

Abstract
Genome-wide association studies (GWAS) testing several hundred thousand SNPs have been performed in multiple sclerosis (MS) and other complex diseases. Typically, the number of markers in which the evidence for association exceeds the genome-wide significance threshold is very small, and markers that do not exceed this threshold are generally neglected. Classical statistical analysis of these datasets in MS revealed genes with known immunological functions. However, many of the markers showing modest association may represent false negatives. We hypothesize that certain combinations of genes flagged by these markers can be identified if they belong to a common biological pathway. Here we conduct a pathway-oriented analysis of two GWAS in MS that takes into account all SNPs with nominal evidence of association (P < 0.05). Gene-wise P-values were superimposed on a human protein interaction network and searches were conducted to identify sub-networks containing a higher proportion of genes associated with MS than expected by chance. These sub-networks, and others generated at random as a control, were categorized for membership of biological pathways. GWAS from eight other diseases were analyzed to assess the specificity of the pathways identified. In the MS datasets, we identified sub-networks of genes from several immunological pathways including cell adhesion, communication and signaling. Remarkably, neural pathways, namely axon-guidance and synaptic potentiation, were also over-represented in MS. In addition to the immunological pathways previously identified, we report here for the first time the potential involvement of neural pathways in MS susceptibility.
INTRODUCTION
The usefulness of genome-wide association studies (GWAS) to discover common genetic variants associated with susceptibility to complex diseases has been empirically demonstrated (1). The aim of these studies is to characterize the genetic architecture of complex genetic traits through the identification of such disease variants against the background of random variation seen in a population as a whole. In a typical GWAS, hundreds of thousands of markers are tested simultaneously in cases and controls and the allelic frequencies of each marker are compared between the two groups. However, because of the exceedingly large multiple testing involved in these studies, very few exceed the genome-wide significance threshold and those that do not exceed this stringent statistical requirement are generally neglected. In many cases where loci with small but measurable genetic effects are involved, it is likely that, accepting the null hypothesis of no association represents a type II error. A notable example of this situation can be illustrated by the confirmed association of PPARG variants in type 2 diabetes (T2D) (2). Due to its modest effect on disease susceptibility (odds ratio 1.2), this true association was overlooked by four out of five studies designed to replicate the initial finding. A similar scenario was more recently found with IL7R in multiple sclerosis (MS) (3). In this paper, we aim to show that while individual modest genetic effects are difficult to ascertain, they can be collectively identified by combining nominally significant evidence of genetic association with current knowledge of biochemical pathways.
MS is the most common acquired neurological disease of young adults with a prevalence of approximately 1:1000 in population groups of northern-European ancestry. MS is characterized by a variable state of relapsing or progressive neurological disability that ensues as the consequence of an autoimmune attack against myelin in the central nervous system (CNS) (4). Compelling data indicate that susceptibility to MS is in part inherited (5–8). In addition to the strong effect of HLA-DRB1, the recently reported GWAS in MS identified and confirmed the involvement of the genes IL2RA and IL7RA in disease susceptibility (9). However, as in other studies of this kind, many associations with markers that were nominally significant but did not reach the genome-wide significance threshold were not pursued any further. It is likely that a significant proportion of these rejected associations are false negatives, and methods of interpretation are needed that allow such associations to be recognized.
We hypothesize that meaningful combination of genes harboring markers with only modest evidence of association can be identified if they belong to the same biological pathway or mechanism. In addition to the single-locus associations identifiable by standard genome-wide analysis, this type of analysis can reveal a statistical enrichment of associations within known biological pathways. The methods presented here may be useful to identify pathways and networks whose involvement in disease susceptibility are consistent with current models of pathogenesis, but most importantly may also identify statistically over-represented but unexpected pathways revealing novel disease mechanisms.
Inspired in part by analytical advances in the study of gene expression, we propose a pathway-oriented analysis for GWAS. We apply a method similar to the one that uses gene ontologies (10) to analyze a list of differentially expressed genes, but replacing the measure of differential expression for each gene by a P-value that indicates the strength for the association of a gene with the disease phenotype. It is important to note that in this adaptation of the method, two sets of P-values are computed: one set for assessment of the association of each SNP with the trait and another set for comparison of the observed and expected number of moderately associated genes in a GO or biological pathway. The second stage requires that evidence of association at all the marker loci genotyped within each gene be reduced to a single, gene-wise P-value.
A limited number of studies have used network-based algorithms to prioritize candidate loci in genetic studies (11–14). However, these studies either do not use actual genetic (genotypic) data or are applied to model organisms. The only study to date that uses pathway-based analysis of GWAS data does not consider a protein interaction network (PIN) to further restrict the possible combinations of causal genes (15).
In this article, we describe a network-based pathway analysis of two GWAS in MS (3,9), where evidence for genetic association is combined with evidence for protein–protein interaction. The rationale for performing a pathway-based analysis in a GWAS lies in the assumption that several genes, each modestly associated with the disease, may interact synergistically to confer susceptibility. We carry out extensive statistical validations and apply the same approach to other published GWAS to demonstrate the potential utility of this method.
RESULTS
GWAS results from two MS studies were analyzed to identify modestly associated variants within genes with related biological functions. The first dataset was produced by the International MS Genetics Consortium (IMSGC), and comprises 931 family trios genotyped with the GeneChip® Human Mapping 500K Array Set (Affymetrix) (9). Quality control for this dataset included sample genotyping efficiency, assessment of marker heterozygosity and allelic frequency, departure from Hardy–Weinberg equilibrium, gender consistency, reproducibility and population genetic structure. A total of 334 923 SNPs survived the quality control protocol and were tested for association with the trait. As expected, a number of markers in the HLA region were strongly associated with the disease phenotype. In addition, 78 markers outside the HLA region were found to exceed the P < 1 × 10−4 genome-wide threshold of significance. The second dataset (the GeneMSA study, (3)] was generated using the Sentrix® HumanHap550 BeadChip (Illumina). After a similar quality control protocol, 551 642 SNPs were used to conduct an association analysis using the genotypic test in 978 cases and 883 controls (3). In addition, the association of each individual marker with the disease was tested by fitting a logistic regression genotypic model in which gender, Center of sample origin and HLA-DRB1*1501 status were included as covariates. In the GeneMSA study, 87 SNPs outside of the HLA region exceeded the genome-wide significance threshold of P < 1 × 10−4. Although there was no full overlapping of associated markers between the two studies, several genes showed evidence of association in both (3). A meta-analysis is being conducted and will be reported in the near future.
To carry out the protein interaction network-based pathway analysis (PINBPA), we computed a single P-value for each gene (the gene-wise P-value, Fig. 1) and overlapped these onto a curated PIN. Many markers map within gene deserts or unannotated genes, and these were excluded from the present analysis. This process resulted in gene-wise P-values for 14 442 and 17 342 genes for the GeneMSA and IMSGC GWAS, respectively. We next conducted sub-network searches on the two MS GWAS using the Cytoscape plugin jActive modules. jActive modules combines the network position and association P-value of each gene to extract potentially meaningful sub-networks or modules. In addition to searching for significant modules using both datasets together, we also conducted individual searches for each study (data not shown). Although the same basic modules were identified in both strategies, higher scores were obtained when both datasets were used together, suggesting a real power gain when larger datasets are used. To assess the specificity of the modules associated with MS, we also performed equivalent analyses on recent GWAS from other autoimmune diseases (rheumatoid arthritis, RA; Crohn's disease, CD; type 1 diabetes, T1D), neurological diseases (Alzheimer's disease, AD; bipolar disorder, BP) and unrelated diseases (coronary artery disease, CAD; hypertension, HT; T2D) (16). We observed statistically significant modules in all diseases (Fig. 2). Interestingly, the largest number of significant modules was observed in MS, suggesting greater genetic heterogeneity in this disease when compared with others. To test to what extent significant network modules could be obtained by chance, we conducted 10 searches randomizing the P-values among the same set of genes (those with association P-values <0.05). With the notable exception of MS, RA and AD, the module scores obtained from the randomized P-values were equal to or even higher than those obtained using the real P-values (Fig. 2A). This observation suggests that many of these modules do not represent bona-fide biological networks and that their high scores may have been obtained by chance. In contrast, significantly fewer modules were identified in the searches based on randomized P-values for MS, RA and AD suggesting that the significant modules obtained from the real P-values in these diseases represent biologically meaningful networks. To examine in detail the magnitude of the scores for real and randomized P-values, we plotted those of the top 20 modules for each set of P-values for each disease (Fig. 2B). As expected from the previous analysis in MS, RA and AD, most of the top 20 modules obtained with real P-values showed higher scores than the average score of the randomized searches. In the case of RA, only the top two modules show significantly higher scores than the average of their randomized searches (Fig. 2B). Notably, these two (partly overlapping) modules are composed exclusively of HLA genes, in which association with the disease is highly significant. Although the total number of modules obtained using real P-values in BD and CD do not differ significantly from those obtained with the randomized P-values (Fig. 2A), 18 of the top 20 scores were higher for the real P-values (Fig. 2B). Altogether, these results suggest that the significant modules found with the original data may represent real effects of interacting proteins on each disease phenotype.
Figure 1.
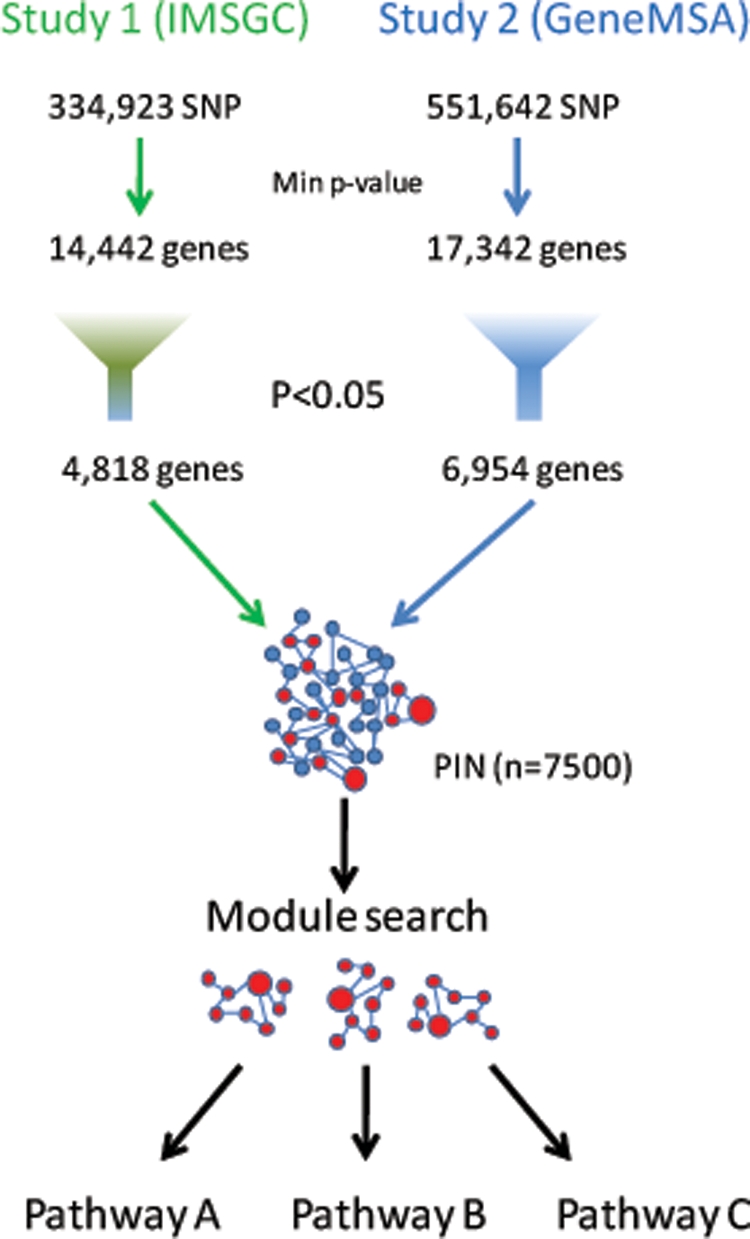
Strategy. A gene-wise P-value for association with MS in two independent studies was computed by selecting the P-value of the most significant marker within each gene. Genes with a P-value less than or equal to 0.05 (red circles) were selected for subsequent analysis. Significant P-values were loaded as attributes of the PIN and visualized using Cytoscape. The size of each network node displayed is proportional to its degree of significance. The plugin Jactive modules was used to identify sub-networks of interacting gene products that were also associated with the disease. Each significant module was tested for enrichment in KEGG pathways.
Figure 2.

Module identification. (A) Number of significant modules (size <50 and score >3) identified by Jactive modules in MS and in a panel of other autoimmune (RA, T1D and CD), other neurological (AD and BD), and other unrelated (T2D, HT and CAD) diseases. Each disease is represented by a different color. Filled bars correspond to the results obtained when real P-values were used to search for modules. Open bars are the results obtained with randomized P-values. (B) Scores of the top 20 modules obtained with real (solid symbols) or randomized (open symbols) P-values for each disease. The average and standard deviation of 10 randomizations is shown for each disease.
Significant modules for MS
We identified 346 significant modules on the basis of their aggregate degree of genetic association with MS. Due to the nature of the search algorithm, several of these modules overlap extensively in their component genes. Thus, to describe modules representative of association with MS, we selected those with the highest scores which also displayed a minimum degree of overlap (Fig. 3). Consistently with all previous genetic studies in MS, the most significant module (MS_I) included several HLA genes (Fig. 3A). Although the only gene consistently found associated to MS in this region is HLA-DRB1, the module shown lists another gene, HLA-DRA, as its most significant node. It is possible that because HLA-DRB1 is highly polymorphic, most SNP markers included in large-capacity arrays are not targeting this gene directly. Indeed, there are three times as many SNPs in HLA-DRA as there are in HLA-DRB1 in the Illumina 550 k platform and the DRA SNP rs313588 tags with high sensitivity the HLA-DRB1*1501 allele. The observed associations with other HLA genes like HLA-DMA/B and HLA-DOA/B may be also due to the extensive linkage disequilibrium (LD) seen in this region. Interestingly, HLA-DRB5, present in the most significant module, is part of the DR15 haplotype and has been identified as a potential modifier of the disease (17,18). The other two HLA genes that are part of the DR15 haplotype (DQA and DQB) are not present in the module shown. Although the P-values for each of these genes exceed the threshold of significance used for this analysis, they are not part of module MS_I because there is no evidence that they physically interact with any of its components. Not surprisingly, a KEGG pathway search with these genes identified the terms ‘antigen processing’, ‘cell adhesion molecules’ and ‘Immune system’ as the most significantly over-represented, relative to the number of genes in these pathways expected in the module by chance (Table 1).
Figure 3.

Representative modules for MS. Nodes represent proteins and connections represent physical interactions as determined by the curated human PID reported in Rual et al. (44). The size of each node is proportional to the −log (10) P-value of association (A, inset). Nodes are colored by chromosome (see key). (A) HLA module. This is the highest scoring module in MS, possibly due to the high significance of HLA-DRA and its interaction with other linked genes in the HLA region. (B) Extended immune module. In addition to HLA genes, this module contains other immune-related genes with more modest P-values of association. The significance of the entire module is possibly the result of the many interactions between these genes. (C) MS neural module 1. Seven genes encoding axon guidance molecules (indicated by asterisks) are part of this small module. (D) MS neural module 2. Seven glutamate receptors (gene symbols starting with GR) and two glutamate-related genes (HOMER1 and DLG1) are included in this module (these nine genes indicated by asterisks).
Table 1.
Significant modules for MS
| Pathway | Annotated genes in module | Observed | Expected | P-value |
|---|---|---|---|---|
| Module MS I | ||||
| Antigen processing and presentation | CD4|HLA-DPA1|HLA-DPB1|HLA-DQA2 | 4/4 (100%) | 43/2361 (1.82%) | 1.05E−06 |
| Cell adhesion molecules (CAMs) | CD4|HLA-DPA1|HLA-DPB1|HLA-DQA2 | 4/4 (100%) | 91/2361 (0.03%) | 1.14E−05 |
| Type I diabetes mellitus | HLA-DPA1|HLA-DPB1|HLA-DQA2 | 3/4 (75%) | 30/2361 (0.01%) | 2.69E−05 |
| Metabolic disorders | HLA-DPA1|HLA-DPB1|HLA-DQA2 | 3/4 (75%) | 78/2361 (0.03%) | 3.73E−04 |
| Immune system | CD4|HLA-DPA1|HLA-DPB1|HLA-DQA2 | 4/4 (100%) | 425/2361 (0.2%) | 2.28E−03 |
| Signaling molecules and interaction | CD4|HLA-DPA1|HLA-DPB1|HLA-DQA2 | 4/4 (100%) | 550/2361 (0.2%) | 5.35E−03 |
| Human diseases | HLA-DPA1|HLA-DPB1|HLA-DQA2 | 3/4 (75%) | 411/2361 (0.2%) | 2.87E−02 |
| Module MS II | ||||
| Cell adhesion molecules (CAMs) | ICAM1|ITGB2|ITGA4|HLA-DMB| | 14/32 (43.8%) | 91/2361 (3.9%) | 9.19E−11 |
| HLA-DQA2|ITGAM|ITGA6|CD58|CD2|CD4| | ||||
| HLA-DPA1|CD226|HLA-DRA|CD28 | ||||
| Immune system | ICAM1|IL2RA|TLR2|ITGB2|ITGA4|HLA-DMB| | 20/32 (62.5%) | 20/2361 (18%) | 6.75E−07 |
| HLA-DQA2|CXCL12|CTNNA3|ITGAM|CTNNB1| | ||||
| TNFRSF10A|PTK2|ITGA6|CD2| | ||||
| HLA-DPA1|CD4|CD14|HLA-DRA|CD28 | ||||
| Environmental information processing | COL2A1|ITGB2|HLA-DMB|CXCL12|PTEN|ITGAM| | 28/32 (87.5%) | 28/2361 (44.9%) | 1.12E−05 |
| CTNNB1|PTK2|ZFYVE9|CD2|CD4|CD28|BMP4| | ||||
| EGFR|ICAM1|IL2RA|SOCS2|TGFBR2|ITGA4| | ||||
| HLA-DQA2|STAT3|TNFRSF10A|ITGA6|CD58| | ||||
| HLA-DPA1|CD226|CD14|HLA-DRA | ||||
| Cellular processes | TLR2|COL2A1|ITGB2|HLA-DMB|CXCL12|PTEN| | 28/32 (87.5%) | 28/2361 (45.6%) | 1.25E−05 |
| ITGAM|CTNNB1|PTK2|ILK|CD2|CD4|CD28|EGFR| | ||||
| ICAM1|IL2RA|SOCS2|MAGI1|TGFBR2|ITGA4| | ||||
| HLA-DQA2|STAT3|CTNNA3|TNFRSF10A|ITGA6| | ||||
| HLA-DPA1|CD14|HLA-DRA | ||||
| Signaling molecules and interaction | EGFR|ICAM1|IL2RA|TGFBR2|ITGB2|COL2A1| | 20/32 (62.5%) | 20/2361 (23.3%) | 2.46E−05 |
| ITGA4|HLA-DMB|HLA-DQA2|CXCL12|ITGAM| | ||||
| TNFRSF10A|ITGA6|CD58|CD2|HLA-DPA1| | ||||
| CD4|CD226|HLA-DRA|CD28 | ||||
| Hematopoietic cell lineage | IL2RA|ITGA6|CD2|CD4|ITGA4|CD14|ITGAM| | 8/32 (25.0%) | 8/2361 (3.0%) | 2.61E−05 |
| HLA-DRA | ||||
| Leukocyte transendothelial migration | ICAM1|PTK2|ITGB2|ITGA4|CXCL12|CTNNA3| | 8/32 (25.0%) | 8/2361 (3.9%) | 1.61E−04 |
| ITGAM|CTNNB1 | ||||
| Type I diabetes mellitus | HLA-DPA1|HLA-DMB|HLA-DQA2|HLA-DRA|CD28 | 5/32 (15.6%) | 5/2361 (1.3%) | 2.73E−04 |
| Antigen processing and presentation | HLA-DPA1|CD4|HLA-DMB|HLA-DQA2|HLA-DRA | 5/32 (15.6%) | 5/2361 (1.8%) | 1.45E−03 |
| Human Diseases | BMP4|EGFR|SOCS2|MAGI1|TGFBR2| | 14/32 (43.8%) | 14/2361 (17.4%) | 2.50E−03 |
| HLA-DMB|PTEN|HLA-DQA2|STAT3|CTNNB1| | ||||
| HLA-DPA1|CD14|HLA-DRA|CD28 | ||||
| Metabolic disorders | SOCS2|HLA-DPA1|HLA-DMB|HLA-DQA2| | 6/32 (18.8%) | 6/2361 (3.3%) | 2.62E−03 |
| HLA-DRA|CD28 | ||||
| Focal adhesion | EGFR|PTK2|ITGA6|ILK|COL2A1|ITGA4|PTEN| | 8/32 (25.0%) | 8/2361 (7.1%) | 6.56E−03 |
| CTNNB1 | ||||
| Cell communication | EGFR|PTK2|ITGA6|MAGI1|TGFBR2|ILK|COL2A1| | 11/32 (14.6%) | 11/2361 (14.6%) | 1.81E−02 |
| ITGA4|PTEN|CTNNA3|CTNNB1 | ||||
| Regulation of actin cytoskeleton | EGFR|PTK2|ITGA6|ITGB2|ITGA4|CD14|ITGAM | 7/32 (21.9%) | 7/2361 (7.1%) | 2.36E−02 |
| Adherens junction | EGFR|TGFBR2|CTNNA3|CTNNB1 | 4/32 (2.8%) | 4/2361 (2.8%) | 4.32E−02 |
| Module MS III | ||||
| Axon guidance | NCK2|EPHA4|FYN|EFNB1|EFNB2|EPHB2|EPHA3 | 7/7 (100%) | 102/2419 (4.3%) | 1.44E−06 |
| Development | NCK2|EPHA4|FYN|EFNB1|EFNB2|EPHB2|EPHA3 | 7/7 (100%) | 119/2419 (5.0%) | 2.14E−06 |
| Module MS IV | ||||
| Neuroactive ligand-receptor interaction | GRIK1|GRIA1|GRIK2|GRIK4|GRID2|GRIN2A|GRIA4 | 7/9 (77.7%) | 201/2419 (8.3%) | 1.00E−05 |
| Signaling molecules and interaction | GRIK1|GRIA1|GRIK2|GRIK4|GRID2|GRIN2A|GRIA4 | 7/9 (77.7%) | 563/2419 (23.2%) | 5.37E−03 |
| Nervous system | GRIA1|GRID2|GRIN2A | 3/9 (33.3%) | 98/2419 (4.0%) | 1.96E−02 |
| Environmental information processing | ERBB4|GRIK1|GRIA1|GRIK2|GRIK4|GRID2| | 8/9 (88.8%) | 1077/2419 (44.5%) | 2.70E−02 |
| GRIN2A|GRIA4 | ||||
| Long-term depression | GRIA1|GRID2 | 2/9 (22.2%) | 61/2419 (2.5%) | 4.77E−02 |
| Long-term potentiation | GRIA1|GRIN2A | 2/9 (22.2%) | 64/2419 (2.6%) | 4.77E−02 |
Figure 3B shows another highly significant module characteristic of MS. In this module (MS_II), several HLA genes are also prominent members, but by virtue of highly connected molecules such as CD4, CD82 and ITGB2, a more extensive immune pattern emerged. Interestingly, two non-HLA susceptibility genes previously associated with MS (IL2Ra and CD58) also appear in this module. We hypothesize that in addition to its own significance, the presence of IL2Ra in this module may result from its physical interaction with STAT3 and ITGB2, which themselves show modest association with MS. CD58 was initially identified as a susceptibility gene in the IMSGC study (9) and its expression was recently found to be upregulated in peripheral blood cells during disease relapses (19). In contrast, IL7Ra, another gene recently identified as a susceptibility factor in MS (20) is not part of this module. We speculate that it may act through an independent pathway. Although several of the other immune-related genes in this module have not been formally associated with MS, their involvement in disease pathology seems plausible. These include several cell adhesion (ITGB2, ITGA4, ITGA6, ITGAM and ICAM1) and signaling molecules (TGFBR2, TNFRSF10A and STAT3). Notably, ITGAM (CD11b) has been recently associated with susceptibility to systemic lupus erythematosus, another autoimmune disease (21). KEGG pathways analysis with genes from module MS_II revealed statistically significant over-representation of the processes of cell adhesion, leukocyte transendothelial migration and antigen processing (Table 1).
Interestingly, the other two modules characteristic of MS (MS_III and MS_IV) suggest a neural component in the susceptibility to the disease. Module MS_III is highly enriched with genes typically expressed in neurons and glia (NCK2, EPHA3, EPHA4, FYN, EFNB1, EFNB2 and EPHB2). Similarly, module MS_IV includes seven glutamate receptors (GluRs) (GRIK1, GRIK2, GRIK4, GRIA1, GRIA4, GRIN2A and GRID2) in addition to HOMER1, DLG1 and DLG2. HOMER1 regulates group 1 metabotropic GluR function, and DLG1 and DLG2 interact at postsynaptic sites to form a multimeric scaffold for the clustering of receptors, ion channels and associated signaling proteins. The identification of the latter two modules in MS suggests for the first time that modestly significant associations in genes involved in neural pathways may contribute to the overall susceptibility to this disease. Indeed, when members of these modules were tested for membership to KEGG pathways, highly significant enrichment in axon guidance pathways (module MS_III) and long-term depression and potentiation pathways (module MS_IV) were detected (Table 1).
As a control for our interpretation of these genes in MS, we next conducted similar analyses on the modules identified for other diseases. Interestingly, for two of the three autoimmune diseases tested (RA and T1D), the most significant modules were exclusively composed of HLA genes (Fig. 4). On the other hand, only genes involved in the JAK-STAT signaling pathway (GRB2, JAK1, STAT3 and IFNAR1), and extracellular matrix-receptor interactions (CD44, COL4A2, COL1A1 and FN1), but not HLA were identified in the third autoimmune disease (CD). The two genes most robustly associated with CD (NOD2 and IL23R) are not part of the selected module. As described for module MS_II, this may be due to the fact that evidence for the interaction between these two genes and the rest of the genes in the module is lacking. As expected, the great majority of pathways identified in the significant modules for AD and BD were neural (Development, Parkinson's disease and long-term depression). Table 2 shows the genes and pathways contained in the statistically significant modules identified for RA, T1D, AD and BD.
Figure 4.

Representative modules for other diseases. Same conventions as in Figure 3. (A) RA; (B) T1D; (C) CD; (D) T2D; (E) CAD; (F) HT; (G) AD; (H) BD.
Table 2.
Significant modules for other autoimmune and neurological diseases
| Pathway | Annotated genes in module | Observed | Expected | P-value |
|---|---|---|---|---|
| RA | ||||
| Cell adhesion molecules (CAMs) | SELL|HLA-DPA1|CD4|HLA-DPB1| | 8/18 (44.4%) | 91/2361 (3.8%) | 6.06E−06 |
| HLA-DMB|SELE|HLA-DQA2|HLA-DRA | ||||
| Antigen processing and presentation | HLA-DPA1|CD4|HLA-DPB1|HLA-DMB| | 6/18 (33.3%) | 43/2361 (1.8%) | 1.07E−05 |
| HLA-DQA2|HLA-DRA | ||||
| Type I diabetes mellitus | HLA-DPA1|HLA-DPB1|HLA-DMB|HLA-DQA2| | 5/18 (27.7%) | 30/2361 (1.2%) | 3.16E−05 |
| HLA-DRA | ||||
| Human diseases | CBLB|RET|MAPK12|GRB2|HLA-DPA1| | 12/18 (66.6%) | 411/2361 (17.4%) | 6.00E−05 |
| HLA-DPB1|PRNP|ABL1|HLA-DMB| | ||||
| HLA-DQA2|PIK3R1|HLA-DRA | ||||
| Metabolic disorders | HLA-DPA1|HLA-DPB1|HLA-DMB| | 6/18 (33.3%) | 78/2361 (3.3%) | 1.54E−04 |
| HLA-DQA2|PIK3R1|HLA-DRA | ||||
| Immune system | CBLB|MAPK12|GRB2|HLA-DPA1|CD4| | 10/18 (55.5%) | 425/2361 (18.0%) | 3.14E−03 |
| HLA-DPB1|HLA-DMB|HLA-DQA2|PIK3R1|HLA-DRA | ||||
| Chronic myeloid leukemia | CBLB|GRB2|ABL1|PIK3R1 | 4/18 (22.2%) | 70/2361 (2.9%) | 1.20E−02 |
| Signaling molecules and interaction | SELL|HLA-DPA1|CD4|HLA-DPB1| | 10/18 (55.5%) | 550/2361 (23.2%) | 2.00E−02 |
| HLA-DMB|SELE|HLA-DQA2|HLA-DRA|KDR|GHR | ||||
| T-cell receptor signaling pathway | CBLB|GRB2|CD4|PIK3R1 | 4/18 (22.2%) | 86/2361 (3.6%) | 2.00E−02 |
| Environmental information processing | GRB2|SELL|HLA-DMB|HLA-DQA2|KDR|CBLB| | 14/18 (77.7%) | 1059/2361 (44.8%) | 2.44E−02 |
| MAPK12|CD4|HLA-DPA1|HLA-DPB1|SELE| | ||||
| PIK3R1|HLA-DRA|GHR | ||||
| VEGF signaling pathway | MAPK12|PIK3R1|KDR | 3/18 (16.6%) | 58/2361 (2.4%) | 4.26E−02 |
| T1D | ||||
| ECM-receptor interaction | LAMA1|COL4A2|COL4A1|HSPG2|COL1A2|ITGA2| | 9/31 (29.0%) | 67/2419 (2.7%) | 3.66E−06 |
| ITGA10|ITGB1|COL11A1 | ||||
| Antigen processing and presentation | TAP1|HLA-DOA|HLA-DMB|HLA-DOB| | 7/31 (22.5%) | 47/2419 (1.9%) | 2.25E−05 |
| HLA-DQA2|HLA-DRA|HLA-F | ||||
| Type I diabetes mellitus | HLA-DOA|HLA-DMB|HLA-DOB|HLA-DQA2| | 6/31 (19.3%) | 30/2419 (1.2%) | 2.25E−05 |
| HLA-DRA|HLA-F | ||||
| Cell Communication | TLN1|MAGI3|COL4A2|COL4A1|ITGA10|ITGA2| | 15/31 (48.3%) | 351/2419 (14.5%) | 8.68E−05 |
| ITPR3|GRM1|ITGB1|ITPR1|PXN|LAMA1|TUBB| | ||||
| COL1A2|COL11A1 | ||||
| Cellular processes | TLN1|ITGA10|BCL2L1|HLA-DMB|ITGB1|PXN| | 26/31 (83.8%) | 1098/2419 (45.3%) | 1.10E−04 |
| TUBB|CASP8|TAP1|HLA-DOA|COL11A1| | ||||
| HLA-DOB|MAGI3|COL4A2|COL4A1|ITGA2| | ||||
| ITPR3|GRM1|HLA-DQA2|ITPR1|HLA-F| | ||||
| LAMA1|RIPK1|COL1A2|APAF1|HLA-DRA | ||||
| Focal adhesion | LAMA1|COL4A2|TLN1|COL4A1|COL1A2|ITGA2| | 10/31 (32.2%) | 170/2419 (7.0%) | 2.43E−04 |
| ITGA10|ITGB1|COL11A1|PXN | ||||
| Cell adhesion molecules (CAMs) | HLA-DOA|HLA-DMB|HLA-DOB|ITGB1| | 7/31 (22.5%) | 93/2419 (3.8%) | 9.26E−04 |
| HLA-DQA2|HLA-DRA|HLA-F | ||||
| Metabolic disorders | HLA-DOA|HLA-DMB|HLA-DOB|HLA-DQA2| | 6/31 (19.3%) | 79/2419 (3.2%) | 2.55E−03 |
| HLA-DRA|HLA-F | ||||
| Signaling molecules and interaction | COL4A2|COL4A1|HSPG2|ITGA2|ITGA10| | 16/31 (51.6%) | 563/2419 (23.2%) | 3.05E−03 |
| HLA-DMB|HLA-DQA2|GRM1|ITGB1| | ||||
| HLA-F|LAMA1|COL1A2|HLA-DOA|COL11A1| | ||||
| HLA-DOB|HLA-DRA | ||||
| Environmental information processing | COL4A2|COL4A1|HSPG2|ITGA2|ITGA10|BCL2L1| | 23/31 (74.1%) | 1077/2419 (44.5%) | 3.88E−03 |
| ITPR3|HLA-DMB|HLA-DQA2|GRM1|ITGB1| | ||||
| ITPR1|PXN|HLA-F|LAMA1|MAP4K4|TAP1|CASP8| | ||||
| COL1A2|HLA-DOA|HLA-DOB|COL11A1|HLA-DRA | ||||
| Human diseases | BCL2L1|HLA-DMB|HLA-DQA2|ITGB1|HLA-F| | 13/31 (41.9%) | 423/2419 (17.4%) | 5.60E−03 |
| LAMA1|APP|TUBB|CASP8|PRNP|HLA-DOA| | ||||
| HLA-DOB|HLA-DRA | ||||
| Neurodegenerative disorders | LAMA1|APP|CASP8|BCL2L1|PRNP | 5/31 (16.1%) | 103/2419 (4.2%) | 3.94E−02 |
| Gap junction | TUBB|ITPR3|GRM1|ITPR1 | 4/31 (12.9%) | 68/2419 (2.8%) | 4.13E−02 |
| Prion disease | LAMA1|PRNP | 2/31 (6.4%) | 13/2419 (0.5%) | 4.30E−02 |
| AD | ||||
| Neurodegenerative disorders | APP|SNCAIP|MAGI1|SNCA|PARK2|ITCH|APBA1 | 7/26 (26.9%) | 103/2419 (4.2%) | 3.77E−03 |
| Parkinson's disease | SNCAIP|SNCA|PARK2 | 3/26 (11.5%) | 13/2419 (0.5%) | 7.93E−03 |
| Development | EGFR|PAK6|CDC42|PAK7|ERBB4|SRGAP1 | 6/26 (23.0%) | 119/2419 (4.9%) | 2.30E−02 |
| Human diseases | EGFR|CDC42|APP|SNCAIP|MAGI1|TGFBR1|SNCA|PARK2|ITCH|APBA1|TGFB2 | 11/26 (42.3%) | 423/2419 (17.4%) | 3.46E−02 |
| BD | ||||
| Adherens junction | EGFR|IGF1R|MAPK1|CREBBP|PTPN1|INSR | 6/28 (21.4%) | 68/2419 (2.8%) | 5.67E−03 |
| MAPK signaling pathway | EGFR|MEF2C|MAPK1|RPS6KA2|NTRK2|GNA12| | 9/28 (32.1%) | 219/2419 (9.0%) | 1.40E−02 |
| PTPRR|NFATC2|CDC25B | ||||
| Nervous system | IGF1R|MAPK1|RPS6KA2|GNAI1|CREBBP|GNA12 | 6/28 (21.4%) | 98/2419 (4.0%) | 1.40E−02 |
| Neurodegenerative disorders | HD|CREBBP|SNCA|PARK2|BCL2L1|INSR | 6/28 (21.4%) | 103/2419 (4.2%) | 1.40E−02 |
| Human diseases | EGFR|IGF1R|MAPK1|HD|HNF4A|CREBBP|SNCA| | 12/28 (42.8%) | 423/2419 (17.4%) | 1.78E−02 |
| PARK2|BCL2L1|RUNX1|PRKCE|INSR | ||||
| Long-term depression | IGF1R|MAPK1|GNAI1|GNA12 | 4/28 (14.2%) | 61/2419 (2.5%) | 4.92E−02 |
Although possibly representing false discoveries, the top modules identified for T2D, CAD and HT are also shown for comparison (Fig. 2B). In T2D, the most significant module contained genes involved in intracellular signaling (EGFR and BCR), apoptosis (IGF1R, AVEN and APAF1) and insulin receptor signaling pathway (IGF1R and IGF2). In HT, the top scoring module listed genes are almost exclusively involved in cell communication (EGFR, VAV3 and RAC1).
To assess module specificity, we compared the performance of each of them in the disease in which they were identified against its performance across all other diseases. This was accomplished by tabulating the gene-wise P-value of association of each gene in the module with every disease. If a module reached significance just because it was composed of large-sized genes, for which relatively low P-values could be obtained by chance, it would be expected that the same module be also significant in several or all other diseases, but modules are significant only in the disease in which they were originally identified, suggesting they were identified because they were disease-specific and not due to chance. As demonstrated in Figure 5, the four most significant modules identified in MS show almost no association with any other disease. However, there are some genes that show strong association with other diseases in addition to MS. In particular, the HLA genes also show highly significant associations with both RA and T1D, two autoimmune diseases. Most notably, several significant genes from modules in AD and BD are also significant in MS. For example, SNCA, CDC42EP3, FHL2 and CRMP1 all show P-values <1 × 10−3 in MS and AD or BD, but consistently higher P-values for all the non-neurological diseases. The maximum number of SNPs tested in these genes ranged from 49 (CRMP1) to 79 (CDC42EP3), slightly above the median number of 40 SNPs/gene across the Illumina 550 k array. In contrast, genes such as PARK2, VAV3, PAK7 and NTRK3 yielded relatively low P-values (P < 1 × 10−3) across most or all diseases, possibly because a larger number of SNPs were tested for these genes, and some achieved significance by chance. Indeed, the number of SNPs for these genes in the Illumina platform ranges from 149 (PAK7) to 455 (PARK2).
Figure 5.

Module specificity. The P-values of genes from the representative modules shown in Figures 3 and 4 are displayed as a heatmap. Each row corresponds to a single gene. Genes are organized by their membership of modules. Genes corresponding to the four modules described for MS (Fig. 3) are at the top, followed by genes corresponding to modules from all other diseases. Because modules from different diseases may share one or more genes (e.g. HLA in autoimmune diseases), these may be represented more than once in the figure. Color-coded bars next to each module mark the genes that the module comprises. The same color code in the column headers indicates the disease for which the P-values are represented below. In general, genes from modules identified in one disease show the highest P-values for that disease, and less significant P-values for most other diseases. A notable exception is the HLA genes which show overlap between MS, RA and T1D, all of which are autoimmune diseases. Interestingly, some of the genes from the AD and BD modules show significant P-values also in MS.
Finally, we identified the 400 genes in which the gene-wise P-values varied most widely across diseases, and performed one-way hierarchical clustering on these P-values to produce a dendrogram identifying diseases with similar patterns of genetic association (Fig. 6). The two MS and the two RA studies clustered almost perfectly with each other and they, in turn, were grouped together in a looser cluster which also included T1D (autoimmune) and AD (neurological), but did not include any of the unrelated diseases. Intriguingly, the study on CD did not cluster with MS and RA, but with the unrelated diseases. One possible explanation for this difference is the lack of a strong association with HLA genes in CD compared with the other autoimmune diseases. Instead, genetic susceptibility to CD appears to be more widely spread across the genome (16,22).
Figure 6.
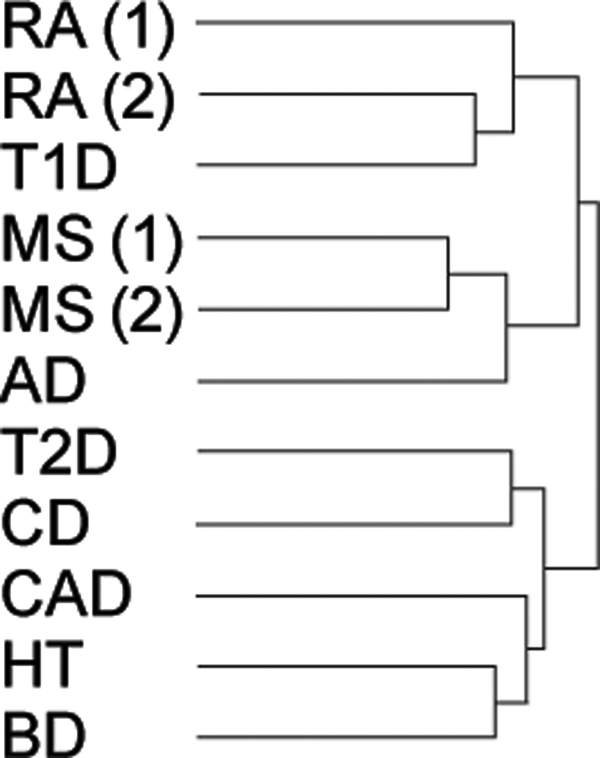
Disease hierarchical tree. The P-values of the genes showing the most variable levels of association across diseases were selected to cluster the GWAS studies. Studies connected by the same branch of the dendrogram are more similar to each other than those in different branches. Notably, the two MS studies cluster together, and with those from RA and T1D. Also, the two RA studies cluster together (and with T1D, another autoimmune disease). MS(1), GeneMSA; MS(2), IMSGC; RA(1), Gregersen; RA(2), WTCCC.
DISCUSSION
There is ongoing debate on the exact mechanism of MS pathogenesis. Some theories support the idea that there is a primary immune disorder targeting the CNS, with subsequent neurodegeneration being a consequence of the initial inflammatory process. A competing theory states that neurodegeneration is the primary cause of the disease, leading to an inflammatory reaction within the CNS (23). Most previous genetic studies in MS (by genome-wide or candidate gene approaches) have involved immune-related genes, thus supporting the first scenario. In this article, we employ a novel network-based pathway analysis using data from two independent GWAS and implicate neural pathways in the susceptibility to MS.
Recently reported pathway-based analyses of GWAS data (15,24,25) relayed exclusively on classical biological pathways as described in KEGG, Biocarta or gene ontology. The article by Torkamani et al. (15) is of particular interest since they analyzed several of the GWAS datasets we used as control. We also tested a similar approach in which a literature-derived network of biological relationships is first assembled and subsequently, an exhaustive search for sub-networks representing particular pathways is conducted (26). Using the two MS datasets, this analysis yielded significant pathways associated with immune and neurological functions including antigen presentation, axon guidance and neurogenesis (Supplementary Material, Table S1). In order to enhance the potential for discovery of biologically relevant circuits, we introduced the PIN to the analysis. The PINBPA approach focuses on the combined effect of associated genes by restricting the search for pathways to only those gene products that actually interact as determined by a high-quality PIN. Aside from significantly reducing the possible total number of interactions, the network-based approach takes advantage of the fact that if two proteins physically interact, they likely belong to the same biological pathway. We acknowledge that restricting the search to only certain interactions also increases the chances of detecting pathways even in the absence of significant P-values for association. To account for this, we performed extensive testing and showed that the module scores obtained for most diseases were lower and less reproducible when randomized datasets were used. Other methods that detect gene–gene interactions have been described (27–30). However, unlike a network-based approach, those methods consider all possible pair-wise interactions, limiting the biological interpretation of the data. Furthermore, implementation of this approach at a whole-genome scale requires extraordinary computational resources. Another advantage of our pathway analysis approach is that it may provide a basis for patient stratification by disease subtype. Whether MS is a single disease or several diseases with a common phenotype is still a matter of debate. Pathway-based genetic analyses may help identify different, and even unrelated, biological mechanisms as responsible for disease pathogenesis. The implications of such potential discovery are broad, as this may lead to targeted therapeutics, and individually tailored disease management.
The most likely cause for the modest P-values seen in all diseases analyzed is insufficient power to detect small genetic effects. It is estimated that the allelic odds ratios of susceptibility variants in most complex diseases lie between 2.0 and 1.2. For an odds ratio of 2.0, a sample size of 500 individuals provides 80% power to detect a causative variant with population frequency of 10% (α = 1 × 10−5, multiplicative model). However, at an odds ratio of 1.2, 9000 cases and controls are needed to achieve the same power (31). All the GWAS analyzed here genotyped between 1000 and 2000 cases, thus only providing adequate power to detect relatively large genetic effects or associations with relatively common disease-specific markers. Under these conditions, a pathway-based analysis may compensate for the lack of statistical power due to insufficient sample size by making use of the much higher prior probability of true associations among certain combinations of genes, determined by their biological relationships, than among other, arbitrary combinations. The added constraint that only associations between genes that physically interact (as determined by the PIN) are taken into account further strengthens the prior.
A necessary step in the search for pathways involves condensing the evidence for association for each marker within a gene into one P-value representative of the gene. Although for our analysis the gene-wise P-value was that of the most significant SNP within that gene (min P-value method), we also explored other methods that would take into account the variable number of markers genotyped within each gene and the extent of LD among them. We examined various techniques for correcting the gene-wise P-value for the number of SNPs per gene, and adjusting the corrected value for LD. We also applied the Fisher's method for combining P-values, followed by an adjustment for LD. However, when these methods were used, a very small number of genes exceeded the threshold of significance, not enough to compute any pathway searches. Although such correction and adjustment are appropriate on the null hypothesis of no associations genome-wide, it may penalize large genes excessively if true (causative) associations are proportionally more common in smaller genes. We also evaluated to what extent larger genes were more likely to be included in our analysis by virtue of being more represented in the array. We found no significant difference in the distribution of gene sizes within a given module when compared with a random set of genes (Supplementary Material, Fig. S1). Altogether, the min P-value method provided the most consistent and balanced results. This method has also been applied by Torkamani et al. (15) in their pathway analysis of the WTCCC dataset with similar results.
Here we implicate neural pathways (e.g. axon guidance and long-term potentiation) in susceptibility to MS. Only one other article has reported a neural gene in MS susceptibility to date (32). Due to the power limitations described above, none of the individual genes in these pathways may exceed the genome-wide threshold of significance in tests of association. However, when an entire pathway is considered, even modest associations in several of its component genes contribute to the overall P-value. For example, seven GluR genes were found to be marginally associated with MS (Module MS_IV). Although each marker may not reach significance when tested in isolation, the probability that several GluRs are identified by chance, if none of them is truly associated with the disease, is exceedingly small. Furthermore, not all GluRs are encoded on the same chromosome, thus reducing the likelihood of inflation of the signal due to LD. The identified associations between GluRs and MS are of substantial biological relevance since glutamate is the principal excitatory neurotransmitter within the CNS. Glutamate acts on neuronal and glial ionotropic receptors coupled to specific ligand-gated cationic channels, and mGluRs, coupled to second messengers. Under normal conditions, astrocytes maintain low extracellular glutamate levels by using transporters to take up glutamate rapidly as high extracellular glutamate levels are neurotoxic. Indeed, elevated extracellular glutamate levels can result in the death of neurons and oligodendrocytes through excitotoxic mechanisms and these have been shown to play a role in the pathology of MS and EAE (33,34). Interestingly, susceptibility to excitotoxicity may be under genetic control (35).
A second neural module (MS_III) contained NCK2 and FYN, in addition to two members of the ephrin family of proteins (EFNB1 and EFNB2) and three or their receptors (EPHA3, EPHA4 and EPHB2). These genes are involved in axon guidance, the process during development by which neurons extend their processes and make connections throughout the CNS. Specifically, Eph/ephrin signaling regulates axon guidance through contact repulsion during development of the CNS, inducing collapse of neuronal growth cones (36). Eph receptors and ephrins continue to be expressed in the adult CNS, although usually at lower levels, but have been found to be upregulated in MS lesions on different cell types, including reactive astrocytes, neurons and oligodendrocytes (37). This upregulated expression may directly inhibit regrowth of regenerating axons, but Eph expression also regulates astrocytic gliosis and formation of the glial scar. Therefore, Eph/ephrin signaling may inhibit regeneration by more than one mechanism and modulation of Eph receptor expression or signaling could prove pivotal in determining the outcome of injury in the adult CNS.
Due to the nature of the searches performed, this is a gene-centered analysis, and thus it is possible that true associations with markers that lie in large intergenic regions were neglected. Also, markers within genes not represented in the PIN were not evaluated in this analysis. Finally, it is reasonable to expect that subgroups of patients with shared risk alleles would be identified by this method. We were unable to subclassify patients because our analysis only takes into account the most significant variant for each gene, and more significant markers may be needed to identify such subgroups. Nevertheless, there is scope for the development of related methods to increase the power to detect associations in these regions and genes. In summary, by following a network-based pathway analysis, we have expanded the immune-related set of genes associated with MS. Furthermore, we have identified neural pathways whose involvement in the disease is biologically plausible. Larger pathway-oriented association studies will ultimately be necessary to validate these findings.
MATERIALS AND METHODS
Genetic association data
In total, 11 GWAS were analyzed (two for MS and nine others as controls). The first of the two studies in MS was a family trio-based analysis recently published by the International MS Genetics Consortium (IMSGC) in which 334 923 SNPs were analyzed in 931 trios by the transmission disequilibrium test (9). The second MS study (GeneMSA) was a multicenter case–control association analysis done collaboratively among the University of California San Francisco, Vrije Universiteit Medical Center in Amsterdam, University Hospital Basel, and the pharmaceutical company Glaxo SmithKline (GSK). The GeneMSA study analyzed 551 642 SNPs in 978 cases and 883 controls (3).
As controls, we used data from four studies in other autoimmune diseases, consisting of two studies in RA, and one each in CD and T1D. Two studies in other neurological diseases included one each in AD and BD. In addition, unrelated diseases included one study each in HT, CAD and T2D. The studies for RA, CD, T1D, BD, HT, CAD and T2D were performed by the Wellcome Trust Case Control Consortium (WTCCC) (16) and the genotypic P-values of association for each tested SNP were obtained from the project's webpage (www.wtccc.org.uk). A second RA study was performed by Plenge and collaborators in which 317 503 SNPs were tested in 1522 cases and 1850 controls (38). P-values for association were obtained from the Supplementary information provided in that article. Processed data for the AD study performed at GSK is publicly accessible from http://www.imgw.com/public/ (39).
Module (sub-network) searches
We first computed the gene-wise significance for association by choosing the lowest P-value of all SNPs mapping to a given gene (min P-value method) without correction. Although this method potentially introduces biases in favor of larger genes (for which more SNPs are generally typed, thus increasing the chances of type I error), the use of the gene-wise P-value as an input variable in a second analysis step (see Introduction) provides protection against spurious findings caused by such bias. Moreover, we implemented rigorous validation steps that included randomized network searches and comparison with similar datasets from other complex diseases. Other measures of gene-wise significance were considered, including Fisher's method of combining P-values (40), and a method that corrects for the number of SNPs tested within each gene and subsequently adjusts for LD (41,42) (data not shown). However, since the most biologically significant findings were obtained with the min P-value method, we only report on these results.
Genes with a gene-wise association P-value of 0.05 or less were considered for further study and loaded into the Cytoscape software, a package for visualization and analysis of networks (43). A curated human PIN (n = 7500) was downloaded and visualized in Cytoscape (44,45). The gene-wise P-values for association with each disease were loaded as node attributes of the PIN and the plugin jActive modules (46) was used to identify sub-networks of modestly associated but interacting gene products. The biological interpretation of a statistically significant module (sub-network) is that the products of a set of genes associated to the disease also interact physically, thus raising the possibility that they belong to the same pathway or biological process. jActive modules grows a network from each node by systematically adding one neighbor at a time and computing an aggregate score (S) based on a given statistical significance, in our case, the gene-wise P-value of association with the disease. Specifically, 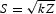 , where each gene P-value is converted to a Z-score (using the inverse normal CDF) and k is the number of genes contributing to the score S. Once S ceases to increase significantly, the sub-network stops growing and is reported as a module (46). Next, the test statistic (S) is compared with an appropriate background distribution. As a background distribution, we used the scores of modules randomly selected from the entire PPI network. Since the background distribution is dependent on module size, jActive modules creates a background distribution by scoring 10 000 random modules of each size (in a Monte Carlo procedure). Furthermore, since the scores distribution is a smooth function of module size, jActive modules applies a sliding window average to the background distribution. As in the original publication, modules with S> 3 (3 SD above the mean of randomized scores) were considered significant. It is interesting to note that scores of up to 12 were obtained for some diseases. If converted back to P-values and corrected for the multiple network searches the algorithm performs (∼107), results would remain highly significant (10−14) even after the correction. P-values from both studies in MS (IMSGC and GeneMSA) and RA (Plenge et al. and WTCCC) were included in the search, resulting in higher confidence in the significance of the modules retrieved. To merge datasets, we included genes with significant P-values in either study, and when a given gene was present in both, the min P-value was considered. To evaluate whether the significant modules obtained were biologically meaningful, we computed their enrichment in human biochemical pathways (i.e. the proportion of genes in a specific module that are in a pathway, compared to the overall proportion of genes described for that pathway) using the plugin BINGO (47) with a custom ontology and annotation files derived from the Kyoto Encyclopedia of Genes and Genomes (KEGG) database. The statistical significance for enrichment of a given module in ontologies and pathways was determined by a chi-square test. Significant KEGG pathways are reported and visualized as directed acyclic graphs similar to those commonly reported for the analysis of GO by many popular gene expression analysis programs. Using the same plugin, we also computed enrichment of significant modules in GO categories. The analysis of several modules resulted in highly significant results with both KEGG pathways and GO. However, although most genes with known functions are categorized in the GO system, this classification is largely based on information retrieved from the literature while KEGG primarily categorizes genes into bona-fide biological pathways. Because biological interpretation of pathways is more straightforward, we report only on KEGG results. In addition, we also consulted commercially curated pathway data from Ingenuity (Redwood City, CA, USA), GeneGo (Encinitas, CA, USA), NetPro Molecular Connections (Singapore) and Jubilant (Berkley Heights, NJ, USA).
, where each gene P-value is converted to a Z-score (using the inverse normal CDF) and k is the number of genes contributing to the score S. Once S ceases to increase significantly, the sub-network stops growing and is reported as a module (46). Next, the test statistic (S) is compared with an appropriate background distribution. As a background distribution, we used the scores of modules randomly selected from the entire PPI network. Since the background distribution is dependent on module size, jActive modules creates a background distribution by scoring 10 000 random modules of each size (in a Monte Carlo procedure). Furthermore, since the scores distribution is a smooth function of module size, jActive modules applies a sliding window average to the background distribution. As in the original publication, modules with S> 3 (3 SD above the mean of randomized scores) were considered significant. It is interesting to note that scores of up to 12 were obtained for some diseases. If converted back to P-values and corrected for the multiple network searches the algorithm performs (∼107), results would remain highly significant (10−14) even after the correction. P-values from both studies in MS (IMSGC and GeneMSA) and RA (Plenge et al. and WTCCC) were included in the search, resulting in higher confidence in the significance of the modules retrieved. To merge datasets, we included genes with significant P-values in either study, and when a given gene was present in both, the min P-value was considered. To evaluate whether the significant modules obtained were biologically meaningful, we computed their enrichment in human biochemical pathways (i.e. the proportion of genes in a specific module that are in a pathway, compared to the overall proportion of genes described for that pathway) using the plugin BINGO (47) with a custom ontology and annotation files derived from the Kyoto Encyclopedia of Genes and Genomes (KEGG) database. The statistical significance for enrichment of a given module in ontologies and pathways was determined by a chi-square test. Significant KEGG pathways are reported and visualized as directed acyclic graphs similar to those commonly reported for the analysis of GO by many popular gene expression analysis programs. Using the same plugin, we also computed enrichment of significant modules in GO categories. The analysis of several modules resulted in highly significant results with both KEGG pathways and GO. However, although most genes with known functions are categorized in the GO system, this classification is largely based on information retrieved from the literature while KEGG primarily categorizes genes into bona-fide biological pathways. Because biological interpretation of pathways is more straightforward, we report only on KEGG results. In addition, we also consulted commercially curated pathway data from Ingenuity (Redwood City, CA, USA), GeneGo (Encinitas, CA, USA), NetPro Molecular Connections (Singapore) and Jubilant (Berkley Heights, NJ, USA).
To account for the possibility that significant modules were obtained by chance, 10 searches with the gene-based P-values randomly permuted over the genes were conducted for each disease. The average score of the randomized searches is compared with the scores obtained with the real (i.e. non-randomized) P-values for each disease.
SUPPLEMENTARY MATERIAL
FUNDING
Funding to pay the Open Access publication charges for this article was provided by The U.S. National Multiple Sclerosis Society.
Supplementary Material
ACKNOWLEDGEMENTS
We thank the MS patients and healthy controls who participated in this study. We also thank the IMSGC for access to their GWAS unpublished data.
Conflict of Interest statement. N.W.G., P.M.M., W.W., R.A.G. and M.R.B. are stock- and option-holding employees of GlaxoSmithKline.
REFERENCES
- 1.Altshuler D., Daly M.J., Lander E.S. Genetic mapping in human disease. Science. 2008;322:881–888. doi: 10.1126/science.1156409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Altshuler D., Hirschhorn J.N., Klannemark M., Lindgren C.M., Vohl M.C., Nemesh J., Lane C.R., Schaffner S.F., Bolk S., Brewer C., et al. The common PPARgamma Pro12Ala polymorphism is associated with decreased risk of type 2 diabetes. Nat. Genet. 2000;26:76–80. doi: 10.1038/79216. [DOI] [PubMed] [Google Scholar]
- 3.Baranzini S.E., Wang J., Gibson R.A., Galwey N., Naegelin Y., Barkhof F., Radue E.W., Lindberg R.L., Uitdehaag B.M., Johnson M.R., et al. Genome-wide association analysis of susceptibility and clinical phenotype in multiple sclerosis. Hum. Mol. Genet. 2009;18:767–778. doi: 10.1093/hmg/ddn388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hauser S.L., Goodin D.S. Multiple sclerosis and other demyelinating diseases. In: Braunwald E., Fauci A.D., Kasper D.L., Hauser S.L., Longo D.L., Jameson J.L., editors. Harrison's Principles in Internal Medicine. 16 edn. New York: McGraw-Hill; 2005. pp. 2461–2471. [Google Scholar]
- 5.Ebers G.C., Kukay K., Bulman D.E., Sadovnick A.D., Rice G., Anderson C., Armstrong H., Cousin K., Bell R.B., Hader W., et al. A full genome search in multiple sclerosis. Nat. Genet. 1996;13:472–476. doi: 10.1038/ng0896-472. [DOI] [PubMed] [Google Scholar]
- 6.Sawcer S., Jones H.B., Feakes R., Gray J., Smaldon N., Chataway J., Robertson N., Clayton D., Goodfellow P.N., Compston A. A genome screen in multiple sclerosis reveals susceptibility loci on chromosome 6p21 and 17q22. Nat. Genet. 1996;13:464–468. doi: 10.1038/ng0896-464. [DOI] [PubMed] [Google Scholar]
- 7.Haines J.L., Ter-Minassian M., Bazyk A., Gusella J.F., Kim D.J., Terwedow H., Pericak-Vance M.A., Rimmler J.B., Haynes C.S., Roses A.D., et al. A complete genomic screen for multiple sclerosis underscores a role for the major histocompatability complex. The Multiple Sclerosis Genetics Group. Nat. Genet. 1996;13:469–471. doi: 10.1038/ng0896-469. [DOI] [PubMed] [Google Scholar]
- 8.Oksenberg J.R., Baranzini S.E., Barcellos L.F., Hauser S.L. Multiple sclerosis: genomic rewards. J. Neuroimmunol. 2001;113:171–184. doi: 10.1016/s0165-5728(00)00444-6. [DOI] [PubMed] [Google Scholar]
- 9.Hafler D.A., Compston A., Sawcer S.J., Lander E.S., Daly M.J., De Jager P.L., de Bakker P.I., Gabriel S.B., Mirel D.B., et al. IMSGC. Risk alleles for multiple sclerosis identified by a Genomewide Study. N. Engl. J. Med. 2007;357:851–862. doi: 10.1056/NEJMoa073493. [DOI] [PubMed] [Google Scholar]
- 10.Ashburner M., Ball C.A., Blake J.A., Botstein D., Butler H., Cherry J.M., Davis A.P., Dolinski K., Dwight S.S., Eppig J.T., et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Franke L., van Bakel H., Fokkens L., de Jong E.D., Egmont-Petersen M., Wijmenga C. Reconstruction of a functional human gene network, with an application for prioritizing positional candidate genes. Am. J. Hum. Genet. 2006;78:1011–1025. doi: 10.1086/504300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lage K., Karlberg E.O., Storling Z.M., Olason P.I., Pedersen A.G., Rigina O., Hinsby A.M., Tumer Z., Pociot F., Tommerup N., et al. A human phenome-interactome network of protein complexes implicated in genetic disorders. Nat. Biotechnol. 2007;25:309–316. doi: 10.1038/nbt1295. [DOI] [PubMed] [Google Scholar]
- 13.Tu Z., Wang L., Arbeitman M.N., Chen T., Sun F. An integrative approach for causal gene identification and gene regulatory pathway inference. Bioinformatics. 2006;22:e489–e496. doi: 10.1093/bioinformatics/btl234. [DOI] [PubMed] [Google Scholar]
- 14.Suthram S., Beyer A., Karp R.M., Eldar Y., Ideker T. eQED: an efficient method for interpreting eQTL associations using protein networks. Mol. Syst. Biol. 2008;4:162. doi: 10.1038/msb.2008.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Torkamani A., Topol E.J., Schork N.J. Pathway analysis of seven common diseases assessed by genome-wide association. Genomics. 2008;92:265–272. doi: 10.1016/j.ygeno.2008.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Consortium T.W.T.C.C. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lang H.L., Jacobsen H., Ikemizu S., Andersson C., Harlos K., Madsen L., Hjorth P., Sondergaard L., Svejgaard A., Wucherpfennig K., et al. A functional and structural basis for TCR cross-reactivity in multiple sclerosis. Nat. Immunol. 2002;3:940–943. doi: 10.1038/ni835. [DOI] [PubMed] [Google Scholar]
- 18.Gregersen J.W., Kranc K.R., Ke X., Svendsen P., Madsen L.S., Thomsen A.R., Cardon L.R., Bell J.I., Fugger L. Functional epistasis on a common MHC haplotype associated with multiple sclerosis. Nature. 2006;443:574–577. doi: 10.1038/nature05133. [DOI] [PubMed] [Google Scholar]
- 19.Arthur A.T., Armati P.J., Bye C., Heard R.N., Stewart G.J., Pollard J.D., Booth D.R. Genes implicated in multiple sclerosis pathogenesis from consilience of genotyping and expression profiles in relapse and remission. BMC Med. Genet. 2008;9:17. doi: 10.1186/1471-2350-9-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gregory S.G., Schmidt S., Seth P., Oksenberg J.R., Hart J., Prokop A., Caillier S.J., Ban M., Goris A., Barcellos L.F., et al. Interleukin 7 receptor alpha chain (IL7R) shows allelic and functional association with multiple sclerosis. Nat. Genet. 2007;39:1083–1091. doi: 10.1038/ng2103. [DOI] [PubMed] [Google Scholar]
- 21.Nath S.K., Han S., Kim-Howard X., Kelly J.A., Viswanathan P., Gilkeson G.S., Chen W., Zhu C., McEver R.P., Kimberly R.P., et al. A nonsynonymous functional variant in integrin-alpha(M) (encoded by ITGAM) is associated with systemic lupus erythematosus. Nat. Genet. 2008;40:152–154. doi: 10.1038/ng.71. [DOI] [PubMed] [Google Scholar]
- 22.Barrett J.C., Hansoul S., Nicolae D.L., Cho J.H., Duerr R.H., Rioux J.D., Brant S.R., Silverberg M.S., Taylor K.D., Barmada M.M., et al. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn's disease. Nat. Genet. 2008;40:955–962. doi: 10.1038/NG.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hauser S.L., Oksenberg J.R. The neurobiology of multiple sclerosis: genes, inflammation, and neurodegeneration. Neuron. 2006;52:61–76. doi: 10.1016/j.neuron.2006.09.011. [DOI] [PubMed] [Google Scholar]
- 24.Lesnick T.G., Papapetropoulos S., Mash D.C., Ffrench-Mullen J., Shehadeh L., de Andrade M., Henley J.R., Rocca W.A., Ahlskog J.E., Maraganore D.M. A genomic pathway approach to a complex disease: axon guidance and Parkinson disease. PLoS Genet. 2007;3:e98. doi: 10.1371/journal.pgen.0030098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang K., Li M., Bucan M. Pathway-Based Approaches for Analysis of Genomewide Association Studies. Am. J. Hum. Genet. 2007;81:1278–1283. doi: 10.1086/522374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rajagopalan D., Agarwal P. Inferring pathways from gene lists using a literature-derived network of biological relationships. Bioinformatics. 2005;21:788–793. doi: 10.1093/bioinformatics/bti069. [DOI] [PubMed] [Google Scholar]
- 27.Ritchie M.D., Hahn L.W., Roodi N., Bailey L.R., Dupont W.D., Parl F.F., Moore J.H. Multifactor-dimensionality reduction reveals high-order interactions among estrogen-metabolism genes in sporadic breast cancer. Am. J. Hum. Genet. 2001;69:138–147. doi: 10.1086/321276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Motsinger A.A., Ritchie M.D., Reif D.M. Novel methods for detecting epistasis in pharmacogenomics studies. Pharmacogenomics. 2007;8:1229–1241. doi: 10.2217/14622416.8.9.1229. [DOI] [PubMed] [Google Scholar]
- 29.Chapman J., Clayton D. Detecting association using epistatic information. Genet. Epidemiol. 2007;31:894–909. doi: 10.1002/gepi.20250. [DOI] [PubMed] [Google Scholar]
- 30.Marchini J., Donnelly P., Cardon L.R. Genome-wide strategies for detecting multiple loci that influence complex diseases. Nat. Genet. 2005;37:413–417. doi: 10.1038/ng1537. [DOI] [PubMed] [Google Scholar]
- 31.Wang W.Y., Barratt B.J., Clayton D.G., Todd J.A. Genome-wide association studies: theoretical and practical concerns. Nat. Rev. Genet. 2005;6:109–118. doi: 10.1038/nrg1522. [DOI] [PubMed] [Google Scholar]
- 32.Aulchenko Y.S., Hoppenbrouwers I.A., Ramagopalan S.V., Broer L., Jafari N., Hillert J., Link J., Lundstrom W., Greiner E., Dessa Sadovnick A., et al. Genetic variation in the KIF1B locus influences susceptibility to multiple sclerosis. Nat. Genet. 2008;40:1402–1403. doi: 10.1038/ng.251. [DOI] [PubMed] [Google Scholar]
- 33.Werner P., Pitt D., Raine C.S. Multiple sclerosis: altered glutamate homeostasis in lesions correlates with oligodendrocyte and axonal damage. Ann. Neurol. 2001;50:169–180. doi: 10.1002/ana.1077. [DOI] [PubMed] [Google Scholar]
- 34.Pitt D., Werner P., Raine C.S. Glutamate excitotoxicity in a model of multiple sclerosis. Nat. Med. 2000;6:67–70. doi: 10.1038/71555. [DOI] [PubMed] [Google Scholar]
- 35.Meldrum B.S. Glutamate as a neurotransmitter in the brain: review of physiology and pathology. J. Nutr. 2000;130:1007S–1015S. doi: 10.1093/jn/130.4.1007S. [DOI] [PubMed] [Google Scholar]
- 36.Gallarda B.W., Bonanomi D., Muller D., Brown A., Alaynick W.A., Andrews S.E., Lemke G., Pfaff S.L., Marquardt T. Segregation of axial motor and sensory pathways via heterotypic trans-axonal signaling. Science. 2008;320:233–236. doi: 10.1126/science.1153758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sobel R.A. Ephrin A receptors and ligands in lesions and normal-appearing white matter in multiple sclerosis. Brain Pathol. 2005;15:35–45. doi: 10.1111/j.1750-3639.2005.tb00098.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Plenge R.M., Seielstad M., Padyukov L., Lee A.T., Remmers E.F., Ding B., Liew A., Khalili H., Chandrasekaran A., Davies L.R., et al. TRAF1-C5 as a risk locus for rheumatoid arthritis—a genomewide study. N. Engl. J. Med. 2007;357:1199–1209. doi: 10.1056/NEJMoa073491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li H., Wetten S., Li L., St Jean P.L., Upmanyu R., Surh L., Hosford D., Barnes M.R., Briley J.D., Borrie M., et al. Candidate single-nucleotide polymorphisms from a genomewide association study of Alzheimer disease. Arch. Neurol. 2008;65:45–53. doi: 10.1001/archneurol.2007.3. [DOI] [PubMed] [Google Scholar]
- 40.Fisher R.A. Combining independent tests of significance. Am. Stat. 1948;2:30. [Google Scholar]
- 41.Li J., Ji L. Adjusting multiple testing in multilocus analyses using the eigenvalues of a correlation matrix. Heredity. 2005;95:221–227. doi: 10.1038/sj.hdy.6800717. [DOI] [PubMed] [Google Scholar]
- 42.Nyholt D.R. A simple correction for multiple testing for single-nucleotide polymorphisms in linkage disequilibrium with each other. Am. J. Hum. Genet. 2004;74:765–769. doi: 10.1086/383251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cline M.S., Smoot M., Cerami E., Kuchinsky A., Landys N., Workman C., Christmas R., Avila-Campilo I., Creech M., Gross B., et al. Integration of biological networks and gene expression data using Cytoscape. Nat. Protoc. 2007;2:2366–2382. doi: 10.1038/nprot.2007.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rual J.F., Venkatesan K., Hao T., Hirozane-Kishikawa T., Dricot A., Li N., Berriz G.F., Gibbons F.D., Dreze M., Ayivi-Guedehoussou N., et al. Towards a proteome-scale map of the human protein-protein interaction network. Nature. 2005;437:1173–1178. doi: 10.1038/nature04209. [DOI] [PubMed] [Google Scholar]
- 45.Goh K.I., Cusick M.E., Valle D., Childs B., Vidal M., Barabasi A.L. The human disease network. Proc. Natl Acad. Sci. USA. 2007;104:8685–8690. doi: 10.1073/pnas.0701361104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ideker T., Ozier O., Schwikowski B., Siegel A.F. Discovering regulatory and signalling circuits in molecular interaction networks. Bioinformatics. 2002;18(Suppl. 1):S233–S240. doi: 10.1093/bioinformatics/18.suppl_1.s233. [DOI] [PubMed] [Google Scholar]
- 47.Maere S., Heymans K., Kuiper M. BiNGO: a Cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics. 2005;21:3448–3449. doi: 10.1093/bioinformatics/bti551. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.