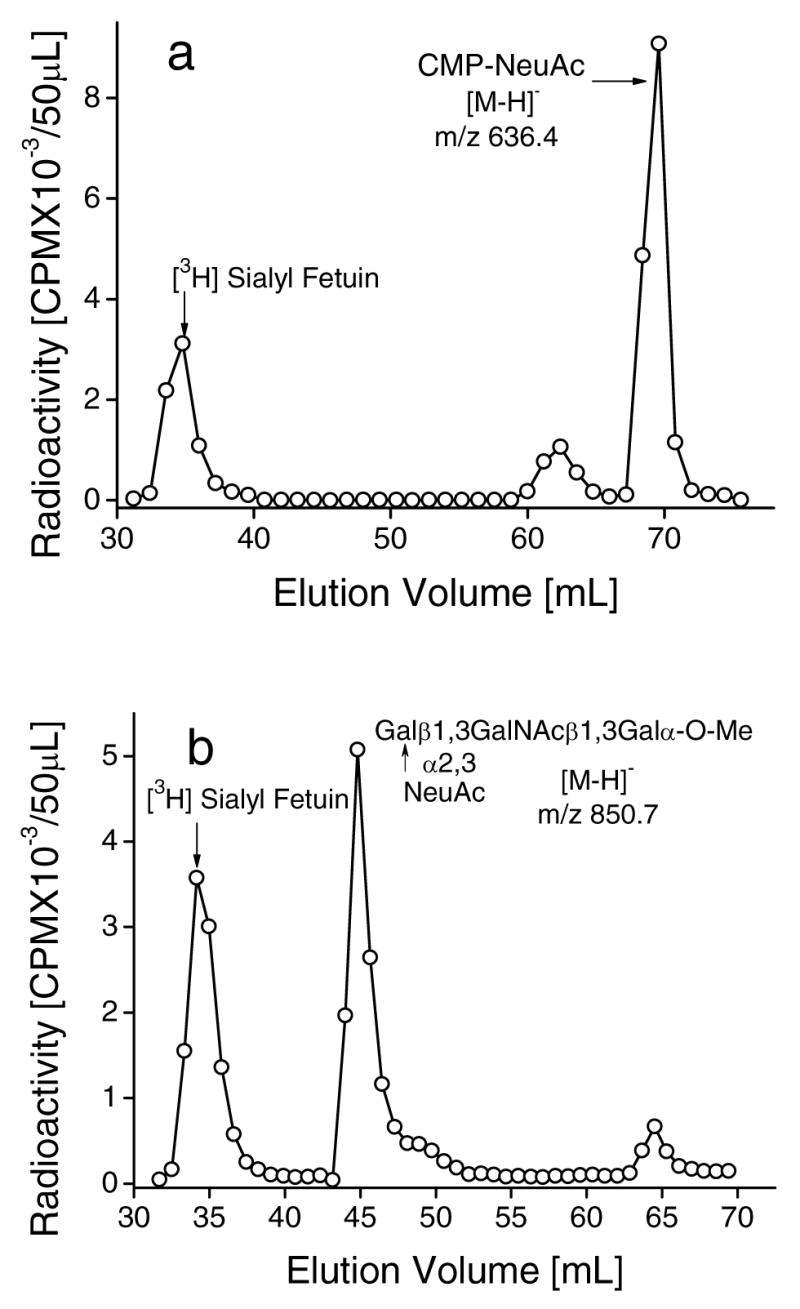
Sialylation of Globo backbone structures by reverse sialylation of ST3Gal-II. Incubation mixtures (500μl) contained [9-3H]Sialyl Fetuin (5 mg), 200mM NaCacodylate pH 6.0, 20mM 5′-CMP and the following: a. 50 mU ST3Gal-II and b. 6.0 mM Galβ1,3GalNAcβ1,3Galα-O-Me and 50mU ST3Gal-II. Reaction products were isolated using Biogel P2 column after 20h at 37ºC and product identity was verified by mass spectrometry as indicated by the molecular weights in the panel. Peak I: Unused [9-3H]Sialyl Fetuin; Peak II: [9-3H]Sialyl product from acceptor; Peak III: CMP-[9-3H]NeuAc from 5′-CMP.
CMP-NeuAc formed by reverse sialylation serves as a sialyl donor for macromolecules in the presence of ST3Gal-II, ST6Gal-I and ST6GalNAc-I
The ability of ST3Gal-II (see Fig 3. Scheme-II) to mediate the formation of sialylated macromolecules was studied using CGM (porcine Cowper’s Gland Mucin, Supplemental Fig. 4a), BSM (Bovine Submaxillary Mucin, Supplemental Fig. 4b), Anti-Freeze glycoprotein, FTG (Fetuin Triantennary glycopeptide, Supplemental Fig. 4c) and FOG (Fetuin O-glycosidic glycopeptides, Supplemental Fig. 6). In the case of CGM (Supplemental Fig. 4a), the acrylamide copolymer Galβ1,3GalNAcα-O-Al/AA-CP (a synthetic macromolecular acceptor) and asialo CGM were incubated separately with donor [9-3H[1]] and ST3Gal-II in the presence of either 5′-CMP or 5′-UMP for 21h at 37ºC, and then subjected to Biogel P2 chromatography to separate [9-3H]sialyl macromolecule from [9-3H]sialyl donor and CMP- or UMP-[9-3H]NeuAc. Both the acrylamide copolymer and asialo CGM served as good acceptors in the presence of 5′-CMP whereas lower amounts of sialylated macromolecules were formed in the presence of 5′-UMP (Supplemental Fig. 4a). UMP-sialic acid was a poor donor of sialic acid for not only ST3Gal-II but also ST3Gal-III and ST6Gal-I (Table 3).
[9-3H] Sialylated CGM could also act as donor to form CMP-[9-3H]NeuAc in presence of 5′-CMP and ST3Gal-II (Supplemental Fig. 5). In this study, [9-3H] sialylated CGM was first synthesized using CGM and CMP-[9-3H]NeuAc in the presence of ST3Gal-II. Subsequently, it was observed that the radiolabeled CGM could donate [9-3H]NeuAc to 5′-CMP in the presence of ST3Gal-II to form new CMP-[9-3H]NeuAc.
Similar to the case of CGM, BSM, asialo BSM and anti freeze glycoprotein also gave rise to the efficient formation of radiolabeled sialylated macromolecules using ST3Gal-II, [9-3H[1]] and 5′-CMP.
α2,6[9-3H] sialylated FTG could be formed using 5′-CMP and asialo FTG upon incubation with donor [9-3H[2]] and two enzymes ST3Gal-II and ST6Gal-I simultaneously (Supplemental Fig. 4c). Here, reverse sialylation using ST3Gal-II and 5′-CMP resulted in the formation of CMP-NeuAc. The newly formed CMP-NeuAc was then utilized by ST6Gal-I to form α2,6 sialylated FTG as shown by its binding to SNA-agarose column.
Similar results were observed with FOG using PNA-agarose chromatography. In these studies, the formation of either [9-3H]NeuAcα2,6(Galβ1,3)GalNAcα-O-Ser/Thr or [9-3H]NeuAcα2,3Galβ1,3GalNAcα-O-Ser/Thr units was detected upon incubation of asialo FOG and CMP-[9-3H] NeuAc with ST6GalNAc-I (Supplemental Fig. 6a) or ST3Gal-II (Supplemental Fig. 6b) respectively. Here, the α2,6-sialylated compound formed in Fig. 9a bound PNA-agarose. The incubation of asialo FOG with 5′-CMP, the donor [9-3H[2]],ST3Gal -II and ST6GalNAc-I also gave rise to product that bound PNA agarose (Supplemental Fig. 6c). This observation is consistent with the notion that CMP-NeuAc can be formed from 5′-CMP by reverse sialylation in the presence of ST3Gal-II. This new CMP-NeuAc can then be utilized by ST6GalNAc-I to form [9-3H]NeuAcα2,6(Galβ1,3)GalNAcα-O-Ser/Thr units. Finally it was observed that sialylated FOG itself could participate in reverse sialylation (Supplemental Fig. 6d). Sialylated FOG for these runs was first enzymatically prepared by reacting asialo-FOG with CMP-[9-3H]NeuAc in the presence of ST3Gal-II. Subsequently, it was observed that this sialylated-FOG could act as a sialic acid donor for D-Fucβ1,3GalNAcα-O-Bn in the presence of 5′-CMP and ST3Gal-II. The formation of NeuAcα2,3D-Fucβ1,3GalNAcα-O-Bn in the above experiment was verified using mass spectrometry.
Reversible sialyltransferase activity in human cells
While the above experiments were performed with cloned rat enzymes, we examined if human cells also exhibited this novel enzyme activity (Table 4). Thus, [9-3H[4]] was used as donor and D-Fucβ1,3GalNAcα-O-Bn [5] as acceptor in the presence of 5′-CMP and solubilized cell extracts. Of the cells tested, human prostate cancer cell lines LNCaP and PC-3 contained significant reversible sialyltransferase activity. These result shows that reverse sialylation occurs in human cells also.
Table 4.
* Reversible sialyltransferase activity in human cancer cell lines
| Cancer Cell Lines | Transfer of [9-3H]NeuAc from [9-3H[4]] to [5]
CPM ×10−3/mg protein |
|---|---|
|
| |
| BREAST: | |
| T47D | 0.52 |
| ZR75-1 | 1.32 |
| MDA-MB231 | 0.09 |
| MDA-MB-435S | 0.69 |
| MCF-7 | 0.57 |
| COLON: | |
| LS180 | 0.01 |
| PROSTATE: | |
| LNCaP | 19.89 |
| PC3 | 4.72 |
| LEUKEMIA | |
| HL60 | 0.76 |
0.15mM [9-3H]NeuAcα2,3Galβ1,3(6-O-sulfo GlcNAcβ1,6) GalNAc-O-Al [9-3H[4]] was added to 3.0mM D-Fucβ1,3GalNAcα-O-Bn [5] in the presence of 1.0mM 5′-CMP and 100 μl Triton-X solubilized cell extract for 16h. at 37ºC at pH 6.0. Total reaction volume was 180 μl. The product [9-3H]NeuAcα2,3D-Fucβ1,3GalNAcα-O-Bn was measured using Sep-Pak C18 separation followed by liquid scintillation counting.
DISCUSSSION
The present paper reports a novel enzymatic reaction mechanism which we term “reverse sialylation”. This enzyme activity is exhibited by rat ST3Gal-II and lysates of human prostate cancer cell lines LNCaP and PC3. This reaction catalyzes the formation of CMP-NeuAc according to scheme-I, in the presence of 5′-CMP and a range of donors including those based on mucin core-2, core-1, glycolipid and macromolecule structures. The reaction mechanism is novel since the reversibility of sialyltransferase activity has not been reported in literature. While previous studies have demonstrated that biochemical reactions can be reversible, the current paper shows that in the case of ST3Gal-II the reaction rate is much more reversible than previously appreciated. The Keq value is less than one when some donors are used, and this suggests that at least in some cases the formation of CMP-NeuAc in the case of this enzyme occurs more readily than the conventional, forward sialylation reaction.
Besides transfer of NeuAc to 5′-CMP using an array of donors, this enzyme activity also transfers sialic acid quite efficiently to 5′-UMP. The newly synthesized UMP-NeuAc, however, was a poor sialyl donor and thus formation of UMP-NeuAc may be an efficient mechanism for depleting sialyl donors.
CMP-NeuAc formed by reverse sialylation above was available for transfer to a range of substrates using sialylTs that catalyzed both α2,3 and α2,6 linkage formation. The enzymes that could transfer NeuAc from CMP-NeuAc formed by reverse sialylation to other acceptors include ST3Gal-II, ST6Gal-I and ST6GlcNAc-I. Together these observations suggest that the reversible sialylation function of ST3Gal-II can be exploited for the synthesis of sialylated glycoconjugates.
The requirements for the formation of CMP-NeuAc using reverse sialylation activity was unlike that of conventional CMP-NeuAc synthetase function (22–26). CMP-NeuAc synthetase utilizes CTP and NeuAc as substrates in the presence of Mg2+ to produce CMP-NeuAc and pyrophosphate (22, 23). In contrast, directly transferred of NeuAc to 5′-CMP proceeded without the need of free sialic acid, divalent metal ions or energy from the breakdown of CTP to CMP and pyrophosphate.
Some of the studies performed in this paper address the possibility: a) that the mechanism of reverse sialylation simply involves a reversal of forward sialylation; and b) that reverse and forward sialylation occur via different reaction coordinates. In this regard, we noted that reverse and forward sialylation are optimum at different, albeit overlapping, pH ranges. While direct/forward sialylation using ST3Gal-II was optimum over a wide pH range from 5.2→7.2, the formation of CMP-NeuAc from 5′-CMP by reverse sialylation occurred between 4.8→6.4. The entire reversible sialylation reaction of ST3Gal-II utilizing CMP-NeuAc produced in situ for sialylation exhibited a sharp peak at a pH of 5.6. These data suggest that direct/forward sialylation and reverse sialylation by ST3Gal-II may be governed by two distinct catalytic mechanisms. In support of this proposition, we also found that: i) While strong inhibition of sialyltransferase activity occurs upon addition of sodium citrate, reverse sialylation (namely the formation CMP-NeuAc from 5′-CMP) was not inhibited by citrate. ii) We also observed that while 5′-CDP was a potent inhibitor of direct/forward sialyltransferase activities, it did not affect the synthesis of CMP-NeuAc from 5′-CMP in the reverse sialylation reaction mediated by ST3Gal-II. iii) While the mucin core 2 compound 3-O-sulfoGalβ1,4GlcNAcβ1,6(Galβ1,3)GalNAcα-O-Bn inhibited the conversion of 5′-CMP to CMP-NeuAc via the reverse sialylation mechanism, the corresponding α2,3-sialyl substituent NeuAcα2,3 Galβ1,4GlcNAcβ1,6(Galβ1,3)GalNAcα-O-Bn did not inhibit reverse sialylation. In previous studies (14), we showed that NeuAcα2,3Galβ1,4 GlcNAcβ1,6(Galβ1,3)GalNAcα-O-Bn was a poor acceptor for ST3Gal-II compared to 3-O-sulfoGalβ1,4GlcNAcβ1,6(Galβ1,3)GalNAcα-O-Bn. Thus the latter acceptor 3-O-sulfoGalβ1,4GlcNAcβ1,6(Galβ1,3)GalNAcα-O-Bn can be sialylated by ST3Gal-II but it inhibits the formation of CMP-NeuAc from 5′-CMP by ST3Gal-II. Taken together the data suggest that forward and reverse sialylation may follow different reaction coordinates and that the enzyme ST3Gal-II may have more than one catalytic activity. Indeed there is precedence in literature for sialyltransferases that exhibit multiple functions (9).
The primary amino acid sequence and protein tertiary structure that contribute to reverse sialylation remains to be determined. All mammalian sialyltransferases characterized to date have type II transmembrane topology and contain highly conserved motifs called sialyl motifs L (long), S (short) and VS (very short) (26–28). Sialyl motif L is characterized by a 45–60 amino acid region in the center of the protein and it has been shown to be involved in the binding of a donor substrate, CMP-NeuAc (29). Sialyl motif S is located in the COOH-terminal region and consists of 20–30 amino acid stretch. It has been shown to be involved in the binding of both the donor and acceptor substrates (30). Sialyl motif VS is also located in the COOH-terminal region, and it is thought to be involved in the catalytic process (28, 31). From the fact that both sialyl motifs, L and S, participate in the binding of the donor substrate CMP-NeuAc, it seems that the two cysteine residues, which are important for the enzyme activity, bring the two sialyl motifs closer together by forming an intra-disulfide linkage and, thus, form the conformation required for binding of the donor substrate (30). A comparison of V and VS motifs between sialyltransferases (30, 31) reveals a substantial difference in the amino acid sequences of the corresponding motifs. This difference could be attributed to a unique conformational difference between ST3Gal-I and ST3Gal-II in the carboxyl terminal portion and thus may explain the unique catalytic ability of the cloned rat liver α2,3(O)ST (ST3Gal-II). It is suggested that subtle differences in the amino acid sequences in the active sites of sialyl motifs may regulate the unique substrate specificities of this family of enzymes, and they may regulate enzyme function in the presence of co-factors, products and transition-state analogs (30–32). This difference may also account for the unique ability of ST3Gal-II, but not ST3Gal-III or ST6Gal-I, to mediate the reversible sialylation reaction. If it is proven that normal/forward and reverse sialylation are distinct processes, these structural features may also contribute to important difference in these activities.
Reverse sialylation provides opportunities for the development of novel glycoconjugate synthetic scheme. For example, since ST3Gal-II readily exchanges sialic acid residues between CMP-NeuAc and the α2,3-sialyl T-hapten unit, such a reaction mechanism may provide a rapid scheme for the synthesis of NeuAc analogs. In such reactions, addition of CMP-NeuAc analogs to mucin glycoproteins containing α2,3-sialyl T-hapten units in the presence of ST3Gal-II can catalyze the formation of modified T-hapten units via the reverse sialylation mechanism. Indeed, a recent report has suggested that reverse glycosyltransferases activity can be exploited for the rapid synthesis of natural products (13). Further, it is noteworthy that if one considers the high cost of CMP-sialic acid and the commercially cheap and easily available Fetuin (a glycoprotein containing sialylated O-glycan chains), the synthesis of CMP-sialic acid using the cheap precursor CMP and ST3Gal-II using the sialyl donor Fetuin appears to be an alternative approach in the in situ production of CMP-sialic acid for an economical synthesis of sialyl oligosaccharides as demonstrated in the present study.
The physiological relevance and biochemical features that contribute to reverse sialylation will be the subject of future investigations. It remains to be determined if reverse sialylation is unique to ST3Gal-II and if this feature is species specific. Since sialyltransferases contain multiple domains for binding acceptors and donors and for catalytic activity (26–31), the exact primary amino acid sequence (30, 31) and protein tertiary structure (32) that contribute to reverse sialylation needs to be determined. Further, numerous studies have documented the existence of soluble glycosyltransferases in body fluids (33–38) and in the growth media of normal and transformed cells (39, 40). Further, in light of the observation that both 5′-UMP and 5′-CMP are efficient acceptors of sialic acid but only CMP-NeuAc is a facile donor for subsequent sialylation, the possible existence of a control mechanism for the level of intra- and extra-cellular sialylation based on the concentration of 5′-UMP and 5′-CMP in the cellular golgi and extracellular milieu needs to be explored.
Supplementary Material
The Supplemental Figures 1–6. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
We are deeply indebted to Dr. Phillips W. Robbins for his critical reading and helpful suggestions on the manuscript. We thank Ms. Charlene Romanello and Ms. Lisa C. Francescone for their excellent secretarial assistance and Mr. Dhananjay Marathe for cell culture.
Footnotes
Abbreviations: sialylT=sialyltransferases; TS=trans-sialidases; AA-CP=Acrylamide copolymer; Al=Allyl; Bn=Benzyl; Me=Methyl; BSM=Bovine Submaxillary Mucin; CGM=Porcine Cowper’s Gland Mucin; CMP=Cytidine 5′-monophosphate; FOG=Fetuin O-glycosidic Glycopeptide; FTG=Fetuin Triantennary Glycopeptide; Fuc=fucose; Gal=galactose; GalNAc=N-acetylgalactosamine; GlcNAc=N-Acetylglucosamine; NeuAc=sialic acid; Sulfo=sulfate ester; T-hapten=Galβ1,3GalNAcα
We acknowledge grant support from NIH (CA35329, HL63014) and DOD W81XWH-06-1-0013.
References
- 1.Haverkamp J, Spoormaker T, Dorland L, Vliegenthart JFG, Schauer R. Determination of the .beta.-anomeric configuration of cytidine 5′-monophospho-N-acetylneuraminic acid by carbon-13 NMR spectroscopy. J Am Chem Soc. 1979;101:4851–4853. [Google Scholar]
- 2.Kolter T, Sandhoff K. Sialic acids-why always α-linked. Glycobiology. 1997;7:vii–ix. doi: 10.1093/glycob/7.7.873. [DOI] [PubMed] [Google Scholar]
- 3.Engstler M, Wirtz E, Cross GAM. Generation of constitutive and inducible trans-sialylation dominant-negative phenotypes in Trypanosoma brucei and Trypanosoma cruzi. Glycobiology. 1997;7:955–964. doi: 10.1093/glycob/7.7.955. [DOI] [PubMed] [Google Scholar]
- 4.Schauer R, Reuter G, Muhlpfordt H, Andrade AF, Pereira ME. The occurrence of N-acetyl- and N-glycoloyineuraminic acid in Trypanosoma cruzi Hoppe-Seylers Z. Physiol Chem. 1983;364:1053–1057. doi: 10.1515/bchm2.1983.364.2.1053. [DOI] [PubMed] [Google Scholar]
- 5.Previato J, Andrade A, Pessolani M, Priviato LM. Incorporation of sialic acid into Trypanosoma cruzi macromolecules. A proposal for a new metabolic route. Mol Biochem Parasitol. 1985;16:85–96. doi: 10.1016/0166-6851(85)90051-9. [DOI] [PubMed] [Google Scholar]
- 6.Zingales B, Carniol C, de Lederkremer RM, Colli W. Direct sialic acid transfer from a protein donor to glycolipids of trypomastigote forms of Trypanosoma cruzi. Mol Biochem Parasitol. 1987;26:135–144. doi: 10.1016/0166-6851(87)90137-x. [DOI] [PubMed] [Google Scholar]
- 7.Scudder P, Doom JP, Chuenkova M, Manger ID, Pereira MEA. Enzymatic characterization of beta-D-galactoside alpha 2,3-trans- sialidase from Trypanosoma cruzi. J Biol Chem. 1993;268:9886–9891. [PubMed] [Google Scholar]
- 8.Cremona ML, Campetella O, Sanchez DO, Frasch ACC. Enzymically inactive members of the trans-sialidase family from Trypanosoma cruzi display beta-galactose binding activity. Glycobiology. 1999;9:581–587. doi: 10.1093/glycob/9.6.581. [DOI] [PubMed] [Google Scholar]
- 9.Yu H, Chokhawala H, Karpel R, Yu H, Wu B, Zhang J, Zhang Y, Jia Q, Chen SA. Multifunctional Pasteurella multocida Sialyltransferase: A Powerful Tool for the Synthesis of Sialoside Libraries. J Am Chem Soc. 2005;127:17618–17619. doi: 10.1021/ja0561690. [DOI] [PubMed] [Google Scholar]
- 10.Hill AC. Reversible zymohydrolysis. J Chem Soc Trans. 1898;73:634–658. [Google Scholar]
- 11.Ellings L, Grothus M, Kula M. Investigation of sucrose synthase from rice for the synthesis of various nucleotide sugars and saccharides. Glycobiology. 1993;3:349–355. doi: 10.1093/glycob/3.4.349. [DOI] [PubMed] [Google Scholar]
- 12.Romer U, Schrader H, Gunther N, Nettelstroth N, Frommer WB, Elling L. Expression, purification and characterization of recombinant sucrose synthase 1 from Solanum tuberosum L. for carbohydrate engineering. J Biotech. 2004;107:135–149. doi: 10.1016/j.jbiotec.2003.10.017. [DOI] [PubMed] [Google Scholar]
- 13.Zhang C, Griffith BR, Fu Q, Albermann C, Fu X, Lee IK, Li L, Thorson JS. Exploiting the Revesibility of natural products Glycosyltransferase-catalyzed Reactions. Science. 2006;313:1291–4. doi: 10.1126/science.1130028. [DOI] [PubMed] [Google Scholar]
- 14.Chandrasekaran EV, Xue J, Xia J, Chawda R, Piskorz C, Locke RD, Neelamegham S, Matta KL. Analysis of the Specificity of Sialytransferases toward Mucin Core 2, Globo, and Related Structures. Identification of the Sialylation Sequence and the Effects of Sulfate, Fucose, Methyl, and Fluoro Substiuents of the Carbohydrate Chain in the Biosynthesis of Selectin and Siglec Ligands, and Novel Sialylation by Cloned 2,3(O)Sialyltransferase. Biochemistry. 2005;44:15619–35. doi: 10.1021/bi050246m. [DOI] [PubMed] [Google Scholar]
- 15.Jain RK, Piskorz CF, Chandrasekaran EV, Matta KL. Synthesis of Gal-β1-(1→4)-GlcNAc-β-(1→6)-[Gal-β-(1→3)]-GalNAc-α-Obn Oligosaccharides Bearing O-methyl or O-sulfo Groups at C-3 of the Gal Residue: Specific Acceptors for Gal: 3-O-sulfotransferases. Glycoconj J. 1998;15:951–959. doi: 10.1023/a:1006977607394. [DOI] [PubMed] [Google Scholar]
- 16.Jain RK, Piskorz CF, Huang BG, Locke RD, Han HL, Koenig A, Varki A, Matta KL. Inhibition of L- and P-selectin by a Rationally Synthesized Novel Core 2-like Branched Structure Containing GalNAc-Lewisx and Neu5Acα2–3Galβ1–3GalNAc Sequences. Glycobiology. 1998;8:707–717. doi: 10.1093/glycob/8.7.707. [DOI] [PubMed] [Google Scholar]
- 17.Chandrasekaran EV, Jain RK, Larsen RD, Wlasichuk K, Matta KL. Selectin-ligands and tumor associated carbohydrate structures: Specificities of α2,3-sialyltransferases in the assembly of 3′-sialyl, 6-sulfo/sialyl Lewis a and x, 3′-sialyl, 6′-sulfo Lewis x and 3′-sialyl, 6-sialyl/sulfo blood group T-hapten. Biochemistry. 1995;34:2925–2936. doi: 10.1021/bi00009a024. [DOI] [PubMed] [Google Scholar]
- 18.Chandrasekaran EV, Jain RK, Vig R, Matta KL. The Enzymatic Sulfation of Glycoprotein Carbohydrate Units: Blood Group T-hapten Specific and Two Other Distinct Gal:3-O-sulfotransferases as Evident from Specificities and Kinetics and the Influence of Sulfate and Fucose Residues Occurring in the Carbohydrate Chain and on C-3 Sulfation of Terminal Gal. Glycobiology. 1997;7:753–768. doi: 10.1093/glycob/7.6.753. [DOI] [PubMed] [Google Scholar]
- 19.Chandrasekaran EV, Chawda R, Locke RD, Piskorz CF, Matta KL. Biosynthesis of the carbohydrate antigenic determinants, Globo H, blood group H, and Lewis b: a role for prostate cancer cell alpha1,2-L-fucosyltransferase. Glycobiology. 2002;12:153–162. doi: 10.1093/glycob/12.3.153. [DOI] [PubMed] [Google Scholar]
- 20.Chandrasekaran EV, Chawda R, Rhodes JM, Locke RD, Piskorz CF, Matta KL. The binding characteristics and utilization of Aleuria aurantia, Lens culinaris and few other lectins in the elucidation of fucosyltransferase activities resembling cloned FT VI and apparently unique to colon cancer cells. Carbohydr Res. 2003;338:887–901. doi: 10.1016/s0008-6215(03)00021-1. [DOI] [PubMed] [Google Scholar]
- 21.Tsuji S, Datta AK, Paulson JC. Letters to the Glyco-Forum: Systematic nomenclature for sialyltransferases. Glycobiology. 1996;6:v–vii. doi: 10.1093/glycob/6.7.647. [DOI] [PubMed] [Google Scholar]
- 22.Roseman S. Enzymatic Synthesis of Cytidine 5′-monophospho-sialic Acids. Proc Natl A cad Sci USA. 1962;48:437–441. doi: 10.1073/pnas.48.3.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Warren L, Blacklow RS. The Biosynthesis of Cytidine 5′-Monophospho-N-acetylneuraminic Acid by an Enzyme from Neisseria meningitidis. J Biol Chem. 1962;237:3527–3534. [PubMed] [Google Scholar]
- 24.Kapitonov D, Yu RK. Conserved domains of glycosyltransferase. Glycobiology. 1999;9:961–978. doi: 10.1093/glycob/9.10.961. [DOI] [PubMed] [Google Scholar]
- 25.Mosimann SC, Gilbert M, Dombrowski D, To R, Wakarchuk W, Strynadka NC. Structure of a Sialic Acid-activating Synthetase, CMP-acylneuraminate Synthetase in the Presence and Absence of CDP. J BioI Chem. 2001;276:8190–8196. doi: 10.1074/jbc.M007235200. [DOI] [PubMed] [Google Scholar]
- 26.Drickamer K. Letters to the Glyco-Forum: A conserved disulphide bond in sialyltransferases. Glycobiology. 1993;3:2–3. doi: 10.1093/glycob/3.1.2. [DOI] [PubMed] [Google Scholar]
- 27.Livingston BD, Paulson JC. Polymerase chain reaction cloning of a developmentally regulated member of the sialyltransferase gene family. J Biol Chem. 1993;268:11504–11507. [PubMed] [Google Scholar]
- 28.Geremia RA, Harduin-Lepers A, Delannoy P. Letters to the Glyco-Forum Identification of two novel conserved amino acid residues in eukaryotic sialyltransferases: implications for their mechanism of action. Glycobiology. 1997;7:v–xi. doi: 10.1093/glycob/7.2.161. [DOI] [PubMed] [Google Scholar]
- 29.Datta AK, Paulson JC. The Sialyltransferase “Sialylmotif” Participates in Binding the Donor Substrate CMP-NeuAc. J Biol Chem. 1995;270:1497–1500. doi: 10.1074/jbc.270.4.1497. [DOI] [PubMed] [Google Scholar]
- 30.Datta AK, Sinha A, Paulson JC. Mutation of the Sialyltransferase S-sialylmotif Alters the Kinetics of the Donor and Acceptor Substrates. J Biol Chem. 1998;273:9608–9614. doi: 10.1074/jbc.273.16.9608. [DOI] [PubMed] [Google Scholar]
- 31.Kitazume-Kawaguchi S, Kabata S, Arita M. Differential Biosynthesis of Polysialic or Disialic Acid Structure by ST8Sia II and ST8Sia IV. J Biol Chem. 2001;276:15696–15703. doi: 10.1074/jbc.M010371200. [DOI] [PubMed] [Google Scholar]
- 32.Erlandsen H, Abola EE, Stevens RC. Combining structural genomics and enzymology: completing the picture in metabolic pathways and enzyme active sites. Curr Opin Struct Biol. 2000;10:719–730. doi: 10.1016/s0959-440x(00)00154-8. [DOI] [PubMed] [Google Scholar]
- 33.DeboseBoyd RA, Nyame AK, Smith DF, Cummings RD. α1,4-Fucosyltransferase Activity in Human Serum and Saliva. Arch Biochem Biophys. 1996;335:109–117. doi: 10.1006/abbi.1996.0487. [DOI] [PubMed] [Google Scholar]
- 34.Fujita-Yamaguchi Y, Yoshida A. Purification and characterization of human serum galactosyltransferase (lactose synthetase A protein. J Biol Chem. 1981;256:2701–2706. [PubMed] [Google Scholar]
- 35.Lammers G, Jamieson JC. The role of a cathepsin D-like activity in the release of Gal beta 1–4GlcNAc alpha 2-6-sialyltransferase from rat liver Golgi. Biochem J. 1988;256:623–631. doi: 10.1042/bj2560623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Paulson JC, Beranek WE, Hill RL. Purification of a sialyltransferase from bovine colostrum by affinity chromatography on CDP-agarose. J Biol Chem. 1997;252:2356–2362. [PubMed] [Google Scholar]
- 37.Piller F, Cartron JP. UDP-GlcNAc:Gal beta 1-4Glc(NAc) beta 1-3N-acetylglucosaminyltransferase. Identification and characterization in human serum. J Biol Chem. 1983;258:12293–12299. [PubMed] [Google Scholar]
- 38.Serafinicessi F, Malagolini N, Dallolio F. Characterization and partial purification of β-N-acetylgalactosaminyltransferase from urine of Sd(a+) individuals. Arch Biochem Biophys. 1988;266:573–582. doi: 10.1016/0003-9861(88)90290-1. [DOI] [PubMed] [Google Scholar]
- 39.Klohs WD, Mastrangelo R, Weiser MM. Release of Glycosyltransferase and Glycosidase Activities from Normal and Transformed Cell Lines. Cancer Res. 1981;41:2611–2615. [PubMed] [Google Scholar]
- 40.Serafinicessi F, Malagolini N, Guerrine S, Turrini I. A soluble form of Sd a-β1,4 –N-acetylgalatosaminyl-transferase is released by differentiated humanecolon carcinoma CaCo-2 cells. Glycoconj J. 1995;12:773–779. doi: 10.1007/BF00731238. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The Supplemental Figures 1–6. This material is available free of charge via the Internet at http://pubs.acs.org.