

Abstract
Herein we report a method for the synthesis of substituted pyridines. The unsaturated ketones and aldehydes derived from the cycloisomerization of primary and secondary propargyl diynols in the presence of [CpRu(CH3CN)3]PF6 (1) are converted to 1-azatrienes which in turn undergo a subsequent electrocyclization-dehydration to provide pyridines with excellent regiocontrol.
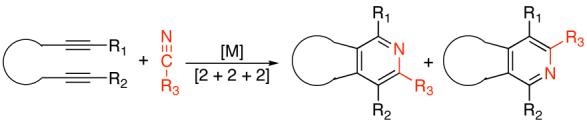
Functionalized pyridines have a range of applications in many areas of chemistry.1 Not surprisingly, a large amount of work has been devoted to the development of methods to provide these products in a straightforward fashion.2,3 New strategies for stiching together pyridines therefore can enhance the flexibility of approaching these important structural entities. The elegant transition metal-catalyzed [2+2+2] cycloaddition of a tethered diyne and a substituted nitrile is one such approach (Eq. 1).4 While recent catalytic methods achieve high levels of regioselectivity for certain classes of substrates (e.g., substrates with two sterically differentiable substituents), the question of regioselectivity remains ambiguous.5,6
 |
(1) |
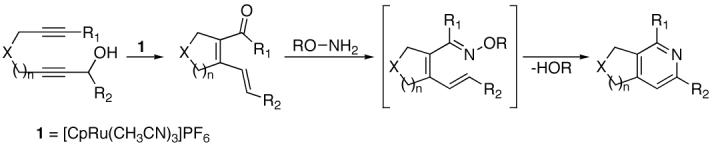
Recently, the cycloisomerization of diynols of type 2 to form α,β,γ,δ-unsaturated ketones and aldehydes in the presence of [CpRu(CH3CN)3]PF6 (1) has been demonstrated (Eq. 2).7 This method quickly builds molecular complexity from readily available starting materials. The following general reaction was proposed as an alternative to the intramolecular aldol condensation.8
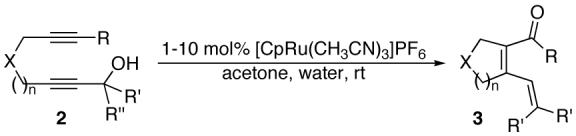 |
(2) |
In an effort to highlight the utility of the ruthenium-catalyzed cycloisomerization, its application in the synthesis of substituted heterocycles was examined. Specifically, it was anticipated that the dienal or dienone formed from the cycloisomerization reaction could be converted to azatriene 4, which, after 6π-cyclization, would afford intermediate dihydropyridine 5. This intermediate would then eliminate water to form pyridine 6 (Scheme 1). While the electrocyclic ring closure of 1,3,5-hexatrienes is widely known, there are only two reports of the analogous electrocyclization of 1-azatrienes to form simple, non-fused pyridine products.9 If successful, this method could be used to construct substituted pyridines with excellent regiocontrol, obviating the problems encountered in the metal-catalyzed [2+2+2] cycloaddtion systems.
Scheme 1.
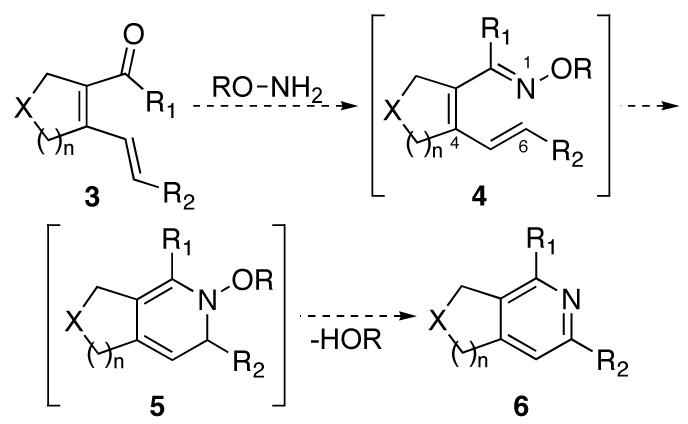
Pyridine formation via dehydrative cyclization of azatriene
To examine the proposed reaction sequence, diynol 2a was prepared, and, upon exposure to catalyst 1, afforded the desired aldehyde (3a).7 Treatment of 3a with hydroxylamine hydrochloride and sodium acetate in refluxing ethanol provided azatriene 4a (Scheme 2). Unfortunately, when 4a was refluxed in toluene for 24 hours, no cyclized material was observed. When subjected to microwave10 irradiation, however, cyclization proceeded efficiently. Ultimately, the desired pyridine (6a) was accessed in 95% yield over two steps from aldehyde 3a.
Scheme 2.
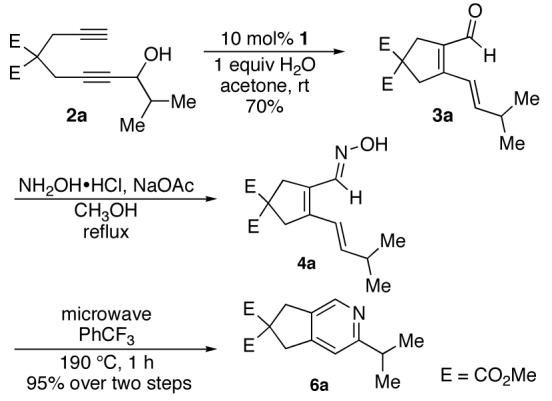
Construction of dienal substrate by Ru-catalyzed cycloisomerization and conditions for formation of pyridine
Encouraged by this positive result, the aryl-substituted diynol 2b was employed to optimize the reaction (Scheme 3). Cycloisomerization of diynol 2b provided aldehyde 3b in 70% yield, which upon heating with hydroxylamine hydrochloride and sodium acetate in ethanol afforded pyridine 6b directly. Oxime 4b presumably forms under these conditions, and spontaneously undergoes the dehydrative cycloisomerization
Scheme 3.
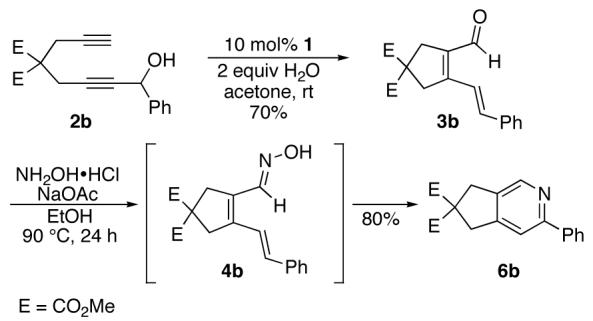
Initial studies: Ru-catalyzed cycloisomerization- 6π-cyclization conditions
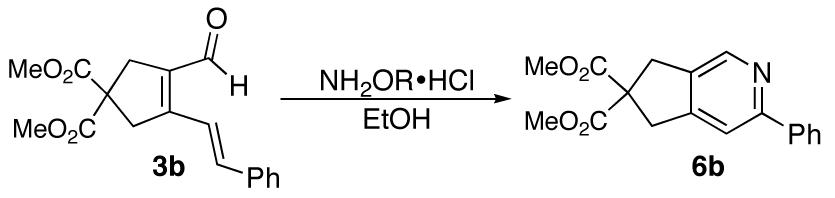
In view of the enhanced reactivity of 4b, we decided to develop general conditions for a one-pot pyridine synthesis from the α,β,γ,δ unsaturated aldehydes. The reaction to form pyridine 6b took 24 h at 90 °C (Table 1, entry 1), and when the temperature was raised to 150 °C, the reaction was complete in 4 h (Table 1, entry 2). Futhermore, when the same reaction was run under microwave irradiation, it was complete in only 1h, affording the desired pyridine in 98% yield (Table 1, entry 3). The effect of the nitrogen substituent (hydroxy versus methoxy) substitution was also examined. The yield dropped to 68% when methoxyamine hydrochloride was used (Table 1, entry 4).
Table 1.
Conditions for pyridine formation from dienal 3b
 | |||||
|---|---|---|---|---|---|
| entry | R | conditions | time | temp (°C) | yield (6b) |
| 1 | H | NaOAc | 24 h | 90 | 80% |
| 2 | H | NaOAc sealed tube | 4 h | 150 | 82% |
| 3 | H | NaOAc microwave | 1 h | 150 | 98% |
| 4 | Me | NaOAc, microwave | 1 h | 150 | 68% |
| 5(a) | H | pyridine then AcCl | 0.5 h | 0-25 | --(b) |
Pyridine used as solvent
Acetoxy derivative of 4b isolated as only product in 69% yield.
Katsumura described the formation of pyridines from N-acetoxy azatrienes at room temperature.9b The substrates examined were limited to those with both a C4-carbonyl group and C6-alkenyl group. In the investigation of pyridine formation from dienal 3b, the 6π-electrocyclization did not proceed under similar conditions (Table 1, entry 5). Reaction did occur at elevated temperatures (>100 °C), but was plagued with impurities formed from undesired side reactions.
Having identified conditions suitable for the one-step conversion of dienals to pyridines, efforts to determine the scope of this synthetic methodology were undertaken. Thus, a variety of diynol substrates of type 2 were constructed, and the cycloisomerization-6π-cyclization sequence applied (Eq. 3).
The use of alkynes linked by heteroatoms were of particular interest, as they can be potentially cleaved at a later stage, allowing for further functionalization of the pyridine skeleton at the 3 and 4 positions. We found that heteroatoms are indeed tolerated, (Table 2, entries 3, 4, and 5). The reactivity of substrates without α-branching at the propargylic position (R2 = Me, Et) was also demonstrated (Table 2, entries 7-9).
Table 2.
Pyridines via the cycloisomerization-6π-electrocyclization sequence
 | ||||
|---|---|---|---|---|
| entry | diene-al/ -one | isolated yield (E/Z) | pyridine | isolated yield |
| 1 | 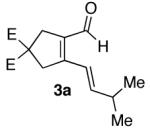 |
70%a) | 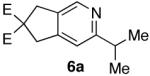 |
95% b) |
| 2 | 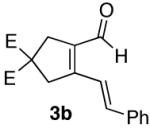 |
70%a) | 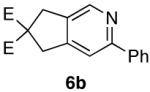 |
99% c) |
| 3 | 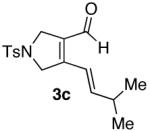 |
70% | 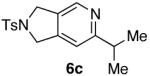 |
40% d) |
| 4 | 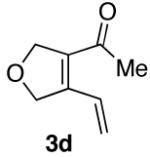 |
68% | 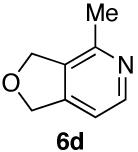 |
75% c) |
| 5 | 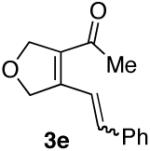 |
60% (5:1) | 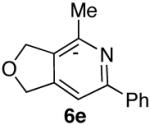 |
85% d) |
| 6 | 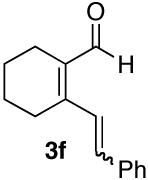 |
80% (7:1) | 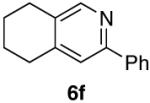 |
84% d) |
| 7 | 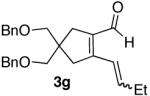 |
54% (8:1) | 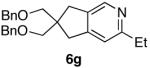 |
73% d) |
| 8 | 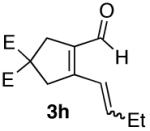 |
70% (12:1) | 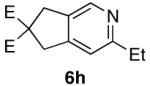 |
97% d) |
| 9 | 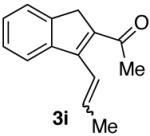 |
60% (2:1) | 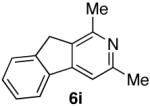 |
85% d) |
| 10 | 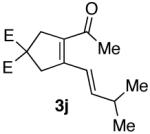 |
74% | 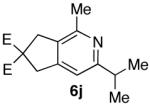 |
85% b) |
| 11 | 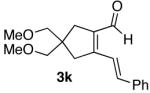 |
70% | 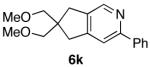 |
87% c) |
E = CO2Me.
See Ref. 7.
methods:i) NH2OH·HCl, NaOAc, EtOH, 90 °C, 30 min, ii) PhCF3, microwave, 220-240 °C, 4 h.
NH2OH·HCl, NaOAc, EtOH, microwave, 150 °C, 1-1.5 h
NH2OH·HCl, NaOAc, EtOH, 90 °C, 5-24 h.
As part of our effort to explore the scope of the cycloisomerization-6π-cyclization sequence, an examination of the effect of ring size was undertaken. Tethers which cycloisomerize to form 6-membered rings have been shown to give slightly lower yields in the ruthenium-catalyzed cycloisomerization than those which form 5-membered rings.7 We were pleased to find that 2f (Table 1, entry 6) underwent the cycloisomerization-6π-cyclization, in high yield for both steps.
Previously, the only isolable products from the cyclization of secondary propargylic alcohols had the E-olefin configuration at the γ,δ-double bond. It was postulated that elimination of the hyroxyl group may occur stereoselectively, where the observed E/Z- ratio is dictated by steric interactions between the R2 group and the cyclopentane ring.7 In a number of the substrates examined, we observed for the first time products containing the external olefin in a mixture of E- and Z-olefin geometries. In some of the examples explored herein the differential A-1,3 strain between the olefin substituent and the annealed ring vs the ligands on Ru appear to be more competitive because of its smaller size. The 6π-cyclizations of these substrates proceeded with complete consumption of starting material (Table 2, entries 5-9), the double bond mixtures having no noticeable effect on the yield of the pyridine obtained in the subsequent 6π-cyclization.
Ketones are known to generate oximes as a mixture of isomers, and we wondered whether this mixture would impact the 6π-cycloisomerization reaction.11 Internal diynols 2d, 2e, 2i and 2j were prepared and submitted to the cycloisomerization reaction to give the desired ketones in high yields (Table 2, entries 4-5, 9-10). The formation of pyridines from dienones 3d, 3e and 3i took place readily, forming the corresponding pyridines 6d, 6e, and 6i in good yields. Using the conditions of Table 1, entry 2, the reaction of dienone 3j required higher temperatures and longer times to provide the corresponding pyridine 6j, in a reduced yield of 67%. The best yields of 6j were obtained when the azatriene was first isolated, and then submitted to the 6π-cyclization in trifluorotoluene, in which case an 85% yield was obtained (Table 2, entry 10).
We also examined substrates containing a terminal olefin (Table 2, entry 4). As reported previously, primary alcohols cycloisomerize to yield both the expected cycloisomerized product 3d and the formally hydrated product 7. When dienal 3d was subjected to our one-step pyridine syntheses compound 6d formed in good yield (Scheme 4).
Scheme 4.
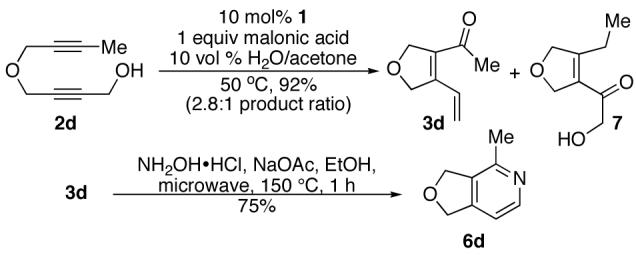
Behavior of primary propargyl alcohols
Finally, to increase the utility of this methodology, we sought to access the pyridine directly from the diynol (2) in a one-pot protocol. Acetone, which is the preferred solvent for the cycloisomerization, poses problems for the reaction with hydroxylamine. This problem could be circumvented, however, by first performing the initial cycloisomerization using our standard conditions (acetone/water). Upon completion, the solvent could be removed, and the crude residue subjected to the previously mentioned pyridine-forming conditions. This mixture was heated in the microwave, to afford pyridine 6e in 54% yield (Scheme 5).
Scheme 5.
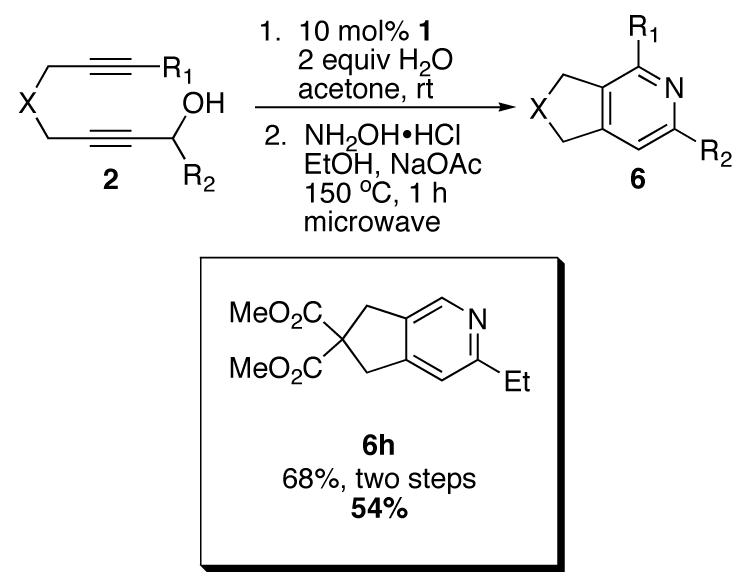
Conditions for one-pot protocol
In conclusion, the method we report here represents a facile route to pyridines which demontrates the utility of the ruthenium-catalyzed cycloisomerization reaction. The reaction sequence tolerates a broad range of functional groups. The double-bond isomeric mixture obtained from the cycloisomerization reaction did not have an effect on the yield of the subsequent electrocyclization. The cycloisomerization-6π-cyclization sequence may be carried out in one pot or in two independent steps. Additionally, the azatriene cyclization may be carried out in 1-3 hours under microwave irradiation or, for more sensitive substrates, at lower temperatures for longer periods of time (6 -24 h at 90 °C). All of the substrates examined underwent the 6π-cyclization to the corresponding pyridines under a variety of conditions and in excellent yields. Our current efforts are directed at improvement of the one-pot protocol and application of this methodology to the synthesis of biologically significant natural products.
Supplementary Material
Acknowledgment
We thank the National Science Foundation and the National Institutes of Health, General Medical Sciences Institute (GM-13598), for their generous support of our programs. We also thank Biotage Inc. for providing the microwave reactor and continuing to provide invaluable help with its service. Mass spectra were provided by the Mass Spectrometry Facility of the University of California-San Francisco, supported by NIH Division of Research Resources.
References
- (1).Joule JA, Mills K. Heterocyclic Chemistry. 4th ed. Blackwell; Oxford: 2000. [Google Scholar]
- (2).Jones G. Pyridines and their benzoderivatives: synthesis. In: Katritzky A, Rees CW, Scriven EF, editors. Comprehensive Heterocyclic Chemistry II. Vol. 5. Pergamon; Oxford, England: 1996. p. 167. [Google Scholar]
- (3).For a recent example, see:Movassaghi M, Hill MD. J. Am. Chem. Soc. 2006;128:4592–4593. doi: 10.1021/ja060626a.
- (4).Saa C, Varela J. Chem. Rev. 2003;103:3787–3801. doi: 10.1021/cr030677f. [DOI] [PubMed] [Google Scholar]
- (5).Louie J, Zuo G, Doung HA, McCormick M. J. Am. Chem. Soc. 2005;127:5030–5031. doi: 10.1021/ja0508931. [DOI] [PubMed] [Google Scholar]
- (6).Yamamoto Y, Kinpara K, Ogawa R, Nishiyama H, Itoh K. Chem. Eur. J. 2006;12:5618–5631. doi: 10.1002/chem.200600176. [DOI] [PubMed] [Google Scholar]
- (7)(a).Trost BM, Rudd MT. J. Am. Chem. Soc. 2002;124:4178–4179. doi: 10.1021/ja012672a. [DOI] [PubMed] [Google Scholar]; (b) Trost BM, Rudd MT. J. Am. Chem. Soc. 2005;127:4763–4776. doi: 10.1021/ja043097o. [DOI] [PubMed] [Google Scholar]
- (8).Wolinsky J, Baker W. J. Am. Chem. Soc. 1960;82:636–638. [Google Scholar]
- (9)(a).Schiess P, Chia H, Ringele P. Tetrahedron Lett. 1972:313. [Google Scholar]; (b) Tanaka K, Mori H, Yamamoto M, Katsumura S. J. Org. Chem. 2001;66:3099–3110. doi: 10.1021/jo005779+. [DOI] [PubMed] [Google Scholar]
- (10).It has been shown that microwave irradiation can provide key advantages over conventional thermal methods, especially in cases when usual methods require forcing conditions or prolonged reaction times.Besson T, Brain C, Westman J. In: Microwave Assisted Organic Synthesis. Tierney JP, Lidstrom P., Eds., editors. Blackwell Publishing; Oxford, UK: 2005. Chapters 3, 5.
- (11).If one considers the disrotatory model for thermal 6π-electrocyclizations, only the E-isomer would position the hydrogen and the N-hydroxyl group which are to be eliminated trans (anti-periplanar) to each other in the resulting dihydropyridine. The Z-isomer might also introduce steric strain that would prevent the substituents from attaining the required planar confirmation.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.