

Abstract
We have developed an efficient versatile in vivo dendritic cell (DC) targeting vector for delivering different classes of antigens such as proteins, peptide, glycolipids and naked DNA for vaccine applications. A single chain antibody (scFv) that recognizes DEC-205 receptor of DC was fused with a core-streptavidin domain and expressed in Escherichia coli using the T7 expression system. The bifunctional fusion protein (bfFp) was expressed as a periplasmic soluble protein and affinity-purified in its monomeric form. The bifunctional activity against DEC-205 and biotin was characterized by ELISA and Western blot. In vivo DC targeting of a diverse group of biotinylated antigens such as viral and bacterial proteins, a cancer peptide, gangliosides and DNA of certain infectious diseases was conducted in mice. Results show that in the presence of bfFp and costimulatory anti-CD40 mAb, both humoral and cell-mediated responses were augmented in either the single antigen or multiple antigen targeting strategy. Lastly, bfFp based DC targeting of antigens in low doses may be a useful strategy for the design of monovalent or polyvalent vaccines for the masses.
Keywords: Bifunctional antibody, recombinant antibody, dendritic cells, DEC-205, antigen targeting, immunotargeting, bioterrorism, cancer
1. Introduction
Dendritic cells (DC) are the most specialized and potent antigen presenting cells in the immune system. DC plays a critical role in innate and adaptive immune responses, especially in priming and activating T cell and B cell immunity. Several clinical trials have been initiated for human tumor immunotherapy based on ex vivo stimulation of DC; however, the clinical responses have been observed in a minority of patients and the procedure is very costly and laborious. In vivo targeting of DC via surface receptors may present an alternative simpler route for targeted immunotherapy.1 DC utilize several surface receptors (Gb3/CD77,2 CD40,3 β2 integrins,4 Fc receptors,5 and C-type lectin receptors6) to internalize, process and present antigens to MHC class I and MHC class II pathways.7 A number of DC receptors have been studied extensively for antigen targeting.1,7 DEC-205 is a C-type lectin receptor that is present on both immature and mature DC in lymphoid tissues, lymph nodes and spleen.8–12 DEC-205 targeting of antigens by an antibody along with costimulatory molecules is an efficient strategy to achieve protection of the host against tumor growth,4 rejection of an existing tumor,13 protection against a virus airway challenge,14 as well as enhanced resistance to an established rapidly growing tumor or viral infection.15 Moreover, DEC-205 targeting of a single protein enables cross-presentation of several peptides more efficiently than CD206 and CD20916 and induces stronger T cell immunity at much lower doses of protein antigen, plasmid DNA or recombinant adenovirus.14 In the absence of costimulation, DEC-205 targeting of antigen can be used to suppress the development of autoimmunity, such as type 1 diabetes.17 Generation of the antibody-mediated antigen targeting system is often complex, costly and a time-consuming process. In addition, it requires either liposomal encapsulation of the antigen,4 chemical cross-linking13,15,17,18 or construction of a new hybrid antibody.14,16,19 These designs may not be suitable for comparison studies with different antigens such as glycolipids and DNA and also in vivo targeting of multiple antigens.
We have previously reported the development of a quadroma (hybrid-hybridoma) based full length bispecific monoclonal antibody (bsmAb) for targeting of biotinylated protein antigens to DEC-205.20 The successful targeting of biotinylated proteins to DC augmented immune responses against the antigen at a ∼500-fold lower dose; however, quadromas express the bsmAb along with parental and unwanted heavy and light chain combinations.21 Furthermore, the antibody-based biotin binding is several orders weaker than streptavidin. Consequently, we have now designed a truncated recombinant construct to demonstrate the versatility of this DC targeting vehicle to deliver four categories of antigens: proteins, a peptide, glycolipids and DNA.
A scFv that recognizes DEC-205 receptor of DC was successfully cloned from the HB290 hybridoma, fused with a truncated core-streptavidin domain and expressed in Escherichia coli using the T7 expression system. The monomeric form of the fusion protein was affinity purified and the bifunctional activity was demonstrated by ELISA and Western blot. In vivo DC targeting and immune response studies in mice were initiated with a variety of biotinylated antigens (Table 2). In the presence of bfFp and costimulatory anti-CD40 mAb, both humoral and cell-mediated responses were estimated.
Table 2.
Summary Table of the Antigens Used for Testing anSD Targeting in the in Vivo Studya
| classification | immunized antigens | testing antigens | references |
|---|---|---|---|
| protein | B-OVA | OVA | 24, 36–39 |
| B-SARS-CoV spike RBD | SARS-CoV spike RBD | ||
| B-EBOV GP1 | EBOV GP1 | ||
| B-anthrax PA | anthrax PA | ||
| peptide | B-MUC-1 | B-MUC-1 | 40, 41 |
| glycolipid | B-GM2 | B-GM2 (IFN-γ assay) | 42, 43 |
| B-GM3 | B-GM3 (IFN-γ assay) | ||
| B-BSA-GM2 (humoral study) | |||
| B-BSA-GM3 (humoral study) | |||
| DNA | B-pVHX-6 (DNA) | WEEV E1 (protein) | 24, 36, 37, 39.44–47 |
| B-pEBOV GP1,2 (DNA) | WEEV E2 (protein) | ||
| B-pSARS-CoV spike (DNA) | EBOV GP1 (protein) | ||
| B-pSARS-CoV membrane (DNA) | EBOV GP2 (protein) | ||
| EBOV GP1,2 (protein) | |||
| SARS-CoV spike S1 (protein) | |||
| SARS-CoV spike S2 (protein) | |||
| SARS-CoV spike RBD (protein) | |||
| SARS-CoV membrane (protein) |
The antigens are divided into four categories, and the antigens immunized into mice are listed in the table. Immunized and unimmunized mice were tested with the antigens for both IFN-γ secretion and serum immune responses.
2. Experimental Section
2.1. Materials
DC 2.4 is a DEC-205 expressing mouse bone marrow DC cell-line transduced with GM-CSF, myc and raf oncogenes.22 HB290, a rat antimouse DEC-205 hybridoma, was obtained from ATCC. BSA (bovine serum albumin), streptavidin-HRPO (horseradish peroxidase), NHS-LC-biotin (biotinamidohexanoic acid 3-sulfo-N-hydroxysuccinimide ester), photobiotin acetate, OVA and goat antimouse-HRPO (GAM-HRPO) were from Sigma (Oakville, Canada). Biotinylated MUC-1 peptide with amino acid sequence of B-GVTSAPDTRGVTSAPDTR (N-terminal biotinylated) was kindly provided by Biomira, Inc. (Edmonton, Alberta, Canada). The streptavidin gene was kindly provided by Dr. T. Sano, Center for Molecular Imaging, Diagnosis and Therapy and Basic Science Laboratory, Boston, MA. HSF (hybridoma serum free media), DMEM (Dulbecco's Modified Eagle Medium), PSG (penicillin, streptomycin and l-glutamine) and FBS (fetal bovine serum) were purchased from Gibco BRL (Burlington, Canada). B-BSA [(biotin)n labeled BSA] was prepared by biotinylation of BSA with NHS-LC-biotin (Sigma) as per vendor's protocol. TMB (3,3′,5,5′-tetramethylbenzidine) peroxidase substrate was purchased from Kirkegaard & Perry Laboratory Inc. (Gaithersburg, MD). Hybond ECL (enhanced chemiluminiscent) nitrocellulose membrane and the ECL Western blotting kit were from Amersham Pharmacia Biotech (BaiedUrfe, Canada). The E. coli strain BL21-CodonPlus (DE3)-RIPL was purchased from Stratagene (Cedar Creek, TX). pVAX1 mammalian expression vector and molecular cloning materials (modifying and restriction enzymes, mRNA isolation kit) were from Invitrogen (Burlington, Canada). Protein assay reagent was purchased from Bio-Rad (Mississauga, Canada). Ni-NTA agarose was purchased from Qiagen (Mississauga, Canada). Anti-His6 mAb (monoclonal antibody) was purchased from Novagen (Madison, WI). Mouse IFN-γ (Interferon gamma) ELISA (enzyme-linked immunosorbent assay) Ready-SET-Go was purchased from eBioscience (San Diego, CA). LAL (limulus amebocyte lysate) PYROGENT Plus Single Test Vials was purchased from Cambrex (Walkersville, MD). Rat antimouse CD40 mAb was prepared from the hybridoma IC10, kindly provided by Dr. M. Gold (University of British Columbia, Canada). pVHX-6, WEEV DNA encoding E1 and E2 proteins was provided by Dr. L. Nagata (Chemical Biological Defense Section, Defense R&D Canada).23 SARS-CoV membrane codon optimized DNA was purchased from GENEART. EBOV GP1,2 DNA,24 EBOV GP1,2 mammalian expressed protein, EBOV GP1,2 DNA (encoding EBOV glycoprotein 1 and 2) and SARS-CoV spike DNA (encoding SARS spike RBD, S1, S2 and transmembrane domain) were from National Microbiology Laboratory, Winnipeg, Canada. Biotinylated gangliosides (GM2 and GM3) and BSA conjugates were courtesy of Dr. D. Bundle (University of Alberta, Canada, unpublished data). Anthrax Protective Antigen (PA), extracted from the S-layer of recombinant Caulobacter crescentus, was gift of Dr. J. Smit (University of British Columbia, Canada).
2.2. Cells and Antigen Preparation
DC 2.4 was cultured at 37 °C with 5% CO2 in DMEM-10 medium [10% (v/v) FBS and 1% (v/v) PSG]. HB290 hybridoma was cultured under the same condition in HSF medium [1% (v/v) FBS and 1% (v/v) PSG]. The various primers and antigens used for the experiments are listed in Tables 1 and 2. SARS-CoV spike DNA, SARS-CoV membrane DNA, EBOV GP1,2 DNA were PCR (polymerase chain reaction) amplified using primers WP011 and WP012, WP013 and WP014, or WP015 and WP016 respectively (Table 1). These primers inserted Kozak translation initiation sequence and initiation codon (ATG) into the restriction sites BamHI and EcoRI. The PCR fragments were gel-purified, double digested with BamHI and EcoRI, ligated to pVAX1 mammalian expression vector and transformed into TOP10 cells. The positive clones were screened by restriction digestion fragment mapping (BamHI and EcoRI) and then biotinylated using photobiotin acetate.25 Core-streptavidin, WEEV E1, WEEV E2 and EBOV GP1 (subfragment D) proteins were prepared based on our published work.26–29 EBOV GP2, SARS-CoV spike S1, S2, RBD and membrane proteins were produced from E. coli expression system (unpublished data). All antigens for immunization were labeled with commercial biotin derivatives. DNA vectors were biotinylated using photobiotin acetate (B-pVHX-6, B-pEBOV GP1,2, B-pSARS-CoV spike, B-pSARS-CoV membrane) by UV photoconjugation.25 All proteins were labeled with NHS-LC-biotin (B-OVA, B-SARS-CoV spike RBD, B-EBOV GP1, B-anthrax PA). Synthetic biotinylated glycolipids (B-GM2, B-GM3, B-BSA-GM2, B-BSA-GM3) and the biotinylated cancer peptide (B-MUC-1) were synthetically labeled with biotin. Biotinylation of all antigens was confirmed by dot blot assay probed with streptavidin-HRPO.
Table 1.
PCR Primers for Cloning of HB290 scFv (VL-VH and VH-VL) and bfFp (Core-Streptavidin-VH-VL, Core-Streptavidin-VL-VH and VL-VH-Core-Streptavidin Orientations) into pET-22b (+) E. coli Periplasmic Expression Vectora
| primer | function | primer sequence (5′–3′) |
|---|---|---|
| WP001 | 5′PCR primer, HB290 VL-CL-p (strep-VL-VH) | ATC AGT GAA TTC GGG AGG TGG CGG ATC AGA CAT CCA GAT GAC ACA GTC T |
| WP002 | 3′PCR primer, HB290 VL-CL-p (strep-VL-VH) | TGG TTT CGC TCA TGC TAG GTC GAC CGT GGA TGG TGG GAA GAT AGA |
| WP003 | 5′PCR primer, HB290 VH (strep-VL-VH) | GTT AAT GTC GAC GAA GTG AAG CTG GTG GAA TCT |
| WP004 | 3′PCR primer, HB290 VH (strep-VL-VH) | TAC TAA GCG GCC GCA AGC TGA GGA GAC TGT GAC |
| WP005 | 5′PCR primer, HB290 VH-CH1-p (strep-VH-VL) | ATC AGT GAA TTC GGG AGG TGG CGG ATC AGA AGT GAA GCT GGT GGA ATC T |
| WP006 | 3′PCR primer, HB290 VH-CH1-p (strep-VH-VL) | TGG TTT CGG TCA TGA TAG GTC GAC AGC AGT TCC AGG AGC CAG T |
| WP007 | 5′PCR primer, HB290 VL (strep-VH-VL) | GTT AGG GTC GAC GAC ATC CAG ATG ACA CAG TCT |
| WP008 | 3′PCR primer, HB290 VL (strep-VH-VL) | TAC TAA GCG GCC GCA AGC CCG TTT CAA TTC CAG C |
| WP009 | 5′PCR primer, core-streptavidin (VL-VH-strep) | TACTAATGCGGCCGCGGAGGTGGCGGATCAGAGGCCGGCATCACCGGCA |
| WP010 | 3′PCR primer, core-streptavidin (VL-VH-strep) | ATTACTCTCGAGGGAGGCGGCGGACGGCTTC |
| WP011 | 5′PCR primer, pSARS-CoV spike | AAGAGGGGGATCCTACCATGGGTAGTGACCTTGACCGGTGCACCACT |
| WP012 | 3′PCR primer, pSARS-CoV spike | CTCGCTCGAGAGAATTCTATTATGTGTAATGTAATTTGACACCCTTGAG |
| WP013 | 5′PCR primer, pSARS-CoV membrane | AAGAGGGTCTTCATATGGGGGATCCTACCATGGCAGACAACGGTACTATTACCGTTGAG |
| WP014 | 3′PCR primer, pSARS-CoV membrane | CTCGCTCGAGAGAATTCTAGTGATGATGGTGGTGATGCTGTACTAGCAAAGCAATATTGT CGTT |
| WP015 | 5′PCR primer, pEBOV GP1,2 | AAGAGGGGGATCCTACCATGGGCGTTACAGGAATATTGCAGTTACCT |
These primers contain the following restriction inserts: EcoRI and SalI for WP001, 002, 005 and 006; SalI and NotI for WP003, 004, 007 and 008; NotI and XhoI for WP009 and WP010. WP011 to 16 are the PCR primers for cloning of viral DNA into the pVAX1 mammalian expression vector. These primers contain the Kozak translation initiation sequence, initiation codon, and BamHI and EcoRI restriction sites.
2.3. Cloning of HB290 scFv and bfFp
The HB290 scFv was generated from the hybridoma cell lines and DNA sequencing was done using established protocol.30 The various cloning designs of HB290 scFv and bfFp are diagrammatically shown in Figure 1. The genes were cloned in different orientations to determine the best expression in E. coli. The heavy and light chain variable regions with or without partial constant regions of anti-DEC205 antibody were amplified from plasmids containing HB290 Fab DNA by PCR and cloned into pET-22b (+) plasmids. The heavy chain variable region gene of HB290 was fused to the 3′ end of the light chain variable region gene with a linker of 15 amino acids from the constant heavy chain region 1 sequence using PCR (PCR primers: WP005 to WP008). Subsequent restriction digest/ligation methods were employed to generate the VH-VL scFv gene (Figure 1A). The PCR primers WP005 and WP006 were inserted into the restriction sites EcoRI and SalI; PCR primers WP007 and WP008 were inserted into the SalI and NotI sites. The PCR fragments were gel-purified, double digested to the respective inserted restriction sites, and ligated to pET-22b (+) plasmids. The VL-VH scFv gene was generated by the same methods utilizing PCR primers WP001 to WP004. WP001 and WP002 were inserted into the EcoRI and SalI and WP003 and WP004 were inserted into the SalI and NotI sites. The light chain variable region gene of HB290 was fused to the 3′ end of the heavy chain variable region gene via 15 amino acids of the constant light chain sequence (Figure 1B). Both scFv orientations were inserted into pET-22b (+) containing the core-streptavidin gene31 in 3′ orientation fusion with the core-streptavidin gene (Figures 1C and 1D). The VL-VH scFv gene was fused to the 5′ terminus with the core-streptavidin gene by the same method described above (Figure 1E). In brief, the core-streptavidin gene was PCR amplified using WP009 and WP010 primers. These primers were inserted into restriction sites NotI and XhoI. The PCR fragment was gel-purified, double digested with NotI and XhoI, and ligated to pET-22b (+) containing the VL-VH scFv gene (Figure 1E). All clones were screened and characterized by both PCR and restriction digestion fragment mapping. The positive cloned fragment was sequenced using T7 promoter and terminator primers employing CEQ2000. The positive clones were designated as follows: (a) pWET5 encoding core-streptavidin-VH-VL, (b) pWET6 encoding core-streptavidin-VL-VH and (c) pWET7 encoding VL-VH-core-streptavidin.
Figure 1.
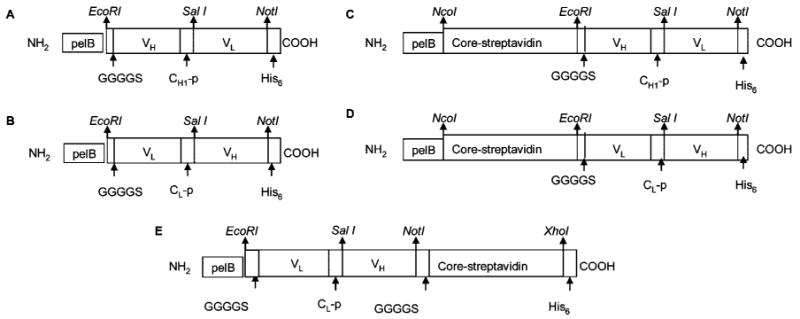
Various constructs of HB290 scFv [(A) VH-VL and (B) VL-VH] and bfFp [(C) pWET5, (D) pWET6 and (E) pWET7]. The genes were cloned in different orientations to select the best production in E. coli. Abbreviations: pelB, bacterial leader sequence; VL, variable domain light chain; VH, variable domain heavy chain; CH1-p, partial constant heavy chain region 1, 15 amino acid linker; CL-p, partial constant light chain region, 15 amino acid linker; G, glycine; S, serine; His6, hexa histidine amino acid tag.
2.4. Expression, Purification and Characterization of HB290 Recombinant Proteins
2.4.1. Expression of HB290 scFv and bfFp
Expressions of scFv and bfFp were done according to the previously published methods.31 The pET-22b (+)-scFv or bfFp genes (pWET5–7) were chemically transformed into BL21-CodonPlus (DE3)-RIPL. The E. coli transformants were cultured, induced for protein expression and the whole-cell bacterial pellets were analyzed by Western blot. The pellets were resuspended in reducing SDS dye (50 mM Tris-HCl, pH 6.8, 2% SDS, 0.1% bromophenol blue, 10% glycerol, 5 mM 2-mercaptoethanol) and heated at 95 °C for 10 min prior to SDS–PAGE. The pellets were electrophoresed and transferred to a Hybond ECL nitrocellulose membrane using the Trans blot apparatus (Bio-Rad). The membrane was then blocked with 5% skim milk, probed with mouse anti-His6 mAb and GAM-HRPO, and revealed by ECL according to the manufacturer's protocol. The bfFp gene with the highest protein expression level was selected for medium scale expression in culture flask and affinity purification was performed from E. coli periplasmic extracts using IMAC purification protocol as described previously.31 The induced periplasm and IMAC purified fractions were analyzed by SDS–PAGE using 10% polyacrylamide gels under reducing conditions following staining with Coomassie brilliant blue. The fractions were heated at 95 °C for 10 min prior to loading on the polyacrylamide gel.
2.4.2. Western Blot: Biotin Binding and Heat Stability of bfFp
IMAC purified bfFp was heated at either 60 or 95 °C for 10 min under reducing conditions and resolved in 10% SDS–PAGE. The resolved proteins were electrophoretically transferred onto a nitrocellulose membrane and probed with B-BSA followed by streptavidin-HRPO.
2.4.3. ELISA: bfFp Bispecificity and Binding to DC 2.4 Cells
DC 2.4 cells were seeded on a 96-well V-bottomed plate (Nunc, Denmark) in quadruplicate (1.0 × 105 cells/well). The plates were washed with PBS (phosphate buffer saline) and blocked with 1% PBS dialyzed BSA (to remove traces of biotin) for 3 h at 4 °C. After incubation, the plates were washed with PBS, and 100 μL of bfFp at 40 μg/mL was added to bind DEC-205 receptors. The plates were incubated at 4 °C for 3 h, then washed with PBS and B-OVA (20 μg/mL in 100 μL volume) was added to each well and incubated for 1 h at 4 °C. After the incubation, the plates were washed and then incubated with streptavidin-HRPO (10 μg/mL in 100 μL volume) for 1 h at 4 °C. Finally, the plates were washed with PBS, and TMB substrate was then added. The OD650 nm was taken after 15 min using an ELISA Vmax kinetic microplate reader (Molecular Devices Corp, CA) and subtracted with control (DC with only streptavidin-HRPO incubated at 4 °C for 1 h). The DEC-205 receptor binding specificity of bfFp was confirmed by competition study with full-length HB290 mAb. Various concentrations of HB290 mAb (50, 100, 200, 300 μg/mL in 100 μL volume) was added after bfFp binding to DEC-205 at 4 °C. The full length HB290 mAb was incubated for 2 h at 4 °C to allow for displacement of bfFp from DC. Following a wash with PBS the bfFp was detected using B-OVA, streptavidin-HRPO and TMB as mentioned above.
2.5. In Vivo Targeting of DC in Mice
2.5.1. Mice and Immunization Protocol
Fifteen groups of female BALB/c mice (5 mice per group, average 6–8 weeks old) were obtained from Health Sciences Laboratory Animals Services of the University of Alberta, Edmonton, Canada. Animal treatment, care and euthanasia were carried out according to the Canadian Council of Animal Care guidelines. Mice were injected subcutaneously near the inguinal lymph node area with 0.1 mL of various formulations of antigens and bfFp in saline. The immunization protocol is listed in Table 3. Three separate animal experiments were conducted and each design has its own control group. The strategy was to test the versatility of bfFp based delivery of protein, peptide, glycolipids and DNA to DC in generating immune responses. Immunization of B-pVHX-6, B-OVA, B-MUC-1, B-GM2 and GM3 were performed in the first experiment (Groups 2–7). Animals in Group 2–4 were injected with various biotinylated DNA formulations to verify the essential requirement for DC targeting vehicle and costimulatory molecule. Group 2 mice were immunized with B-pVHX-6 and bfFp without the presence of anti-CD40 mAb to verify the essential role of anti-CD40 mAb costimulation. Group 3 mice were coimmunized with anti-CD40 mAb without the bfFp targeting vector to determine the nontargeted immune responses. Group 4 mice were coimmunized with both bfFp and anti-CD40 mAb. Protein antigen delivery was studied in Group 5 mice and peptide delivery was focused in Group 6 mice. Lastly, glycolipid antigens were investigated in Group 7. The second experiment was designed to confirm the versatility of the naked DNA delivery strategy. A variety of infectious disease viral DNA were targeted as a single antigen to DC using the bfFp mediated delivery system (Group 9, EBOV GP1,2 DNA; Group 10, SARS-CoV spike DNA; Group 11, SARS-CoV membrane DNA). Multivalent immune responses against a mixture of different class antigens were studied in the third experiment. Groups 13–15 were designed to evaluate the multivalent immune responses against proteins and peptide, B-OVA, B-EBOV GP1, B-SARS-CoV spike RBD, B-MUC-1 and B-anthrax PA immunized as a mixture in saline. Group 13 mice were coimmunized with anti-CD40 mAb and core-streptavidin without the bfFp to determine the level of nontargeted immune responses. Group 14 mice were coimmunized with bfFp without the presence of anti-CD40 mAb to verify the essential role of anti-CD40 mAb costimulation. Group 15 mice were immunized with antigens, bfFp and anti-CD40 mAb. Groups 1, 8 and 12 were the control groups for the three separate animal experiments. All mice were boosted with the same concentration of antigen(s) in each category in PBS 12 days following primary immunization. Prior to immunization, every reagent (antigens, bfFp and mAb) was checked using LAL PYROGENT Plus Single Test Vials kits to identify lipopolysaccharide (LPS) contamination if any (endotoxin sensitivity at 0.125 EU/mL). The immunization reagents were premixed for 30 min at RT prior to immunization. The mice were sacrificed after 9 days of boost, and the blood and the spleen were collected. The serum was isolated using standard procedures32 and used to evaluate humoral immune responses. Spleens were used to study IFN-gamma immune responses. The spleens from the 15 different groups of immunized mice were aseptically removed and each group was pooled. The responder cells were isolated using nylon wool columns and the stimulator cells from a separate group of naïve mouse spleens and were treated with mitomycin C as previously described.20
Table 3.
Immunization Protocol for Evaluating Humoral and Cell-Mediated Immune Responses to Biotinylated Antigens in Mice (n = 5 for Each Group)a
| groups | day 0 | day 12 | day 21 | day 24 | |
|---|---|---|---|---|---|
| 1 | control | PBS | PBS | spleen (T cell) | IFN-γ assay |
| serum (humoral) | |||||
| 2 | B-pVHX-6 | 500 ng | 500 ng | spleen (T cell) | IFN-γ assay |
| bfFp | 20 μg | 0 | serum (humoral) | ||
| 3 | B-pVHX-6 | 500 ng | 500 ng | spleen (T cell) | IFN-γ assay |
| anti-CD40 mAb | 25 μg | 0 | serum (humoral) | ||
| 4 | B-pVHX-6 | 500 ng | 500 ng | spleen (T cell) | IFN-γ assay |
| bfFp | 20 μg | 0 | serum (humoral) | ||
| anti-CD40 mAb | 25 μg | 0 | |||
| 5 | B-OVA | 200 ng | 200 ng | spleen (T cell) | IFN-γ assay |
| bfFp | 20 μg | 0 | serum (humoral) | ||
| anti-CD40 mAb | 25 μg | 0 | |||
| 6 | B-MUC-1 | 200 ng | 200 ng | spleen (T cell) | IFN-γ assay |
| bfFp | 20 μg | 0 | serum (humoral) | ||
| anti-CD40 mAb | 25 μg | 0 | |||
| 7 | B-GM3 | 1 μg | 1 μg | spleen (T cell) | IFN-γ assay |
| B-GM2 | 1 μg | 1 μg | serum (humoral) | ||
| bfFp | 20 μg | 0 | |||
| anti-CD40 mAb | 25 μg | 0 | |||
| groups | day 0 | day 12 | day 21 | day 24 | |
| 8 | control | PBS | PBS | spleen (T cell) | IFN-γ assay |
| serum (humoral) | |||||
| 9 | B-pEBOV GP1,2 | 500 ng | 500 ng | spleen (T cell) | IFN-γ assay |
| bfFp | 20 μg | 0 | serum (humoral) | ||
| anti-CD40 mAb | 25 μg | 0 | |||
| 10 | B-pSARS-CoV spike | 500 ng | 500 ng | spleen (T cell) | IFN-γ assay |
| bfFp | 20 μg | 0 | serum (humoral) | ||
| anti-CD40 mAb | 25 μg | 0 | |||
| 11 | B-pSARS-CoV membrane | 500 ng | 500 ng | spleen (T cell) | IFN-γ assay |
| bfFp | 20 μg | 0 | serum (humoral) | ||
| anti-CD40 mAb | 25 μg | 0 | |||
| groups | day 0 | day 12 | day 21 | day 24 | |
| 12 | control | PBS | PBS | spleen (T cell) | IFN-γ assay |
| serum (humoral) | |||||
| 13 | B-OVA | 200 ng | 200 ng | spleen (T cell) | IFN-γ assay |
| B-EBOV GP1 | 200 ng | 200 ng | serum (humoral) | ||
| B-SARS-CoV spike RBD | 200 ng | 200 ng | |||
| B-MUC-1 | 200 ng | 200 ng | |||
| B-anthrax PA | 200 ng | 200 ng | |||
| anti-CD40 mAb | 25 μg | 0 | |||
| core-streptavidin | 10 μg | 0 | |||
| 14 | B-OVA | 200 ng | 200 ng | spleen (T cell) | IFN-γ assay |
| B-EBOV GP1 | 200 ng | 200 ng | serum (humoral) | ||
| B-SARS-CoV spike RBD | 200 ng | 200 ng | |||
| B-MUC-1 | 200 ng | 200 ng | |||
| B-anthrax PA | 200 ng | 200 ng | |||
| bfFp | 20 μg | 0 | |||
| 15 | B-OVA | 200 ng | 200 ng | spleen (T cell) | IFN-γ assay |
| B-EBOV GP1 | 200 ng | 200 ng | serum (humoral) | ||
| B-SARS-CoV spike RBD | 200 ng | 200 ng | |||
| B-MUC-1 | 200 ng | 200 ng | |||
| B-anthrax PA | 200 ng | 200 ng | |||
| bfFp | 20 μg | 0 | |||
| anti-CD40 mAb | 25 μg | 0 | |||
The amounts of antigens, antibodies and bfFp are either in μg or ng per mouse. All mice were injected subcutaneously near the inguinal lymph node. Three separate experiments were conducted in 15 groups of mice and each experiment had its own control group. Groups 1, 8 and 12 were the control groups. The first experiment demonstrated the versatility of the bfFp based delivery of protein, peptide, glycolipids and DNA to DC in generating immune responses (Group 2–7 mice). The second experiment was designed to confirm the DNA delivery strategy of targeting varies infectious disease viral DNA to DC (Groups 9–11). Multivalent immune responses against a mixture of antigens (proteins and peptide) were studied in the third set of experiments (Groups 13–15). All mice were boosted with the same concentrations of antigen(s) in PBS 12 days following primary immunization. The mice were sacrificed after 9 days of boost.
2.5.2. Evaluation of Humoral Immune Responses and IFN-γ
Antibody titers were measured individually in each mouse by ELISA following immunization of the respective antigen (Table 2). The ELISA method was done by overnight coating of the specific antigen in the Nunc 96-well ELISA microplates (10 μg/mL in 100 μL volume). After overnight coating, the plates were washed with PBST (0.1% Tween 20 in PBS, pH 7.3) and the plates were blocked with BSA for 3 h at RT. After incubation, the plates were washed with PBST and the 1:1000 diluted serum from each mouse in quadruplicate was added, incubated for 2 h at RT. Plates were then washed with PBST and incubated with GAM-HRPO for 1 h at RT. After 1 h, the plates were washed again with PBST and TMB was then added to each well and OD650 nm was taken after 10 min using the microplate reader. IFN-γ activity was determined by the IFN-γ concentration generated after 3 days of incubation of the responder cells (2.5 × 105 cells) and/or stimulator cells (3 × 105 cells) with the respective antigens: OVA, SARS-CoV spike RBD, EBOV GP1, EBOV GP2, EBOV GP1,2, SARS-CoV spike S1, SARS-CoV spike S2, SARS-CoV spike RBD, SARS-CoV membrane antigen, anthrax PA, B-MUC-1, B-GM2, B-GM3, WEEV E1 and WEEV E2 (10 μg of each antigen was separately incubated). IFN-γ ELISA Ready-SET-Go kit was used to determine the IFN-γ concentration.
2.5.3. Immune Responses toward Biotin, bfFp and Core-Streptavidin
The 15 groups of mice serum were tested against B-BSA, bfFp and core-streptavidin using ELISA method. The serum reactivity was determined by the same method as described in section 2.5.2. The plates were coated with either B-BSA, bfFp or core-streptavidin (10 μg/well). The rest of the procedures followed the preceding sections.
3. Results
3.1. Construction, Expression and Purification of HB290 scFv and bfFp
The genes encoding HB290 Fab were generated by RT-PCR and TD-PCR. The sequence generated was compared with the known mAb sequences in the NCBI BLAST database indicating the sequence is antibody related. The HB290 VL amino acid sequence from our recombinant clone was 100% identical to a previous report;33 however, HB290 VH amino acid sequence (EVKLVESGGGLVQPGGSLRLSCAASGFTFNDFYMNWIRQPPGQAPEWLGVIRNKGNGYTTEVNTSVKGRFTISRDNTQNILYLQMNSLRAEDTAIYYCARGGPYYYSGDDAPYWGQGVMVTVSS) shared only 46% homology. The VH amino acid sequence appears to be unique from other published amino acid sequences in the NCBI protein database; whereas, the amino acid sequence published by Demangel et al. is 99% identical (only a two amino acid difference in the 5′ terminal) with the single chain antibody against rice stripe virus protein P20 (accession: AAG28706). The HB290 VL-VH and VH-VL genes were successfully cloned into separate pET-22b (+) plasmids by PCR, restriction digest and DNA ligation methods. The plasmid vectors pWET5 and pWET6 were constructed by inserting the sequence encoding the HB290 single-chain anti-DEC205 antibody (VL-VH or VH-VL respectively) next to the pelB leader sequence of core-streptavidin containing pET22b (+) plasmids (Figure 1A–D). The plasmid vector pWET7 was constructed by inserting the core-streptavidin sequence into HB290 VL-VH scFv containing pET22b (+) plasmids (Figure 1B and 1E). The core-streptavidin sequence is at the N-terminus of the scFv in pWET5 and pWET6 constructs; whereas, in pWET7 the core-streptavidin is at the C-terminal. The scFv and bfFp vectors were transformed into E. coli, cultured, induced and the whole-cell bacterial pellets were analyzed by Western blot using the anti-His6 mAb (Figure 2A). The optimal scFv and bfFp orientation for expression were determined by Western blot. HB290 VL-VH scFv, pWET6 and pWET7 were successfully expressed and the proteins are shown at the desired molecular weight band either at ∼30 kDa or ∼46 kDa (Figure 2A). pWET7 had a higher level of protein expression compared to pWET6; therefore, pWET7 was subjected to medium scale expression. The bfFp in the periplasmic space was extracted and affinity purified by IMAC column. Both the periplasmic fraction and the affinity purified fraction were analyzed on SDS–PAGE (Figure 2B). IMAC purification of the pWET7 bfFp was successful; a clear band was seen at 46 kD in the affinity purified fraction (Figure 2B). Approximately 1.0 mg of pWET7 bfFp was affinity purified from a 2 L culture.
Figure 2.
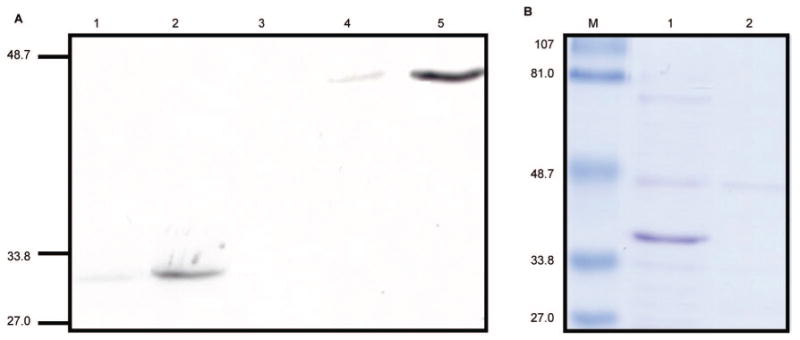
Expression and purification of recombinant proteins. The scFv and bfFp genes were chemically transformed, cultured, induced and the whole-cell bacterial pellets were analyzed by Western blot. (A) Western blot probed with anti-His6 mAb for analysis of scFv and bfFp expression. Lane 1: HB290 scFv VH-VL. Lane 2: scFv VL-VH. Lane 3: bfFp pWET5, core-streptavidin-VH-VL. Lane 4: bfFp pWET6, core-streptavidin-VL-VH. Lane 5: bfFp pWET7, VL-VH-core-streptavidin. pWET7 had the highest protein expression level. (B) SDS–PAGE of pWET7 before and after IMAC purification. Lane 1: pWET7 periplasmic protein. Lane 2: IMAC-purified pWET7. M stands for molecular weight marker in kDa.
3.2. Characterization of bfFp
E. coli expressed bfFp appeared in 3 isoforms: monomeric, dimeric and tetrameric form.34 Following affinity purification by either IMAC or iminobiotin column, the fusion protein was subjected to size exclusion chromatography for isolation of the tetrameric fusion protein.34,35 Fusion proteins are known to be heat sensitive and heating at 95 °C dissociates the tetramer completely into monomeric forms. The tetrameric form remains stable at temperature below 60 °C.34 The bfFp isoforms and the biotin binding activity of the bfFp were analyzed by Western blot probed with B-BSA. pWET7 bfFp was detected using B-BSA and streptavidin-HRPO in Western blot and the bfFp appear predominantly in monomeric form after heating at either 60 or 95 °C (Figure 3A). The predominant monomeric form may be due to the difference in the linker between core-streptavidin and scFv.31
Figure 3.
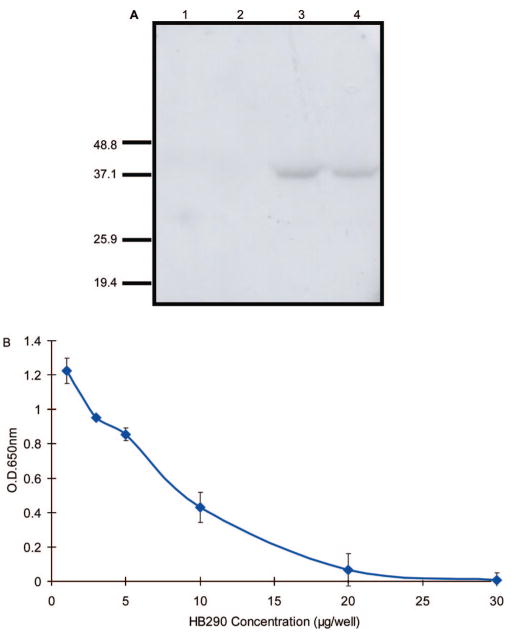
Demonstration of IMAC-purified bfFp bifunctional activity and thermal stability. (A) IMAC purified bfFp was incubated either at 60 °C (lane 3) or 95 °C (lane 4) for 10 min and probed with B-BSA followed by streptavidin-HRPO. Lanes 1 (HB290 scFv VH-VL) and 2 (HB290 scFv VL-VH) are controls. The IMAC-purified bfFp bifunctional activity was tested against DC 2.4 cells and B-OVA by ELISA. (B) Competition binding studies of HB290 full length mAb and bfFp for binding to DC. The binding of bfFp is confirmed by B-OVA and streptavidin-HRPO. The error is depicted by the standard deviations.
The bispecificity of the bfFp was confirmed by cell ELISA. The anti-DEC-205 activity was demonstrated on DC 2.4 cells employing B-OVA with streptavidin-HRPO for detection. In addition, specific DEC-205 receptor binding activity was also confirmed by competitive displacement of bfFp with increasing concentrations of HB290 mAb (Figure 3B). In addition confocal study was done to demonstrate DC binding (unpublished data).
3.3. bfFp Mediated Immune Responses: Humoral and IFN-γ
Four different classes of antigens were chosen to demonstrate the versatility of in vivo antigen targeting. Both humoral and cell-mediated responses were investigated using a variety of antigens [proteins,24,36–39 peptide,40,41 glycolipid42,43 and DNA24,36,37,39,44–47] listed in Table 2. The immunization protocol is described in Table 3, and the results of humoral and cell-mediated responses are shown in Figures 4 and 5. Humoral responses were measured by the antibody titers against the immunized antigens or its respective proteins from the DNA vectors in each mouse by ELISA method. The magnitude of cell-mediated immune responses was determined by the amount of IFN-γ secreted from spleen T cells in response to the antigens. The spleen cells from each group were pooled to average the quality of data for IFN-γ secretion. Responder cells from both immunized and nonimmunized mice without stimulator cells had minimal IFN-γ secretion (data not shown). The 15 groups of mice were divided into three different experiments to demonstrate the ability and efficacy of DC targeting strategies. Group 1, 8 and 12 were the control groups for the three separate animal experiments and these groups were immunized and boosted with PBS only. In these groups both humoral and cell-mediated immune responses were minimal (Figure 4 and 5).
Figure 4.
Analysis of humoral immune responses to biotinylated antigens in vivo. Groups of five mice were immunized with different antigen combinations along with a PBS control. The specific components, amounts, and the schedule of immunizations are outlined in Table 3. A simplified form of the immunization components is listed in the figure legend. (A) Versatility of bfFp based delivery of protein, peptide, glycolipids and DNA to DC. (B) bfFp mediated delivery of other infectious diseases viral DNA to DC. (C) Multiple antigen delivery to DC using bfFp. The mice were analyzed individually, and the data was pooled. The humoral response, as quantified by serum antibody titers on day 21 post immunization, was measured by ELISA method against the respective antigens shown in X-axis (listed in Table 2). The method involved coating of 100 μL of 10 μg/mL antigen in microtiter plates followed by addition of 1:1000 diluted serum antibody, and detection using GAM-HRPO. The ELISA measurements were done in quadruplicate for each mouse. The mean ELISA values obtained for each individual mouse were further averaged. The error bars represent the standard deviation in a group of 5 mice.
Figure 5.
Analysis of cell-mediated immune responses based on IFN-γ estimation. (A) Versatility of bfFp based delivery of protein, peptide, glycolipids and DNA to DC. (B) bfFp mediated delivery of other infectious diseases viral DNA to DC. (C) Multiple antigen delivery to DC using bfFp. Spleen T cells (responder cells) from the groups of mice immunized with different antigens (a simplified form of the immunization components is listed in the figure legend, for details see Table 2 and 3) were isolated and purified using a nylon wool column. The five spleens in each group were pooled and mixed in DMEM prior to nylon wool purification. Stimulator cells prepared from naïve mice spleen cells were isolated and treated with mitomycin C. Purified responder cells from both immunized and nonimmunized mice were aliquoted in quadruplicate with or without stimulator cells. The cells were then incubated with 10 μg of antigen (shown in X-axis) for 3 days at 37 °C in a CO2 atmosphere. After incubation, the IFN-γ concentration in the supernatant was determined using mouse IFN-γ ELISA Ready-SET-Go kit. Each data set is shown following subtraction of the corresponding ELISA values obtained without stimulator cells. The error bars are the standard deviations.
3.3.1. bfFp Mediated Protein, Peptide, Ganglioside and DNA Targeting to DC
Groups 2–7 were designed to demonstrate the versatility of bfFp based delivery of distinct classes of antigens to DC in generating immune responses. Group 2–4 mice focused on the naked DNA delivery to DC and to verify the essential requirement of bfFp and anti-CD40 mAb. Group 2 mice were immunized with B-pVHX-6 and bfFp without the presence of anti-CD40 mAb. Group 3 mice were coimmunized with anti-CD40 mAb without the bfFp based targeting. Group 4 mice were coimmunized with both bfFp and anti-CD40 mAb. The results indicate that Group 4 had the highest antibody titer and augmented IFN-γ secretion against the WEEV E1 and WEEV E2 proteins compared to Groups 1–3 (Figures 4A and 5A). Immune responses against WEEV E2 appeared to be higher than WEEV E1 in Group 4. Immune responses in Groups 2 and 3 were minimal toward WEEV E1 and E2 protein antigens, and were probably due to LPS, anti-CD40 mAb or bfFp effect (Figures 4A and 5A). Anti-CD40 mAb and bfFp appeared to be essential for DNA targeting strategy. The similar immunization strategy was applied to the synthetic biotinylated peptide MUC-1 and glycolipids (GM2 and GM3). Ovalbumin was also included in our experiments as a model antigen and control. Humoral responses and cell-mediated immune responses were shown in mice immunized with protein (Group 5), peptide (Group 6) and glycolipids (Group 7) in the presence of bfFp and anti-CD40 mAb (Figures 4A and 5A). In summary, the results show that anti-CD40 mAb and bfFp are required to achieve good immune responses based on DC delivery of DNA, protein, peptide and even gangliosides.
Direct biotinylated DNA targeting to DC also generated strong immune responses against the respective DNA encoding proteins. The same targeting strategy was applied for a variety of infectious disease DNA. Groups 9–11 were immunized with different naked DNA vectors encoding genes for different viral proteins (Group 9, EBOV GP1,2 DNA; Group 10, SARS-CoV spike DNA; Group 11, SARS-CoV membrane DNA). pEBOV GP1,2 encoded the EBOV GP1 and GP2 proteins, pSARS-CoV spike encoded the SARS-CoV S1, S2, RBD and the transmembrane domain, and pSARS-CoV membrane encoded the SARS-CoV membrane protein. Strong humoral and cell-mediated responses were achieved against the encoded viral proteins by targeting the viral DNA to DC (Figures 4B and 5B). EBOV GP1 generated the highest immune responses compared to GP2 and GP1,2. The immune responses against mammalian expressed GP1,2 protein were lower than the E. coli expressed fragments (GP1 and GP2) (Figures 4B and 5B). The immune response against EBOV GP1,2 was relatively lower than its E. coli fragment. This may be due to the glycosylation masking of the epitopes.48 There was no significant difference in serum titer between SARS-CoV spike proteins, but higher IFN-γ concentrations were found in SARS-CoV RBD.
3.3.2. bfFp Mediated Multiple Antigen Targeting Strategy for Combo-Vaccines
Groups 13–15, were designed to evaluate multivalent immune responses by simultaneously targeting a mixture of biotinylated proteins and peptide antigen in nanogram concentrations. Group 13 reflects the possible involvement of core-streptavidin targeting of biotinylated proteins and peptide to DC. Groups 14 and 15 demonstrated the efficacy of the bfFp in the absence and presence of the anti-CD40 mAb costimulator respectively. The results show that Group 15 has the highest antibody titer and augmented IFN-γ secretion against OVA, SARS-CoV RBD, anthrax PA and MUC-1 compared to Groups 12, 13 and 14 (Figures 4C and 5C). Groups 13–15 showed minimal immune responses against EBOV GP1 (Figures 4C and 5C). In Group 15, the highest antibody titer was found against OVA and the lowest against anthrax PA (Figure 4C); however, the highest IFN-γ secretion was found against anthrax PA (Figure 5C). Minimal humoral or cell-mediated immune responses were generated from Groups 12–14 (Figures 4C and 5C). MUC-1 and OVA immune responses were compared between the single antigen and multiple antigen targeting strategies. Higher IFN-γ secretion and serum titer against MUC-1 were achieved in the single antigen targeting strategy of MUC-1 peptide antigen (Group 6) compared to the multiple antigen targeting strategy (Group 15) (Figures 4A, 4C, 5A, 5C). Although the serum titer against OVA was not different between Groups 5 and 15 (Figures 4A and 4C), Group 5 had higher IFN-γ secretion compared to Group 15 (Figures 5A and 5C). To summarize, both bfFp and anti-CD40 mAb are required to achieve strong immune responses against protein and peptide delivered to DC. Single antigen targeting appears to achieve strong humoral and cell-mediated immune responses in comparison to multiple antigen targeting strategy. Immune responses were shifted or selective in the multiantigen targeting method, possibly toward immunodominant antigens.
3.4. Immunogenicity of bfFp and Biotin Hapten
Serum titer against biotin, bfFp and core-streptavidin were analyzed from every group to evaluate the immunogenicity of the bfFp targeting vehicle. Identical concentrations of bfFp and core-streptavidin were coated onto the plate. Serum reactivity was found to some extent against core-streptavidin (OD ∼0.25) and bfFp (OD ∼0.15). However, this level of serum reactivity is not different compare to the control groups (Figure 6). The serum reactivity toward B-BSA was minimal in all groups (OD ∼0.01).
Figure 6.
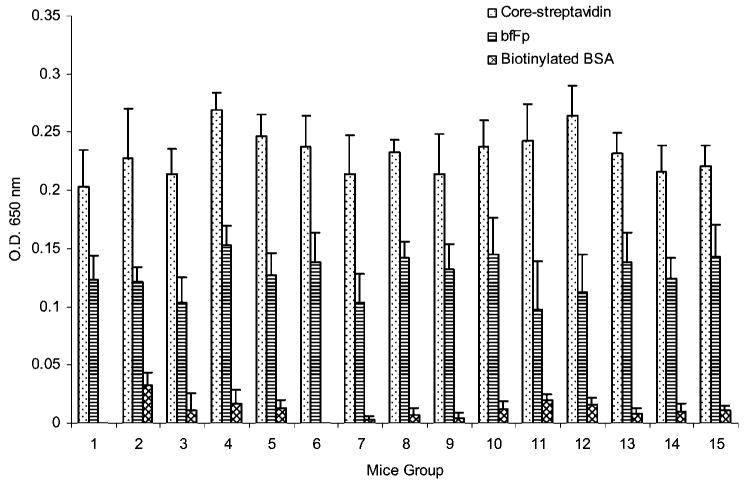
Analysis of serum reactivity toward biotin, bfFp and core-streptavidin. The mice (5 mice per group) were analyzed individually, and the data was pooled. The mouse groups are listed on the X-axis and correspond to Table 3. Groups 1, 8 and 12 were the control groups with only PBS immunization. Other groups have been immunized in the presence or absence of the bfFp (according to Table 3). The serum reactivity, as quantified by serum antibody titers on day 21 post immunization, was measured by ELISA method against B-BSA, bfFp and core-streptavidin (shown in the figure legend). The method involved coating of 100 μL of 10 μg/mL antigen (shown in the figure legend) in microtiter plates followed by addition of 1:1000 diluted serum antibody, and detection using GAM-HRPO. The ELISA measurements were done in quadruplicate for each mouse. The mean ELISA values obtained for each individual mouse were further averaged. The error bars represent the standard deviation in a group of 5 mice.
4. Discussion
In vivo targeting of the DC DEC-205 receptor has been shown to be effective in generating immune responses to protect the host against cancer, viral infection and autoimmune disease. Four types of DEC-205 targeting systems have been reported thus far. These include (a) HB290 scFv coated liposome,4 (b) chemical cross-linking of HB290 with antigen,13,15,17,18 (c) HB290 hybrid antibody14,16,19 and (d) bsmAb targeting system.20 Several limitations are seen in these systems. Currently, the above strategies have only demonstrated efficient delivery of peptides or proteins to DC. These delivery systems may not be versatile or flexible enough for clinical applications, comparison studies with different antigens, and in vivo targeting of multiple antigens such as DNA and glycolipids. Limitations of the scFv coated liposome targeting strategy include instability of the scFv on the liposome surface (scFv could be lost during circulation), liposome instability, scale-up issues, relative complexity in the encapsulation process and only certain class of antigens can be encapsulated efficiently. Additional issues include batch to batch variation (cross-linking and purification) and Fc domain mediated effects, which are limitations of the chemical cross-linking of the whole mAb with antigen. Hybrid antibody or recombinant antigen fusion protein generation using a mammalian expression system is labor intensive requiring careful monitoring of cell productivity, post-translational chemical modifications, degradation and aggregation of the final product.49 Furthermore, a new antibody fusion protein is required for each antigen (protein or peptide) and the process is often time-consuming and costly. The bsmAb produced from a quadroma is not ideal for clinical application and the limitations include the yield and purity.20 The major disadvantage for these systems could be dose-limiting toxicity (prolonged circulation time) and immunogencity against the targeting system, which can alter pharmacokinetics (biodistribution and clearance), block receptor-antigen interactions, and induce hypersensitivity reactions and injection site reactions.49 Consequently, a truncated monomeric single-chain antibody-core-streptavidin fusion protein (bfFp) that can form a complex with any biotinylated antigen (Kd ∼ 10−15 M) with one paratope and deliver the antigen to the DC via the DEC-205 receptor with its second paratope may be a desired alternative for in vivo DC targeting. This system has several unique properties over the other targeting systems. Almost any class of antigen can be biotinylated by chemical conjugation (for instance, NHS-LC-biotin), photoactivation (photobiotin acetate) or incorporation by synthetic strategies. Biotinylation avoids the need of encapsulation, chemical cross-linking, or construction of a new hybrid fusion antibody. The bfFp lacks the Fc domain and the E. coli based production is consistent and economical. Faster clearance rate (kidney glomerular filtration cutoff is 70 kDa) and lower immunogenicity is expected due to the smaller molecular weight (∼46 kDa) of the vector. If required, bfFp stability and half-life can be increased by polyethylene glycol linkage50 or by isolation of the tetrameric form of the bfFp. Moreover, such a targeting vehicle can be translated into clinical applications.51,52
In this communication, we have developed a versatile bfFp that can bind any biotinylated antigen and target them to DC DEC-205 receptors using recombinant antibody technology. DEC-205 receptor binding specificity was also confirmed via competitive binding replacement of bfFp with the full length HB290 mAb study. In vivo studies in mice with biotinylated DNA (WEEV, EBOV GP1,2, SARS-CoV spike, SARS-CoV membrane), protein (OVA), peptide (MUC-1) or glycolipid (GM2 and GM3) have shown that in the presence of bfFp and anti-CD40 mAb as costimulator, both humoral and cell-mediated responses can be augmented. In the multiple antigen targeting strategy, we have also achieved humoral and cell-mediated responses for OVA, SARS-CoV spike RBD, MUC-1 and anthrax PA. Most antigens were administered at the very low doses of 200 ng by our DC targeting strategy. With respect to the above data, an in vivo fluorescence imaging would decisively establish the specificity for in vivo targeting and we propose to do this in the future. Such antigen sparing strategies are important to pandemic disease management if large populations need vaccination. We have made theoretical calculations that suggest that in the event of a SARS or H5N1 avian influenza outbreak in Canada, the DC targeted vaccine needed to cover 33 M subjects require only 7 g (at 200 ng dose) of the vaccine antigen. In comparison, a recent commercial influenza vaccine clinical trial used 90 μg/patient with modest immune responses requires 3 kg of antigen (450-fold more). Our speculation is that DC targeting more efficiently delivers the antigen to the DC than the conventional subcutaneous vaccine that could encounter prolonged exposure to enzymes and uptake by other none DC cells. Another important application is the delivery of multiple pathogen antigens for combination vaccines that could provide sufficient protection for first emergency responders and biodefense applications.
In summary, our DC targeting approach achieves strong immune responses against all classes of antigen. Single antigen targeting exhibited strong humoral and cell-mediated immune responses. In comparison, multiantigen targeting strategy likely exhibited immune responses that are shifted toward the most immunogenic antigen. Higher immune responses were achieved for weak immunogens (MUC-1 and OVA) in single antigen targeting strategy compared to multivalent strategy. We have also demonstrated that DEC-205 targeting of bioterrorism agent DNA's (EBOV GP1,2, SARS-CoV spike, WEEV structural DNA) induces strong humoral and cell-mediated immune responses against the DNA encoded proteins. In this targeting formulation, low concentrations of biotinylated DNA (500 ng) in saline were adequate to achieve a strong immune response in mice. In contrast, conventional DNA vaccine or therapy strategy often requires high concentration and multiple immunizations, and cannot efficiently induce immune responses. More importantly, the widely used experimental viral vector based gene targeting strategies have caused severe adverse side-effects including death. We speculate that in the absence of such viral vector the naked biotinylated DNA targeting could be a safer alternative. In vitro and in vivo neutralization study of SARS-CoV and EBOV and challenge study of WEEV are in progress. The immune responses toward either the hapten biotin (B-BSA) or the carrier bfFp is minimal and can be further mitigated by deimmunization strategies. The slight reactivity toward bfFp appears to be against the core-streptavidin portion of the fusion protein. The bfFp targeting of biotinylated antigens to DC could be a convenient, versatile method to deliver single or multiple antigens to DC. Such formulations could induce immune responses toward peptide, protein, glycolipid, and DNA vaccine based expression products. bfFp targeting of DC may be a potential candidate for the development of vaccine or therapy for cancer (GM3, GM2, MUC-1) and bioterrorism agents (WEEV, EBOV, SARS-CoV, anthrax).
Translation of the in vivo targeting strategy to humans is likely to be more complex since DEC-205 receptors in humans may also target B cells, NK cells, T cells, monocytes and thymic epithelial cells. In addition, multiple immunizations may be required to achieve adequate immune responses in human. In fact even in our mouse model, the bfFp is only injected once followed by nontargeted booster dose.
Acknowledgments
W.W.W. would like to thank Rx&D/CIHR and Dr. Mike Wolowyk for financial support. M.R.S. would like to thank CIHR and NIH (No. 1U01A1061-233-01) for grant support and various donors of the antigens.
Abbreviations Used
- bsmAb
bispecific or bifunctional monoclonal antibody
- scFv
single chain antibody
- bfFp
bifunctional fusion protein
- EBOV
Ebola virus
- SARS-CoV
severe acute respiratory syndrome-coronavirus
- GP
glycoprotein
- RBD
receptor binding domain
- PA
protective antigen
- MUC-1
epithelial mucin 1
- ATCC
American type culture collection
- B-BSA
(biotin)n labeled BSA
- B-OVA
(biotin)n labeled OVA
- B-MUC-1
synthetically biotin labeled MUC-1
- B-SARS-CoV spike RBD
(biotin)n labeled SARS-CoV spike protein
- B-EBOV GP1
(biotin)n labeled EBOV GP1
- B-anthrax PA
(biotin)n labeled anthrax PA
- B-GM2
synthetically biotin labeled GM2
- B-BSA-GM2
synthetically biotin labeled BSA and GM2
- B-GM3
synthetically biotin labeled GM3
- B-BSA-GM3
synthetically biotin labeled BSA and GM3
- B-pVHX-6
photoactiviated biotin labeled WEEV DNA vector
- B-pEBOV GP1,2
photoactiviated biotin labeled EBOV GP1,2 DNA vector
- B-pSARS-CoV spike
photoactiviated biotin labeled SARS-CoV spike DNA vector
- B-pSARS-CoV membrane
photoactiviated biotin labeled SARS-CoV membrane DNA vector
- CL
constant light chain region
- CH2
constant heavy chain region 2
- ECL
enhanced chemiluminescence
- GAM-HRPO
goat antimouse-HRPO
- His
histidine
- HRPO
horseradish peroxidase
- HSF
hybridoma serum free media
- IMAC
immobilized metal affinity chromatography
- LAL
limulus amebocyte lysate
- MMR
macrophage mannose receptor
- MHC
major histocompatibility complex
- Ni
nickel
- NCBI
National Center for Biotechnology Information
- ng
nanogram
- NHS-LC-biotin
bioti-namidohexanoic acid 3-sulfo-N-hydroxysuccinimide ester
- PBST
PBS with 0.1% Tween 20
- PSG
penicillin, streptomycin and l-glutamine
- RT
room temperature
- RT-PCR
reverse transcription polymerase chain reaction
- TD-PCR
touch down polymerase chain reaction
- VL
variable light chain
- VH
variable heavy chain
- WEEV
Western equine encephalitis virus
- E
envelope protein
- TMB
3,3′,5,5′-tetramethylbenzidine
References
- 1.Tacken PJ, de Vries IJ, Torensma R, Figdor CG. Dendritic-cell immunotherapy: from ex vivo loading to in vivo targeting. Nat Rev Immunol. 2007;7(10):790–802. doi: 10.1038/nri2173. [DOI] [PubMed] [Google Scholar]
- 2.Haicheur N, Bismuth E, Bosset S, Adotevi O, Warnier G, Lacabanne V, Regnault A, Desaymard C, Amigorena S, Ricciardi-Castagnoli P, Goud B, Fridman WH, Johannes L, Tartour E. The B subunit of Shiga toxin fused to a tumor antigen elicits CTL and targets dendritic cells to allow MHC class I-restricted presentation of peptides derived from exogenous antigens. J Immunol. 2000;165(6):3301–8. doi: 10.4049/jimmunol.165.6.3301. [DOI] [PubMed] [Google Scholar]
- 3.Zhang L, Tang Y, Akbulut H, Zelterman D, Linton PJ, Deisseroth AB. An adenoviral vector cancer vaccine that delivers a tumor-associated antigen/CD40-ligand fusion protein to dendritic cells. Proc Natl Acad Sci U S A. 2003;100(25):15101–6. doi: 10.1073/pnas.2135379100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van Broekhoven CL, Parish CR, Demangel C, Britton WJ, Altin JG. Targeting dendritic cells with antigen-containing liposomes: a highly effective procedure for induction of antitumor immunity and for tumor immunotherapy. Cancer Res. 2004;64(12):4357–65. doi: 10.1158/0008-5472.CAN-04-0138. [DOI] [PubMed] [Google Scholar]
- 5.Deo YM, Graziano RF, Repp R, van de Winkel JG. Clinical significance of IgG Fc receptors and Fc gamma R-directed immunotherapies. Immunol Today. 1997;18(3):127–35. doi: 10.1016/s0167-5699(97)01007-4. [DOI] [PubMed] [Google Scholar]
- 6.Figdor CG, van Kooyk Y, Adema GJ. C-type lectin receptors on dendritic cells and Langerhans cells. Nat Rev Immunol. 2002;2(2):77–84. doi: 10.1038/nri723. [DOI] [PubMed] [Google Scholar]
- 7.Tacken PJ, Torensma R, Figdor CG. Targeting antigens to dendritic cells in vivo. Immunobiology. 2006;211(6–8):599–608. doi: 10.1016/j.imbio.2006.05.021. [DOI] [PubMed] [Google Scholar]
- 8.Pack M, Trumpfheller C, Thomas D, Park CG, Granelli-Piperno A, Munz C, Steinman RM. DEC-205/CD205+ dendritic cells are abundant in the white pulp of the human spleen, including the border region between the red and white pulp. Immunology. 2008;123(3):438–46. doi: 10.1111/j.1365-2567.2007.02710.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Witmer-Pack MD, Swiggard WJ, Mirza A, Inaba K, Steinman RM. Tissue distribution of the DEC-205 protein that is detected by the monoclonal antibody NLDC-145. II. Expression in situ in lymphoid and nonlymphoid tissues. Cell Immunol. 1995;163(1):157–62. doi: 10.1006/cimm.1995.1110. [DOI] [PubMed] [Google Scholar]
- 10.Inaba K, Swiggard WJ, Inaba M, Meltzer J, Mirza A, Sasagawa T, Nussenzweig MC, Steinman RM. Tissue distribution of the DEC-205 protein that is detected by the monoclonal antibody NLDC-145. I. Expression on dendritic cells and other subsets of mouse leukocytes. Cell Immunol. 1995;163(1):148–56. doi: 10.1006/cimm.1995.1109. [DOI] [PubMed] [Google Scholar]
- 11.den Haan JM, Lehar SM, Bevan MJ. CD8(+) but not CD8(-) dendritic cells cross-prime cytotoxic T cells in vivo. J Exp Med. 2000;192(12):1685–96. doi: 10.1084/jem.192.12.1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Granelli-Piperno A, Pritsker A, Pack M, Shimeliovich I, Arrighi JF, Park CG, Trumpfheller C, Piguet V, Moran TM, Steinman RM. Dendritic cell-specific intercellular adhesion molecule 3-grabbing nonintegrin/CD209 is abundant on macrophages in the normal human lymph node and is not required for dendritic cell stimulation of the mixed leukocyte reaction. J Immunol. 2005;175(7):4265–73. doi: 10.4049/jimmunol.175.7.4265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mahnke K, Qian Y, Fondel S, Brueck J, Becker C, Enk AH. Targeting of antigens to activated dendritic cells in vivo cures metastatic melanoma in mice. Cancer Res. 2005;65(15):7007–12. doi: 10.1158/0008-5472.CAN-05-0938. [DOI] [PubMed] [Google Scholar]
- 14.Trumpfheller C, Finke JS, Lopez CB, Moran TM, Moltedo B, Soares H, Huang Y, Schlesinger SJ, Park CG, Nussenzweig MC, Granelli-Piperno A, Steinman RM. Intensified and protective CD4+ T cell immunity in mice with anti-dendritic cell HIV gag fusion antibody vaccine. J Exp Med. 2006;203(3):607–17. doi: 10.1084/jem.20052005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bonifaz LC, Bonnyay DP, Charalambous A, Darguste DI, Fujii S, Soares H, Brimnes MK, Moltedo B, Moran TM, Steinman RM. In vivo targeting of antigens to maturing dendritic cells via the DEC-205 receptor improves T cell vaccination. J Exp Med. 2004;199(6):815–24. doi: 10.1084/jem.20032220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bozzacco L, Trumpfheller C, Siegal FP, Mehandru S, Markowitz M, Carrington M, Nussenzweig MC, Piperno AG, Steinman RM. DEC-205 receptor on dendritic cells mediates presentation of HIV gag protein to CD8+ T cells in a spectrum of human MHC I haplotypes. Proc Natl Acad Sci U S A. 2007;104(4):1289–94. doi: 10.1073/pnas.0610383104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bruder D, Westendorf AM, Hansen W, Prettin S, Gruber AD, Qian Y, von Boehmer H, Mahnke K, Buer J. On the edge of autoimmunity: T-cell stimulation by steady-state dendritic cells prevents autoimmune diabetes. Diabetes. 2005;54(12):3395–401. doi: 10.2337/diabetes.54.12.3395. [DOI] [PubMed] [Google Scholar]
- 18.Bonifaz L, Bonnyay D, Mahnke K, Rivera M, Nussenzweig MC, Steinman RM. Efficient targeting of protein antigen to the dendritic cell receptor DEC-205 in the steady state leads to antigen presentation on major histocompatibility complex class I products and peripheral CD8+ T cell tolerance. J Exp Med. 2002;196(12):1627–38. doi: 10.1084/jem.20021598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hawiger D, Inaba K, Dorsett Y, Guo M, Mahnke K, Rivera M, Ravetch JV, Steinman RM, Nussenzweig MC. Dendritic cells induce peripheral T cell unresponsiveness under steady state conditions in vivo. J Exp Med. 2001;194(6):769–79. doi: 10.1084/jem.194.6.769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang WW, Das D, Tang XL, Budzynski W, Suresh MR. Antigen targeting to dendritic cells with bispecific antibodies. J Immunol Methods. 2005;306(1–2):80–92. doi: 10.1016/j.jim.2005.07.023. [DOI] [PubMed] [Google Scholar]
- 21.Suresh MR, Cuello AC, Milstein C. Bispecific monoclonal antibodies from hybrid hybridomas. Methods Enzymol. 1986;121:210–28. doi: 10.1016/0076-6879(86)21019-8. [DOI] [PubMed] [Google Scholar]
- 22.Shen Z, Reznikoff G, Dranoff G, Rock KL. Cloned dendritic cells can present exogenous antigens on both MHC class I and class II molecules. J Immunol. 1997;158(6):2723–30. [PubMed] [Google Scholar]
- 23.Nagata LP, Hu WG, Masri SA, Rayner GA, Schmaltz FL, Das D, Wu J, Long MC, Chan C, Proll D, Jager S, Jebailey L, Suresh MR, Wong JP. Efficacy of DNA vaccination against western equine encephalitis virus infection. Vaccine. 2005;23(17–18):2280–3. doi: 10.1016/j.vaccine.2005.01.032. [DOI] [PubMed] [Google Scholar]
- 24.Wahl-Jensen VM, Afanasieva TA, Seebach J, Stroher U, Feldmann H, Schnittler HJ. Effects of Ebola virus glycoproteins on endothelial cell activation and barrier function. J Virol. 2005;79(16):10442–50. doi: 10.1128/JVI.79.16.10442-10450.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McInnes JL, Forster AC, Skingle DC, Symons RH. Preparation and uses of photobiotin. Methods Enzymol. 1990;184:588–600. doi: 10.1016/0076-6879(90)84323-9. [DOI] [PubMed] [Google Scholar]
- 26.Das D, Nagata LP, Suresh MR. Immunological evaluation of Escherichia coli expressed E2 protein of Western equine encephalitis virus. Virus Res. 2007;128(1–2):26–33. doi: 10.1016/j.virusres.2007.03.030. [DOI] [PubMed] [Google Scholar]
- 27.Wang WW, Das D, Suresh MR. Biotin carboxyl carrier protein co-purifies as a contaminant in core-streptavidin preparations. Mol Biotechnol. 2005;31(1):29–40. doi: 10.1385/MB:31:1:029. [DOI] [PubMed] [Google Scholar]
- 28.Das D, Gares SL, Nagata LP, Suresh MR. Evaluation of a Western Equine Encephalitis recombinant E1 protein for protective immunity and diagnostics. Antiviral Res. 2004;64(2):85–92. doi: 10.1016/j.antiviral.2004.06.008. [DOI] [PubMed] [Google Scholar]
- 29.Das D, Jacobs F, Feldmann H, Jones SM, Suresh MR. Differential expression of the Ebola virus GP(1,2) protein and its fragments in E. coli. Protein Expression Purif. 2007;54(1):117–25. doi: 10.1016/j.pep.2007.02.004. [DOI] [PubMed] [Google Scholar]
- 30.O'Brien MP, Aitken R. Antibody phage display: methods and protocols. Vol. 178. Humana Press; Totowa, NJ: 2002. pp. 1–401. [Google Scholar]
- 31.Wang WW, Das D, McQuarrie SA, Suresh MR. Design of a bifunctional fusion protein for ovarian cancer drug delivery: single-chain anti-CA125 core-streptavidin fusion protein. Eur J Pharm Biopharm. 2007;65(3):398–405. doi: 10.1016/j.ejpb.2006.12.012. [DOI] [PubMed] [Google Scholar]
- 32.Coligan JE, Kruisbeek AM, Margulies DH, Shevach EM, Strober W. Current Protocols in Immunology. Greene Publishing and Wiley-Interscience; New York: 1992. [Google Scholar]
- 33.Demangel C, Zhou J, Choo AB, Shoebridge G, Halliday GM, Britton WJ. Single chain antibody fragments for the selective targeting of antigens to dendritic cells. Mol Immunol. 2005;42(8):979–85. doi: 10.1016/j.molimm.2004.09.034. [DOI] [PubMed] [Google Scholar]
- 34.Kipriyanov SM, Little M, Kropshofer H, Breitling F, Gotter S, Dubel S. Affinity enhancement of a recombinant antibody: formation of complexes with multiple valency by a single-chain Fv fragment-core streptavidin fusion. Protein Eng. 1996;9(2):203–11. doi: 10.1093/protein/9.2.203. [DOI] [PubMed] [Google Scholar]
- 35.Schultz J, Lin Y, Sanderson J, Zuo Y, Stone D, Mallett R, Wilbert S, Axworthy D. A tetravalent single-chain antibody-streptavidin fusion protein for pretargeted lymphoma therapy. Cancer Res. 2000;60(23):6663–9. [PubMed] [Google Scholar]
- 36.Han Z, Licata JM, Paragas J, Harty RN. Permeabilization of the plasma membrane by Ebola virus GP2. Virus Genes. 2007;34(3):273–81. doi: 10.1007/s11262-006-0009-4. [DOI] [PubMed] [Google Scholar]
- 37.Brindley MA, Hughes L, Ruiz A, McCray PB, Jr, Sanchez A, Sanders DA, Maury W. Ebola virus glycoprotein 1: identification of residues important for binding and postbinding events. J Virol. 2007;81(14):7702–9. doi: 10.1128/JVI.02433-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Young JA, Collier RJ. Anthrax toxin: receptor binding, internalization, pore formation, and translocation. Annu Rev Biochem. 2007;76:243–65. doi: 10.1146/annurev.biochem.75.103004.142728. [DOI] [PubMed] [Google Scholar]
- 39.Du L, Zhao G, He Y, Guo Y, Zheng BJ, Jiang S, Zhou Y. Receptor-binding domain of SARS-CoV spike protein induces long-term protective immunity in an animal model. Vaccine. 2007;25(15):2832–8. doi: 10.1016/j.vaccine.2006.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ramanathan RK, Lee KM, McKolanis J, Hitbold E, Schraut W, Moser AJ, Warnick E, Whiteside T, Osborne J, Kim H, Day R, Troetschel M, Finn OJ. Phase I study of a MUC1 vaccine composed of different doses of MUC1 peptide with SB-AS2 adjuvant in resected and locally advanced pancreatic cancer. Cancer Immunol Immunother. 2005;54(3):254–64. doi: 10.1007/s00262-004-0581-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gilewski T, Adluri S, Ragupathi G, Zhang S, Yao TJ, Panageas K, Moynahan M, Houghton A, Norton L, Livingston PO. Vaccination of high-risk breast cancer patients with mucin-1 (MUC1) keyhole limpet hemocyanin conjugate plus QS-21. Clin Cancer Res. 2000;6(5):1693–701. [PubMed] [Google Scholar]
- 42.Biswas K, Richmond A, Rayman P, Biswas S, Thornton M, Sa G, Das T, Zhang R, Chahlavi A, Tannenbaum CS, Novick A, Bukowski R, Finke JH. GM2 expression in renal cell carcinoma: potential role in tumor-induced T-cell dysfunction. Cancer Res. 2006;66(13):6816–25. doi: 10.1158/0008-5472.CAN-06-0250. [DOI] [PubMed] [Google Scholar]
- 43.Azuma Y, Ishikawa Y, Kawai S, Tsunenari T, Tsunoda H, Igawa T, Iida S, Nanami M, Suzuki M, Irie RF, Tsuchiya M, Yamada-Okabe H. Recombinant human hexamer-dominant IgM monoclonal antibody to ganglioside GM3 for treatment of melanoma. Clin Cancer Res. 2007;13(9):2745–50. doi: 10.1158/1078-0432.CCR-06-2919. [DOI] [PubMed] [Google Scholar]
- 44.Schlesinger S, Schlesinger MJ. Togaviridae: the viruses and their replication. Lippincott Williams & Wilkins; Philadelphia: 2001. pp. 2265–300. [Google Scholar]
- 45.Wu JQ, Barabe ND, Chau D, Wong C, Rayner GR, Hu WG, Nagata LP. Complete protection of mice against a lethal dose challenge of western equine encephalitis virus after immunization with an adenovirus-vectored vaccine. Vaccine. 2007;25(22):4368–75. doi: 10.1016/j.vaccine.2007.03.042. [DOI] [PubMed] [Google Scholar]
- 46.Chan CM, Ma CW, Chan WY, Chan HY. The SARS-Coronavirus Membrane protein induces apoptosis through modulating the Akt survival pathway. Arch Biochem Biophys. 2007;459(2):197–207. doi: 10.1016/j.abb.2007.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zeng F, Hon CC, Yip CW, Law KM, Yeung YS, Chan KH, Malik Peiris JS, Leung FC. Quantitative comparison of the efficiency of antibodies against S1 and S2 subunit of SARS coronavirus spike protein in virus neutralization and blocking of receptor binding: implications for the functional roles of S2 subunit. FEBS Lett. 2006;580(24):5612–20. doi: 10.1016/j.febslet.2006.08.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dowling W, Thompson E, Badger C, Mellquist JL, Garrison AR, Smith JM, Paragas J, Hogan RJ, Schmaljohn C. Influences of glycosylation on antigenicity, immunogenicity, and protective efficacy of ebola virus GP DNA vaccines. J Virol. 2007;81(4):1821–37. doi: 10.1128/JVI.02098-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Filpula D. Antibody engineering and modification technologies. Biomol Eng. 2007;24(2):201–15. doi: 10.1016/j.bioeng.2007.03.004. [DOI] [PubMed] [Google Scholar]
- 50.Holliger P, Hudson PJ. Engineered antibody fragments and the rise of single domains. Nat Biotechnol. 2005;23(9):1126–36. doi: 10.1038/nbt1142. [DOI] [PubMed] [Google Scholar]
- 51.Zhang M, Zhang Z, Garmestani K, Schultz J, Axworthy DB, Goldman CK, Brechbiel MW, Carrasquillo JA, Waldmann TA. Pretarget radiotherapy with an anti-CD25 antibody-streptavidin fusion protein was effective in therapy of leukemia/lymphoma xenografts. Proc Natl Acad Sci U S A. 2003;100(4):1891–5. doi: 10.1073/pnas.0437788100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Graves SS, Dearstyne E, Lin Y, Zuo Y, Sanderson J, Schultz J, Pantalias A, Gray D, Axworthy D, Jones HM, Auditore-Hargreaves K. Combination therapy with Pretarget CC49 radioimmunotherapy and gemcitabine prolongs tumor doubling time in a murine xenograft model of colon cancer more effectively than either monotherapy. Clin Cancer Res. 2003;9(10 Part 1):3712–21. [PubMed] [Google Scholar]