

Abstract
A variety of lipophilic 2-oxoamides containing sulfonamide analogues of γ-amino acids as well as acyl sulfonamides of γ-aminobutyric acid were synthesized. Their ability to inhibit intracellular GIVA cPLA2 and GVIA iPLA2 as well as secreted GV sPLA2 was evaluated. The sulfonamide group seems a bioisosteric group suitable to replace the carboxyl group in 2-oxoamide inhibitors of GVIA cPLA2.
Keywords: acyl sulfonamides, inhibitors, 2-oxoamides, phospholipase A2, sulfonamides
Introduction
The phospholipase A2 (PLA2) superfamily of enzymes consists of a broad range of enzymes defined by their ability to catalyze the hydrolysis of the ester bond at the sn-2 position of phospholipids, yielding free fatty acids, including arachidonic acid, and lysophospholipids.1-4 Historically, a broad classification divides the PLA2 classes into three types: secretory (sPLA2), cytosolic Ca2+-dependent (cPLA2) and cytosolic Ca2+-independent (iPLA2). PLA2 enzymes have been systematically classified into 15 groups and subgroups on the basis of their nucleotide and amino acid sequence.1,3,4 Among the various phospholipases, the Group IVA cPLA2 (GIVA cPLA2) is a particularly attractive target for drug development, since it is the rate-limiting provider of arachidonic acid and lysophospholipids that can be converted into prostaglandins, leukotrienes and PAF, respectively.5 The role of the other intracellular PLA2, calcium-independent PLA2 (GVIA iPLA2), in the inflammatory process is still unclear, and it appears to be the primary PLA2 for basal metabolic functions within the cell.6,7 Both intracellular enzymes share the same catalytic mechanism utilizing a serine residue as the nucleophile. The other major class of PLA2 enzymes includes the small, secreted sPLA2s and is characterized by a catalytic His/Asp dyad and catalytic Ca2+.1,3,4,8 A well-studied, simple example of this class is the human Group V secreted phospholipase A2 (GV sPLA2).9,10 In many cases the activity of GV sPLA2 is dependent on or linked to the activity of GIVA cPLA2.1,11,12
A great variety of compounds have been studied for their activity against GIVA cPLA2 and the synthetic inhibitors of GIVA cPLA2 have been summarized in a recent review.13 Within the last five years, we have been developing a novel class of GIVA cPLA2 inhibitors that are based on the 2-oxoamide reactive functionality.14-20 Lipophilic 2-oxoamides based on γ-aminobutyric acid (compound 1a, Figure 1) or the non-natural amino acid γ-norleucine (compound 1b, Figure 1) are potent inhibitors of GIVA cPLA2 presenting in vivo anti-inflammatory and analgesic activity.14,15 AX048 (compound 1c, Figure 1), the ethyl ester derivative of compound 1a, is the first systemically bioavailable compound with a significant affinity for GIVA cPLA2, which produces potent anti-hyperalgesia.17 We also synthesized 2-oxoamides based on long chain β-amino acids, however such derivatives were found inactive.16 On the contrary, 2-oxoamide AX109 based on the non-natural amino acid δ-norleucine (compound 1d, Figure 1) exhibit slightly higher inhibitory activity than the corresponding analogue based on γ-norleucine.19
Figure 1.

Structures of known 2-oxoamide inhibitors of GIVA cPLA2 based on γ- and δ-amino acids and inhibitors containing sulfonamide groups designed in this study.
The aim of this work was to synthesize lipophilic 2-oxoamides containing sulfonamide groups bioisosteric to the carboxyl group and to evaluate their activity on three human PLA2 isoforms.
Results and Discussion
We have previously shown that 2-oxoamides based on γ- or δ-amino acids containing a free carboxyl group are selective inhibitors of GIVA cPLA2, affecting the activity of neither GVIA iPLA2 nor GV sPLA2.15,19 Ethyl ester AX048 inhibits in vitro not only GIVA cPLA2, but also calcium-independent GVIA iPLA2, which is the main other intracellular PLA2 isoform.17 In addition, methyl esters of 2-oxoamides may inhibit in vitro both GIVA cPLA2 and GVIA iPLA2.18 Bioisosterism represents an interesting approach used in medicinal chemistry for the rational modification of lead compounds into agents exhibiting improved properties.21 Non-classical bioisosteres for the carboxyl group may involve replacement of either only the hydroxyl portion or both the hydroxyl and carbonyl group of this functional group. We designed the replacement of the hydroxyl group of the carboxylic acid by a sulfonamide group, resulting in the formation of acyl sulfonamide (Figure 1). In addition, we decided to replace the entire carboxyl functional group by a sulfonamide group. As known, the pKa values for sulfonamides are similar to that of an aryl carboxylic acid.21 Numerous examples in literature report such replacements leading to compounds with improved biological activities.22-25 Furthermore, we and others have recently demonstrated that the conversion of the proline carboxyl group to acyl sulfonamides is successful for the preparation of improved organocatalysts.26,27
For the synthesis of 2-oxoamides containing a sulfonamide group, two different synthetic routes were studied using 1,3-propanediamine and 1,4-butanediamine as starting materials. Monotosylation of diamines 2a,b led to derivatives 3a,b, which were coupled with 2-hydroxy-hexadecanoic acid using N-ethyl-N′-dimethylaminopropylcarbodiimide (WSCI)28 as a coupling agent in the presence of N-hydroxybenzotriazole (HOBt) to afford 2-hydroxyamides 4a,b (Figure 2). However, this route led to low yields and impure products for monomesylated derivatives. Thus, monoacylated derivatives 6a,b were prepared by coupling of diamines 2a,b with 2-hydroxyhexadecanoic acid using the WSCI/HOBt method (Figure 3). Then, the amino group of 6a,b was mesylated by treatment with methanesulfonyl chloride to give compounds 7a,b. Oxidation of compounds 4a,b and 7a,b to 2-oxoamides 5a,b and 8a,b was carried out by the Dess-Martin periodinane or the PDC reagent (Figure 2 and 3).
Figure 2.

(a) TsCl, NMM THF; (b) CH3(CH2)13CHOHCOOH, WSCI, HOBt, Et3N, CH2Cl2; (c) Dess-Martin reagent, CH2Cl2.
Figure 3.

(a) CH3(CH2)13CHOHCOOH, WSCI, HOBt, Et3N, CH2Cl2; (b) MsCl, NMM THF; (c) PDC, CH3COOH.
2-Oxoamides 14a,b containing a short linear side chain were synthesized from Boc-norleucinol (9), which was prepared by reduction of Boc-norleucine29,30 as depicted in Figure 4. As we have previously shown, N-protected amino aldehydes are useful intermediates for the synthesis of γ-amino acids and other non-natural amino acids.31,32 Thus, Boc-norleucinol (9) was oxidized to aldehyde by the NaOCl/AcNH-TEMPO method and reacted with (triphenyl phosphoranylidene)acetonitrile to produce unsaturated nitrile 10. After hydrogenation of the double bond, the nitrile group was reduced to amino group by NiCl2·6H2O/NaBH433 and immediately reacted with methane or toluenesulfonyl chloride. Removal of the Boc group, coupling with 2-hydroxyhexadecanoic acid and oxidation produced 2-oxoamides 14a,b.
Figure 4.

(a) NaOCl, AcNH-TEMPO, NaBr, NaHCO3, EtOAc/PhCH3/H2O (3:3:0.5), -5 °C; (b) CNCH=P(C6H5)3, THF, reflux; (c) H2, 10% Pd/C, MeOH; (d) NiCl2·6H2O, NaBH4, MeOH; (e) RSO2Cl, NMM, THF; (f) 5N HCl/Et2O; (g) CH3(CH2)13CHOHCOOH, WSCI, HOBt, Et3N; (h) Dess-Martin reagent, CH2Cl2.
The synthesis of acyl sulfonamides based on γ-aminobutyric acid is presented in Figure 5. Boc-γ-aminobutyric acid (15) was coupled with methane or toluene sulfonamides by the DCC/DMAP method.26 By procedures similar to those described above, derivatives 18a,b were prepared. The acidic hydrogen of the sulfonamide group of compound 17b was replaced by a methyl group by treatment with methyl iodide in the presence of Na2CO3. Then, oxidation of compound 19 led to compound 20.
Figure 5.

(a) RSO2NH2, DCC, DMAP, CH2Cl2, r.t; (b) 5N HCl/Et2O; (c) CH3(CH2)13CHOHCOOH, WSCI, HOBt, Et3N, CH2Cl2; (d) Dess-Martin reagent, CH2Cl2; (e) Na2CO3, CH3I, THF, r.t.
Compounds 5a,b, 8a,b, 14a,b, 18a,b and 20 were tested for their ability to inhibit human GIVA cPLA2 in a GIVA cPLA2-specific assay, which uses mixed micelles of substrate 1-palmitoyl-2-arachidonyl phosphatidylcholine, phosphatidylinositol 4,5-bisphosphate and detergent Triton X-100 (97: 3: 400 μM), as previously described.15 In addition, their activity against GVIA iPLA2 and the secreted PLA2 isoform GV sPLA2 was determined. The in vitro assay systems for GVIA iPLA2 and GV sPLA2 have been previously described.18,19 The compounds synthesized in this work were initially tested at a 0.091 mole fraction. When the inhibitory potency was higher than 80%, XI(50) values were determined. The XI(50) is the mole fraction of inhibitor in the total substrate interface required to inhibit the enzyme by 50%. The results are summarized in Table 1.
Table 1.
In vitro inhibition of human PLA2 by 2-oxoamides containing sulfonamide groups. Average percent inhibition and standard error (n=3) reported for each compound at 0.091 mole fraction. XI(50) values determined for inhibitors with greater than 80% inhibition at 0.091 mole fraction. ND signifies compounds with less than 25% inhibition (or no detectable inhibition).
| Compound | Structure | GIVA cPLA2 | GVIA iPLA2 | GV sPLA2 |
|---|---|---|---|---|
| 5a |
|
XI(50)
0.055 ± 0.006 |
XI(50)
0.076 ± 0.020 |
ND |
| 5b |
|
52 ± 4 | ND | ND |
| 8a |
|
ND | ND | ND |
| 8b |
|
ND | ND | ND |
| 14a |
|
XI(50)
0.029 ± 0.019 |
XI(50)
0.060 ± 0.037 |
66 ± 13 |
| 14b |
|
XI(50)
0.033 ± 0.018 |
57 ± 6 | 61 ± 12 |
| 18a |
|
58 ± 2 | ND | 38 ± 6 |
| 18b |
|
42 ± 8 | ND | ND |
| 20 |
|
XI(50)
0.033 ± 0.018 |
53 ± 15 | 68 ± 9 |
| AX006 |
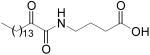
|
XI(50)
0.024 ± 0.015 a |
ND a | |
| AX048 |
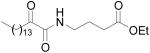
|
XI(50)
0.031 ± 0.017 a |
XI(50)
0.026 ± 0.014 a |
|
| AX007 |

|
XI(50)
0.009 ± 0.004 b |
ND b |
Data taken from reference 17.
Data taken from reference 15.
Among the 2-oxoamides based on monomesylated or monotosylated 1,3-propanenediamine and 1,4-butanediamine (compounds 5a,b and 8a,b, Table 1), only compound 5a showed moderate inhibition of intracellular GIVA cPLA2 and GVIA iPLA2. However, when a short linear side chain corresponding to the norleucine side chain was introduced at the neighboring carbon atom near the 2-oxoamide functionality, both sulfonamide derivatives 14a and 14b presented significant inhibition of GIVA cPLA2. In addition, compound 14a exhibited moderate inhibition of GVIA iPLA2. Acyl sulfonamide derivatives 18a and 18b are very week inhibitors of GIVA cPLA2. On the contrary, derivative 20 significantly inhibited GIVA cPLA2. Among the compounds studied in this work only four derivatives (14a, 14b, 18a and 20) presented some weak inhibition of GV sPLA2.
From the above results, it seems that sulfonamides containing a short linear side chain (14a and 14b) are good inhibitors of GIVA cPLA2 [XI(50) values 0.029 and 0.033, respectively]. None of the acyl sulfonamides 18a and 18b considerably inhibited GIVA cPLA2 and GVIA iPLA2. Suprisingly, derivative 20 lacking an acidic hydrogen preferentially inhibited GIVA cPLA2 [XI(50) 0.033]. The three derivatives 14a, 14b and 20 inhibited GIVA cPLA2 at a level comparable to the lead compounds AX006 and AX048 [XI(50) values 0.024 and 0.031 respectively].17 Furthermore, their potency is comparable to that of the reference inhibitor of GIVA cPLA2 arachidonyl trifluoromethyl ketone [XI(50) 0.036].14 In addition, compounds 14b and 20 preferentially inhibit GIVA cPLA2, in comparison to GVIA iPLA2. We have recently reported that 2-oxoamide derivatives containing a tetrazole or a phosphonate group in replacement of the carboxyl group presented very weak activity on GIVA cPLA2.20 On the contrary, in the present study we demonstrate that 2-oxoamides 14a, 14b and 20 containing sulfonamide groups efficiently inhibit GIVA cPLA2.
We have proposed a model for the binding of 2-oxoamide inhibitors containing a free carboxyl group to GIVA cPLA2.15 According to that model the carboxyl group of the inhibitor interacts with the polar binding site (Arg 200) in a manner similar to that of the phosphate group of the substrate phospholipid. It should be noticed that compound 20 lacks an acidic hydrogen able to interact through electrostatic interactions. Thus, it seems that an alternative interaction of the acyl sulfonamide group with the active site of GIVA cPLA2 is possible. The oxygen atoms or the nitrogen atom of the sulfonamide group may act as hydrogen bond acceptors.
In conclusion, we synthesized a variety of lipophilic 2-oxoamides containing a sulfonamide or an acyl sulfonamide group. Three of them inhibited GIVA cPLA2 at a level similar to that of the lead compounds AX006 and AX048. Thus, the bioisosteric sulfonamide group seems a group suitable to replace the carboxyl group in lipophilic 2-oxoamides inhibitors of GIVA cPLA2.
Materials and Methods
Melting points are uncorrected. Specific rotations were measured on a Perkin Elmer 841 polarimeter using a 10 cm cell. NMR spectra were recorded on a 200-MHz spectrometer. TLC plates (silica gel 60 F254) and silica gel 60 (70-230 or 230-400 mesh) for column chromatography were purchased from Merck. Visualization of spots was effected with UV light and/or phosphomolybdic acid and/or ninhydrin, both in EtOH stain. THF was dried by standard procedures and stored over molecular sieves. All other solvents and chemicals were reagent grade and used without further purification. Fast atom bombardment (FAB) mass spectra were recorded using a VG analytical ZAB-SE instrument.
General Procedure for the Reaction of Diamines with Toluenesulfonyl Chloride
A solution of TsCl (0.15 g, 0.8 mmol) in dry THF (0.76 mL) was added drop-wise to a stirred solution of diamine 2a,b (1 mmol) and NMM (0.11 mL, 1 mmol) in THF (1mL) over a period of 30 min. The volatile components were removed under reduced pressure and the residue was partitioned between 5% NaHCO3 (5 mL) and CH2Cl2 (5 mL). The organic layer was separated, and the aqueous layer was extracted with CH2Cl2 (2 × 5 mL). Combined CH2Cl2 solutions were dried (Na2SO4) and concentrated to afford an oil that was extracted with 1 N HCl (5 mL) and precipitated material was filtered off. The filtrate was extracted with Et2O (2 × 3 mL) and was then basified (pH 11) by the addition of NH3 (25%) and extracted with CH2Cl2 (2 × 10 mL). The CH2Cl2 extracts were combined, dried over Na2SO4 and evaporated under reduced pressure.
N-(3-Aminopropyl)-4-methylbenzenesulfonamide (3a)
Yield 35%; white solid; mp 112-114 °C. 1H NMR (200 MHz, CDCl3): δ 7.73 (2H, d, J = 6.6 Hz, Ph), 7.28 (2H, d, J = 7.6 Hz, Ph), 3.05 (2H, t, J = 5.4 Hz, CH2NH), 2.77 (2H, t, J = 5.2 Hz, H2NCH2), 2.41 (3H, s, C6H4CH3), 1.62-1.45 (2H, m, H2NCH2CH2). 13C NMR (50 MHz, CDCl3): δ 143.2, 136.9, 129.9, 127.3, 43.0, 40.9, 31.1, 21.8. Anal. calcd for C10H16N2O2S: C, 52.61; H, 7.06; N, 12.27. Found: C, 52.45; H, 7.23; N, 12.41.
N-(4-Aminobutyl)-4-methylbenzenesulfonamide (3b)
Yield 38%; yellow oil; used without any purification in the next step.
General Procedure for the Coupling Reaction of Diamines with 2-Hydroxyhexadecanoic Acid
To a stirred solution of 2-hydroxy hexadecanoic acid (0.27g, 1 mmol) and the diamine (1 mmol) in CH2Cl2 (5 mL), Et3N (0.15 mL, 1.1 mmol) and subsequently WSCI (0.21 g, 1.1 mmol) and HOBt (0.15 g, 1 mmol) were added at 0 °C. The reaction mixture was stirred for 1 h at 0 °C and overnight at room temperature. The solvent was evaporated under reduced pressure and the residue was used in the next step without any purification.
A solution of MsCl (0.08 mL, 1 mmol), in dry THF (1 mL) was added drop-wise to a stirred solution of the amine (1 mmol) and NMM (0.11 mL, 1 mmol) in THF (0.76 mL) over a period of 30 min. The organic solvent was evaporated under reduced pressure and the residue was dissolved in AcOEt. The organic layer was washed consecutively with brine, 10% aqueous KHSO4, brine, dried over Na2SO4, and evaporated under reduced pressure. The residue was purified by column chromatography.
2-Hydroxy-N-(3-(methylsulfonamido)propyl)hexadecanamide (7a)
Yield 38%; white solid; mp 134-137 °C. 1H NMR (200 MHz, DMSO-d6): δ 7.74 (1H, t, J = 5.4 Hz, NH), 6.94 (1H, t, J = 5.4 Hz, NH), 5.40 (1H, d, J = 4.8 Hz, OH), 3.82-3.75 (1H, m, CHOH), 3.31-3.06 (2H, m, CH2NHCO), 2.91-2.88 (2H, m, CH2NHSO2), 2.88 (3H, s, CH3SO2), 1.62-1.45 (4H, m, 2 × CH2), 1.29-1.18 (24H, m, 12 × CH2), 0.85 (3H, t, J = 6.8 Hz, CH3). 13C NMR (50 MHz, DMSO-d6): δ 175.2, 71.4, 36.1, 34.8, 31.9, 29.3, 25.1, 22.7, 14.6. Anal. calcd for C20H42N2O4S: C, 59.08; H, 10.41; N, 6.89. Found: C, 58.87; H, 10.63; N, 6.98.
2-Hydroxy-N-(4-(methylsulfonamido)butyl)hexadecanamide (7b)
Yield 36%; white solid; mp 139 °C. 1H NMR (200 MHz, DMSO-d6): δ 7.74 (1H, t, J = 5.4 Hz, NH), 6.94 (1H, t, J = 5.4 Hz, NH), 5.42 (1H, d, J = 4.8 Hz, OH), 3.80-3.76 (1H, m, CHOH), 3.30-3.05 (2H, m, CH2NHCO), 2.90-2.89 (2H, m, CH2NHSO2), 2.88 (3H, s, CH3SO2), 1.62-1.45 (6H, m, 3 × CH2), 1.30-1.15 (24H, m, 12 × CH2), 0.85 (3H, t, J = 6.8 Hz, CH3). 13C NMR (50 MHz, DMSO-d6): δ 175.1, 71.3, 36.1, 34.7, 31.9, 29.6, 29.3, 25.1, 22.7, 14.6. Anal. calcd for C21H44N2O4S: C, 59.96; H, 10.54; N, 6.66. Found: C, 59.80; H, 10.78; N, 6.78.
(S,E)-tert-Butyl 1-cyanohept-1-en-3-ylcarbamate (10)
To a solution of compound 9 (0.22g, 1 mmol) in a mixture of toluene-EtOAc (6 mL), a solution of NaBr (0.11 g, 1.1 mmol) in water (0.5 mL) was added, followed by TEMPO (0.21 mg. 0.01 mmol). To the resulting biphasic system, which was cooled at −5 °C, an aqueous solution of 0.35 M NaOCl (2.2 mL, 1.1 mmol) containing NaHCO3 (0.24 g, 3 mmol) was added drop-wise while stirring vigorously at −5 °C over a period of 1 h. After the mixture had been stirred for a further 15 min at 0 °C, EtOAc (10 mL) and H2O (5 mL) were added. The aqueous layer was separated and washed with EtOAc (10 mL). The combined organic layers were washed consecutively with 5% aqueous citric acid (10 mL) containing KI (0.18 g), 10% aqueous Na2S2O3 (10 mL), and brine, dried over Na2SO4 and evaporated under reduced pressure. The residue was used immediately in the next step without any purification.
To the solution of (S)-tert-butyl 1-oxohexan-2-ylcarbamate (0.22g, 1 mmol) in dry THF (10 mL), NCCH=P(C6H5)3 (0.33 g, 1.1 mmol) was added and the reaction mixture was refluxed for 1 h. The organic solvent was evaporated under reduced pressure and the residue was purified by column chromatography [EtOAc-petroleum ether (bp 40-60 °C) 3:7].Yield 74%; white solid; mp 50-53 °C; [α]D -11.8 (c 1.0 CHCl3). 1H NMR (200 MHz, CDCl3): δ 6.63 (1H, dd, J = 5.4 Hz, J = 16.4 Hz, CH=CHCN), 5.48 (1H, d, J = 16.4 Hz, CH=CHCN), 4.52 (1H, d, J = 6.6 Hz, OCONH), 4.25-4.05 (1H, m, CHNH), 1.52-1.45 [11H, m, C(CH3)3, CH2CH], 1.45-1.10 (4H, m, 2 × CH2), 0.91 (3H, t, J = 6.0 Hz, CH3). 13C NMR (50 MHz, CDCl3): δ 155.2, 154.9, 117.1, 99.4, 79.5, 52.1, 33.7, 28.2, 27.6, 22.2, 13.8. Anal. calcd for C13H22N2O2: C, 65.51; H, 9.30; N, 11.75. Found: C, 65.43; H, 9.54; N, 11.81.
(S)-tert-Butyl 1-cyanoheptan-3-ylcarbamate (11)
To a solution of the unsaturated nitrile 10 (0.24g, 1 mmol) in MeOH (10 mL) (through which N2 had been passed for 5 min), 10% Pd/C catalyst was added. The reaction mixture was stirred under H2 atmosphere overnight at room temperature. The catalyst was removed by filtration through a pad of Celite and the organic solvent was evaporated under reduced pressure. The residue was purified by column chromatography [EtOAc-petroleum ether (bp 40-60 °C) 3:7]. Yield 84%; white solid; mp 38-40 °C; [α]D +0.7 (c 1.0 CHCl3). 1H NMR (200 MHz, CDCl3): δ 4.42 (1H, d, J = 8.8 Hz, OCONH), 3.62-3.48 (1H, m, NHCH), 2.38 (2H, t, J = 7.6 Hz, CH2CN), 1.94-1.58 (2H, m, CH2CH2CN), 1.41-1.28 [11H, m, C(CH3)3, CH2CH], 1.28-1.18 (4H, m, 2 × CH2), 0.87 (3H, t, J = 6.0 Hz, CH3). 13C NMR (50 MHz, CDCl3): δ 155.7, 119.7, 79.4, 50.0, 34.8, 31.6, 28.2, 27.9, 22.3, 14.1, 13.9. Anal. calcd for C13H24N2O2: C, 64.97; H, 10.07; N, 11.66. Found: C, 64.75; H, 10.22; N, 11.48.
General Procedure for the Synthesis of Compounds 12a,b
To a stirred solution of nitrile 11 (0.24g, 1 mmol) in MeOH (8 mL) was added nickel chloride hexahydrate (1.19 g, 5 mmol) at 0 °C, followed by sodium borohydride (0.30 g, 8 mmol) in small portions. After stirring for 30 min at room temperature, water was added and the mixture neutralized with 0.5 M H2SO4 and the organic solvent was removed under reduced pressure. The aqueous phase was extracted with ethyl acetate (5 × 15 mL). After drying (Na2SO4) the solvent was evaporated to dryness and the amine was used directly for the reaction with sulfonyl chlorides as described above.
(S)-tert-Butyl 1-(methylsulfonamido)octan-4-ylcarbamate (12a)
Yield 40%; white solid; mp 75-77 °C; [α]D -5.0 (c 1.0 CHCl3). 1H NMR (200 MHz, CDCl3): δ 5.17-5.13 (1H, m, NH), 4.39 (1H, d, J = 8.8 Hz, NH), 3.60-3.35 (1H, m, CHNH), 3.10-3.02 (2H, m, CH2NHSO2), 2.92 (3H, s, SO2CH3), 1.62-1.52 (2H, m, CH2CH2NHSO2), 1.47-1.27 [17H, m, C(CH3)3, 4 × CH2], 0.86 (3H, t, J = 6.5 Hz, CH3). 13C NMR (50 MHz, CDCl3): δ 155.8, 78.9, 50.0, 43.0, 39.9, 35.3, 32.6, 28.3, 27.9, 26.3, 22.4, 13.9. Anal. calcd for C14H30N2O4S: C, 52.15; H, 9.38; N, 8.69. Found: C, 52.00; H, 9.49; N, 8.63.
(S)-tert-Butyl 1-(4-methylphenylsulfonamido)octan-4-ylcarbamate (12b)
Yield 38%; white solid; mp 88-90 °C; [α]D -2.7 (c 1.0 CHCl3). 1H NMR (200 MHz, CDCl3): δ 7.70 (2H, d, J = 8.0 Hz, Ph), 7.25 (2H, d, J = 8.0 Hz, Ph), 5.05-4.98 (1H, m, NH), 4.22 (1H, d, J = 9.2 Hz, NH), 3.48-3.23 (1H, m, CHNH), 2.98-2.76 (2H, m, CH2NHSO2), 2.38 (3H, s, PhCH3), 1.43-1.10 [19H, m, 5 × CH2, C(CH3)3], 0.82 (3H, t, J = 6.2 Hz, CH3CH2). 13C NMR (50 MHz, CDCl3): δ 155.9, 143.2, 137.0, 129.6, 127.0, 79.1, 50.1, 43.0, 35.2, 32.7, 28.3, 25.8, 22.5, 21.5, 14.0. Anal. calcd for C20H34N2O4S: C, 60.27; H, 8.60; N, 7.03. Found: C, 60.02; H, 8.82; N, 6.95.
General Procedure for the Coupling of Boc-γ-Aminobutyric Acid with Sulfonamides
To a solution of compound 15 (0.20g, 1 mmol) in CH2Cl2 (20 mL) MsCl or TsCl (1 mmol) was added followed by DCC (0.21 g, 1 mmol) and 4-dimethylaminopyridine (0.12 g, 1 mmol). The mixture was stirred for 24 h and the dicyclohexylurea was removed by filtration through a pad of Celite. The organic solvent was evaporated under reduced pressure and the residue was purified by column chromatography [CHCl3-MeOH 9:1].
tert-Butyl 4-(methylsulfonamido)-4-oxobutylcarbamate (16a)
Yield 64%; white solid; mp 105-107 °C. 1H NMR (200 MHz, CDCl3): δ 4.98 (1H, t, J = 5.6 Hz, NH), 3.26 (3H, s, SO2CH3), 3.20-3.05 (2H, m, NHCH2), 2.38 (2H, t, J = 7.2 Hz, CH2CO), 1.85-1.62 (2H, m, CH2CH2CO), 1.42 [9H, s, (CH3)3]. 13C NMR (50 MHz, CDCl3): δ 172.9, 156.8, 79.8, 41.2, 39.2, 33.4, 28.3, 22.4. Anal. calcd for C10H20N2O5S: C, 42.84; H, 7.19; N, 9.99. Found: C, 42.65; H, 7.36; N, 10.05.
tert-Butyl 4-(4-methylphenylsulfonamido)-4-oxobutylcarbamate (16b)
Yield 68%; white solid; mp 144-146 °C. 1H NMR (200 MHz, CDCl3): δ 7.87 (2H, d, J = 8.4 Hz, Ph), 7.25 (2H, d, J = 8.0 Hz, Ph), 4.93 (1H, t, J = 5.6 Hz, NH), 3.12-2.95 (2H, m, NHCH2), 2.36 (3H, s, PhCH3), 2.26 (2H, t, J = 6.6 Hz, CH2CO), 1.78-1.58 (2H, m, CH2CH2CO), 1.36 [9H, s, C(CH3)3]. 13C NMR (50 MHz, CDCl3): δ 171.4, 156.7, 144.7, 135.7, 129.4, 128.1, 79.5, 39.1, 33.2, 28.2, 25.2, 21.5. Anal. calcd for C16H24N2O5S: C, 53.91; H, 6.79; N, 7.86. Found: C, 53.82; H, 6.87; N, 7.83.
General Procedure for the Removal of Boc Group
N-Boc-protected compound (1 mmol) was added to a solution of 5 N HCl in MeOH (7 mL, 35 mmol). After being stirred at room temperature for 30 min, the mixture was evaporated, dry Et2O was added, and the product was filtered and used for coupling with 2-hydroxy acid.
General Method for the Coupling of 2-Hydroxy-hexadecanoic Acids with Amines
To a stirred solution of 2-hydroxy-hexadecanoic acid (0.27g, 1 mmol) and the diamine (1 mmol) in CH2Cl2 (5 mL), Et3N (0.15 mL, 1.1 mmol) and subsequently WSCI (0.21 g, 1.1 mmol) and HOBt (0.15 g, 1 mmol) were added at 0 °C. The reaction mixture was stirred for 1 h at 0 °C and overnight at room temperature. The solvent was evaporated under reduced pressure and the residue was used immediately in the next step without any purification.
2-Hydroxy-N-[3-(4-methylphenylsulfonamido)propyl]hexadecanamide (4a)
Yield 52%; white solid; mp 82-85 °C. 1H NMR (200 MHz, CDCl3): δ 7.68 (2H, d, J = 7.8 Hz, Ph), 7.25 (2H, d, J = 7.6 Hz, Ph), 7.05 (1H, t, J = 5.6 Hz, NH), 5.98 (1H, t, J = 7.8 Hz, NH), 4.12-3.98 (1H, m, CHOH), 3.75-3.59 (1H, m, CHOH), 3.38-3.22 (2H, m, CH2NHCO), 3.00-2.78 (2H, m, CH2NHSO2), 2.38 (3H, s, 3H, C6H4CH3), 1.80-1.40 (4H, m, 2 × CH2), 1.38-1.12 (24H, m, 12 × CH2), 0.85 (3H, t, J = 6.6 Hz, CH3). 13C NMR (50 MHz, CDCl3): δ 175.4, 143.2, 136.9, 129.6, 126.9, 72.0, 39.9, 35.6, 34.7, 31.9, 29.7, 29.6, 29.4, 29.3, 25.1, 22.6, 21.5, 14.1. Anal. calcd for C26H46N2O4S: C, 64.69; H, 9.61; N, 5.80. Found: C, 64.55; H, 9.68; N, 5.86.
2-Hydroxy-N-(4-(4-methylphenylsulfonamido)butyl)hexadecanamide (4b)
Yield 48%; white solid; mp 90-91 °C. 1H NMR (200 MHz, CDCl3): δ 7.68 (2H, d, J = 7.8 Hz, Ph), 7.24 (2H, d, J = 9.2 Hz, Ph), 6.83 (1H, t, J = 5.4 Hz, NH), 5.43 (1H, t, J = 7.8 Hz, NH), 4.08-3.98 (1H, m, CHOH), 3.72-3.57 (1H, m, CHOH), 3.45-3.02 (2H, m, CH2NHCO), 2.98-2.79 (2H, m, CH2NHSO2), 2.37 (3H, s, C6H4CH3), 1.81-1.48 (6H, m, 3 × CH2), 1.40-1.05 (24H, s, 12 × CH2), 0.82 (3H, t, J = 6.6 Hz, CH3). 13C NMR (50 MHz, CDCl3): δ 175.0, 143.7, 136.9, 130.0, 127.3, 72.4, 43.0, 38.6, 35.0, 32.2, 29.9, 29.8, 29.7, 29.6, 26.9, 25.4, 22.9, 21.8, 14.4. Anal. calcd for C27H48N2O4S: C, 65.28; H, 9.74; N, 5.64. Found: C, 65.37; H, 9.58; N, 5.73.
2-Hydroxy-N-[(S)-1-(methylsulfonamido)octan-4-yl]hexadecanamide (13a)
Yield 45%; white solid; mp 98-101 °C. 1H NMR (200 MHz, CDCl3): δ 6.52-6.32 (1H, m, NH), 5.37-5.02 (1H, m, NH), 4.10-4.02 (1H, s, CHOH), 4.00-3.81 (1H, s, CHNH), 3.60-3.22 (1H, b, CHOH), 3.20-3.10 (2H, m, CH2NHSO2), 2.95 (3H, s, SO2CH3), 1.78-1.26 (36H, m, 18 × CH2), 0.88 (6H, t, J = 6.0 Hz, 2 × CH3). 13C NMR (50 MHz, CDCl3): δ 174.5, 72.3, 48.7, 48.2, 43.0, 39.9, 35.0, 34.7, 32.3, 32.0, 29.7, 29.5, 28.1, 26.3, 25.1, 22.7, 22.5, 14.1, 14.0. Anal. calcd for C25H52N2O4S: C, 62.98; H, 10.99; N, 5.88. Found: C, 62.73; H, 11.14; N, 5.99.
2-Hydroxy-N-[(S)-1-(4-methylphenylsulfonamido)octan-4-yl]hexadecanamide (13b)
Yield 45%; white solid; 1H NMR (200 MHz, CDCl3): δ 7.73 (2H, d, J = 7.6 Hz, Ph), 7.29 (2H, d, J = 7.8 Hz, Ph), 6.63-6.40 (1H, m, NH), 5.74-5.51 (1H, m, NH), 4.18-4.00 (1H, m, CHOH), 3.90-3.75 (1H, m, CHNH), 3.02-2.75 (2H, m, CH2NHSO2), 2.42 (3H, s, PhCH3), 1.84-1.15 (36H, m, 18 × CH2), 0.87 (6H, s, 2 × CH3). 13C NMR (50 MHz, CDCl3): δ 174.5, 143.3, 136.7, 129.6, 127.0, 72.1, 48.8, 48.3, 42.9, 34.9, 32.2, 31.9, 29.7, 29.3, 28.0, 25.7, 25.0, 22.6, 22.5, 21.4, 14.1, 13.9. Anal. calcd for C31H56N2O4S: C, 67.35; H, 10.21; N, 5.07. Found: C, 67.46; H, 10.09; N, 5.03.
2-Hydroxy-N-[4-(methylsulfonamido)-4-oxobutyl]hexadecanamide (17a)
Yield 45%; white solid; mp 94-96 °C. 1H NMR (200 MHz, CDCl3): δ 6.79 (1H, t, J = 5.4 Hz, NH), 4.07 (1H, dd, J = 3.6 Hz, J = 7.8 Hz, CHOH), 3.68 (3H, s, SO2CH3), 3.41-3.24 (2H, m, NHCH2), 2.88 (1H, b, CHOH), 2.37 (2H, t, J = 7.2 Hz, CH2CONH), 1.90-1.75 (3H, m, CHHCHOH, CH2CH2CO), 1.73-1.42 (1H, m, CHHCHOH), 1.38-1.10 (24H, s, 12 × CH2), 0.87 (3H, t, J = 6.4 Hz, CH3). 13C NMR (50 MHz, CDCl3): δ 174.2, 173.8, 72.1, 51.7, 38.4, 34.9, 31.9, 31.4, 29.7, 29.6, 29.5, 29.4, 29.3, 23.0, 24.7, 14.1. Anal. calcd for C21H42N2O5S: C, 58.03; H, 9.74; N, 6.45. Found: C, 57.83; H, 9.93; N, 6.41.
2-Hydroxy-N-[4-(4-methylphenylsulfonamido)-4-oxobutyl]hexadecanamide (17b)
Yield 39%; white solid; mp 78-80 °C. 1H NMR (200 MHz, CDCl3): δ 7.87 (2H, d, J = 8.0 Hz, Ph), 7.25 (2H, d, J = 8.0 Hz, Ph), 7.15 (1H, t, J = 5.4 Hz, NH), 4.17-4.08 (1H, m, CHOH), 3.58-3.32 (1H, m, CHHNHCO), 3.27-3.05 (1H, m, CHHNHCO), 2.36 (3H, s, PhCH3), 2.24 (2H, t, J = 6.0 Hz, CH2CO), 1.95-1.65 (3H, m, CH2CH2CO, CHHCHOH), 1.60-1.09 (25H, m, 12 × CH2, CHHCHOH), 0.82 (3H, t, J = 6.2 Hz, CH3). 13C NMR (50 MHz, CDCl3): δ 176.3, 172.3, 144.8, 135.7, 129.4, 128.3, 72.2, 38.4, 34.4, 33.9, 31.9, 29.7, 29.5, 29.3, 25.3, 24.7, 22.6, 21.6, 14.1. Anal. calcd for C27H46N2O5S: C, 63.50; H, 9.08; N, 5.48. Found: C, 63.24; H, 9.21; N, 5.63.
Synthesis of N-[4-(N,4-Dimethylphenylsulfonamido)-4-oxobutyl)-2-hydroxyhexadecanamide (19)
To a stirred solution of 17b (0.51g, 1 mmol) in dry THF (10 mL), Na2CO3 (0.21g, 2 mmol) and CH3I (0.48mL, 7.7 mmol) were added at room temperature. The reaction mixture was stirred for 24 h at room temperature. The organic solvent was evaporated under reduced pressure and water was added. The mixture extracted with CHCl3 (3 × 10 mL), the organic layers were combined, dried over Na2SO4 and evaporated under reduced pressure. The residue was purified by column chromatography [CHCl3-MeOH 95:5].
Yield 41%; white solid; mp 60-62 °C. 1H NMR (200 MHz, CDCl3): δ 7.78 (2H, d, J = 8.4 Hz, Ph), 7.34 (2H, d, J = 8.4 Hz, Ph), 6.76 (1H, t, J = 5.8 Hz, NH), 4.05 (1H, dd, J = 3.2 Hz, J = 7.2 Hz, CHOH), 3.42-3.00 (5H, m, CONHCH2, NCH3), 2.67 (2H, t, J = 6.6 Hz, CH2CO), 2.44 (3H, s, PhCH3), 1.95-1.68 (3H, m, CONHCH2CH2, CHHCHOH), 1.65-1.53 (1H, m, CHHCHOH), 1.45-1.05 (24H, m, 12 × CH2), 0.88 (3H, t, J = 7.0 Hz, CH3). 13C NMR (50 MHz, CDCl3): δ 174.4, 173.0, 145.1, 135.9, 129.9, 127.5, 72.1, 38.3, 34.7, 33.8, 33.1, 31.9, 29.6, 29.5, 29.4, 29.3, 25.1, 24.3, 22.6, 21.6, 14.1. Anal. calcd for C28H48N2O5S: C, 64.09; H, 9.22; N, 5.34. Found: C, 63.92; H, 9.34; N, 5.39.
General Procedures for the Oxidation of 2-Hydroxyamides to 2-oxoamides
Method A
A solution of 2-hydroxyamide (1 mmol) in dry CH2Cl2 (1 mL) was added drop-wise to a stirred solution of the Dess-Martin reagent (0.47 g, 1.1 mmol) in CH2Cl2 (3 mL) over a period of 2 min. After stirring for 20 min at room temperature the mixture was diluted with Et2O (20 mL) and poured into a saturated aqueous NaHCO3 (10 mL) containing 1.5 g of Na2S2O3. The mixture was stirred for 5 min. Et2O (20 mL) was added, the layers were separated and the ether layer was washed with saturated aqueous NaHCO3, water and dried (Na2SO4). The organic solvent was evaporated under reduced pressure and the residue was purified by column chromatography.
N-(3-(4-Methylphenylsulfonamido)propyl)-2-oxohexadecanamide (5a)
Yield 73%; white solid; 80-81 °C. 1H NMR (200 MHz, CDCl3): δ 7.75 (2H, d, J = 8.0 Hz, Ph), 7.30 (2H, d, J = 8.0 Hz, Ph), 7.20-7.08 (1H, m, NH), 5.26 (1H, t, J = 7.8 Hz, NH), 3.40-3.32 (2H, m, CH2NHCO), 2.98-2.84 (4H, m, CH2NHSO2, CH2COCO), 2.42 (3H, s, C6H4CH3), 1.81-1.15 (26H, m, 13 × CH2), 0.88 (3H, t, J = 6.6 Hz, CH3). 13C NMR (50 MHz, CDCl3): δ 198.8, 160.9, 143.4, 137.0, 129.7, 127.0, 39.8, 36.8, 35.8, 31.9, 29.7, 29.6, 29.4, 29.3, 29.1, 23.1, 22.7, 21.5, 14.1. MS(FAB): m/z = 481 (100%) [M + H]+. Anal. calcd for C26H44N2O4S: C, 64.96; H, 9.23; N, 5.83. Found: C, 65.03; H, 9.44; N, 5.78.
N-(4-(4-Methylphenylsulfonamido)butyl)-2-oxohexadecanamide (5b)
Yield 78%; white solid; 100-103 °C. 1H NMR (200 MHz, CDCl3): δ 7.68 (2H, d, J = 8.2 Hz, Ph), 7.25 (2H, d, J = 8.0 Hz, Ph), 6.95 (1H, t, J = 5.4 Hz, NH), 4.61 (1H, t, J = 6.6 Hz, NH), 3.21-3.12 (2H, m, CH2NHCO), 2.95-2.70 (4H, m, CH2NHSO2, CH2COCO), 2.37 (3H, s, C6H4CH3), 1.72-1.06 (28H, m, 14 × CH2), 0.82 (3H, t, J = 6.6 Hz, CH3). 13C NMR (50 MHz, CDCl3): δ 199.5, 160.5, 143.7, 137, 130.0, 127.3, 42.8, 38.9, 37.0, 32.1, 29.9, 29.7, 29.6, 29.3, 27.0, 26.5, 23.4, 22.9, 21.7, 14.4. Anal. calcd for C27H46N2O4S: C, 65.55; H, 9.37; N, 5.66. Found: C, 65.34; H, 9.52; N, 5.78.
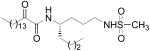
(S)-N-[1-(Methylsulfonamido)octan-4-yl]-2-oxohexadecanamide (14a)
Yield 79%; white solid; mp 88-91 °C. 1H NMR (200 MHz, CDCl3): δ 6.76 (1H, d, J = 9.2 Hz, NH), 4.72 (1H, t, J = 5.8 Hz, NH), 3.92-3.81 (1H, m, CHNH), 3.20-3.04 (2H, m, CH2NHSO2), 2.96 (3H, s, SO2CH3), 2.92 (2H, t, J = 7.8 Hz, CH2COCO), 1.80-1.05 (34H, m, 17 × CH2), 0.88 (6H, t, J = 6.6 Hz, 2 × CH3). 13C NMR (50 MHz, CDCl3): δ 199.5, 160.0, 49.1, 42.8, 40.1, 36.8, 34.7, 32.1, 31.9, 29.6, 29.4, 29.3, 29.0, 28.0, 26.4, 23.1, 22.7, 22.4, 14.1, 13.9. MS (FAB): m/z = 475 (100%) [M + H]+. Anal. calcd for C25H50N2O4S: C, 63.25; H, 10.62; N, 5.90. Found: C, 63.08; H, 10.77; N, 5.96.
(S)-N-[1-(4-Methylphenylsulfonamido)octan-4-yl]-2-oxohexadecanamide (14b)
Yield 83%; white solid; mp 99-101 °C. 1H NMR (200 MHz, CDCl3): δ 7.69 (2H, d, J = 7.6 Hz, Ph), 7.25 (2H, d, J = 8.0 Hz, Ph), 6.62 (1H, d, J = 10.0 Hz, NH), 4.75 (1H, t, J = 6.0 Hz, NH), 3.79-3.67 (1H, m, CHNH), 2.98-2.75 (4H, m, CH2NH, CH2COCO), 2.37 (3H, s, PhCH3), 1.74-1.02 (34H, m, 17 × CH2), 0.82 (6H, s, 2 × CH3). 13C NMR (50 MHz, CDCl3): δ 199.5, 160.0, 143.3, 136.9, 129.7, 127.1, 49.1, 42.9, 36.8, 34.7, 32.2, 31.9, 29.6, 29.4, 29.3, 29.1, 28.0, 25.9, 23.2, 22.7, 22.4, 21.5, 14.1, 13.9. MS (FAB): m/z = 551 (100%) [M + H]+. Anal. calcd for C31H54N2O4S: C, 67.59; H, 9.88; N, 5.09. Found: C, 67.45; H, 9.95; N, 5.14.
N-[4-(Methylsulfonamido)-4-oxobutyl]-2-oxohexadecanamide (18a)
Yield 90%; white solid; mp 70-72 °C. 1H NMR (200 MHz, CDCl3): δ 7.18 (1H, t, J = 5.8 Hz, NH), 3.64 (3H, s, SO2CH3), 3.69-3.25 (m, 2H, NHCH2), 2.87 (2H, t, J = 7.0 Hz, CH2COCO), 2.34 (2H, t, J = 7.4 Hz, CH2CONH), 1.86 (2H, quintet, J = 6.8 Hz, NHCH2CH2), 1.61-1.50 (2H, m, CH2CH2COCO), 1.38-1.12 (22H, m, 11 × CH2), 0.84 (3H, t, J = 6.2 Hz, CH3). 13C NMR (50 MHz, CDCl3): δ 199.1, 173.3, 160.3, 51.6, 38.6, 36.6, 31.8, 31.2, 29.5, 29.3, 29.2, 29.0, 24.3, 23.1, 22.6, 14.0. Anal. calcd for C21H40N2O5S: C, 58.30; H, 9.32; N, 6.48. Found: C, 58.18; H, 9.41; N, 6.61.
N-[4-(4-Methylphenylsulfonamido)-4-oxobutyl]-2-oxohexadecanamide (18b)
Yield 96%; white solid; mp 124-126 °C. 1H NMR (200 MHz, CDCl3): δ 7.86 (2H, d, J = 8.0 Hz, Ph), 7.31-7.20 (3H, m, Ph, NH), 3.27-3.15 (2H, m, NHCH2), 2.83 (2H, t, J = 7.4 Hz, CH2COCO), 2.36 (3H, s, PhCH3), 2.26 (2H, t, J = 6.6 Hz, CH2CONHSO2), 1.83-1.63 (2H, m, NHCH2CH2), 1.58-1.35 (2H, m, CH2CH2COCO), 1.35-1.05 (22H, s, 11 × CH2), 0.80 (3H, t, J = 5.8 Hz, CH3). 13C NMR (50 MHz, CDCl3): δ 198.7, 171.0, 160.9, 144.9, 135.7, 129.5, 128.2, 38.3, 36.8, 33.4, 31.8, 29.6, 29.4, 29.3, 29.0, 24.4, 23.0, 22.6, 21.6, 14.1. MS (FAB): m/z = 509 (90%) [M + H]+. Anal. calcd for C27H44N2O5S: C, 63.75; H, 8.72; N, 5.51. Found: C, 63.42; H, 8.99; N, 5.63.
N-[4-(N,4-Dimethylphenylsulfonamido)-4-oxobutyl)-2-oxohexadecanamide (20)
Yield 75%; light yellow solid; mp 80-82 °C. 1H NMR (200 MHz, CDCl3): δ 7.76 (2H, d, J = 8.4 Hz, Ph), 7.36 (2H, d, J = 8.2 Hz, Ph), 7.07 (1H, t, J = 5.8 Hz, NH), 3.41-3.18 (5H, m, NHCH2, NCH3), 2.89 (2H, t, J = 7.4 Hz, CH2COCO), 2.73 (2H, t, J = 7.0 Hz, CH2CONCH3), 2.45 (3H, s, PhCH3), 1.87 (2H, quintet, J = 6.6 Hz, NHCH2CH2), 1.78-1.52 (2H, m, CH2CH2COCO), 1.45-1.15 (22H, m, 11 × CH2), 0.88 (3H, t, J = 6.2 Hz, CH3). 13C NMR (50 MHz, CDCl3): δ 199.1, 172.5, 160.3, 145.0, 136.1, 130.0, 127.3, 38.5, 36.7, 33.8, 33.0, 31.9, 29.6, 29.4, 29.3, 29.1, 24.2, 23.2, 22.7, 21.6, 14.1. Anal. calcd for C28H46N2O5S: C, 64.33; H, 8.87; N, 5.36. Found: C, 64.49; H, 8.78; N, 5.29.
Method B
To a solution of 2-hydroxyamide (1 mmol), in CH3COOH (5 mL), PDC (1.13 g, 3 mmol) was added and the mixture was stirred for 30 min at room temperature. The mixture was then neutralized with 5% NaHCO3 and extacted with AcOEt (3 × 10 mL). The organic layers were washed with brine and dried (Na2SO4). The organic solvent was evaporated under reduced pressure and the residue was purified by column chromatography.
N-(3-(Methylsulfonamido)propyl)-2-oxohexadecanamide (8a)
Yield 66%; white solid; mp 107-110 °C. 1H NMR (200 MHz, CDCl3): δ 7.35-7.27 (1H, m, NH), 5.10 (1H, t, J = 6.2 Hz, NH), 3.51-3.38 (2H, m, CH2NHCO), 3.20-3.08 (2H, m, CH2NHSO2), 2.97 (3H, s, SO2CH3), 2.94 (2H, t, J = 7.8 Hz, COCOCH2), 1.85-1.70 (2H, m, CH2), 1.68-1.43 (4H, m, 2 × CH2), 1.40-1.05 (20H, m, 10 × CH2), 0.88 (3H, t, J = 6.9 Hz, CH3). 13C NMR (50 MHz, CDCl3): δ 199.1, 161.3, 40.7, 40.1, 37.0, 36.0, 32.1, 30.4, 29.9, 29.7, 29.6, 29.3, 23.4, 23.0, 14.4. Anal. calcd for C20H40N2O4S: C, 59.37; H, 9.96; N, 6.92. Found: C, 59.14; H, 10.15; N, 6.98.
N-(4-(Methylsulfonamido)butyl)-2-oxohexadecanamide (8b)
Yield 62%; white solid; mp 110-113 °C. 1H NMR (200 MHz, CDCl3): δ 7.15-7.00 (1H, m, NH), 4.62-4.51 (1H, m, NH), 3.42-3.30 (2H, m, CH2NHCO), 3.20-3.14 (2H, m, CH2NHSO2), 2.97 (3H, s, SO2CH3), 2.95-2.82 (2H, m, CH2COCO), 1.75-1.15 (28H, m, 14 × CH2), 0.88 (3H, t, J = 6.6 Hz, CH3). 13C NMR (50 MHz, CDCl3): δ 199.0, 161.4, 42.7, 40.3, 38.6, 36.7, 31.9, 29.6, 29.4, 29.3, 29.0, 27.3, 26.4, 23.1, 22.7, 14.1. Anal. calcd for C21H42N2O4S: C, 60.25; H, 10.11; N, 6.69. Found: C, 60.01; H, 10.33; N, 6.74.
Acknowledgments
This project was supported by NIH Grant GM 20,501 (E.A.D) and by AnalgesiX/UC Discovery Biotechnology Grant #B1002-10303 (E.A.D.). The project is co-funded by the European Social Fund and National Resources (EPEAEK II)(G.K.).
Abbreviations
- AcNH-TEMPO
4-acetamido-2,2,6,6-tetramethyl-1-piperidinyloxy free radical
- Boc
N-(tert-butoxycarbonyl)
- Dess-Martin reagent
1,1,1-tris(acetyloxy)-1,1-dihydro-1,2-benziodoxol-3-(1H)-one
- DCC
N,N′-dicyclohexylcarbodiimide
- DMAP
4-dimethylaminopyridine
- DMSO
dimethyl sulfoxide
- EtOAc
ethyl acetate
- GIVA cPLA2
Group IVA cytosolic phospholipase A2
- GV sPLA2
Group V secreted phospholipase A2
- GVIA iPLA2
Group VIA calcium-independent phospholipase A2
- HOBt
N-hydroxybenzotriazole
- MsCl
methanesulfonyl chloride
- NMM
N-methylmorpholine
- PAF
platelet-activating factor
- PDC
pyridinium dichromate
- THF
tetrahydrofuran
- TsCl
p-toluenesulfonyl chloride
- WSCI
N-ethyl-N′-dimethylaminopropylcarbodiimide
References
- 1.Schaloske RH, Dennis EA. The phospholipase A(2) superfamily and its group numbering system. Biochim Biophys Acta. 2006;1761:1246–1259. doi: 10.1016/j.bbalip.2006.07.011. [DOI] [PubMed] [Google Scholar]
- 2.Kudo I, Murakami M. Phospholipase A2 enzymes. Prostaglandins Other Lipid Mediat. 2002;68-69:3–58. doi: 10.1016/s0090-6980(02)00020-5. [DOI] [PubMed] [Google Scholar]
- 3.Balsinde J, Winstead MV, Dennis EA. Phospholipase A2 regulation of arachidonic acid mobilization. FEBS Lett. 2002;531:2–6. doi: 10.1016/s0014-5793(02)03413-0. [DOI] [PubMed] [Google Scholar]
- 4.Six DA, Dennis EA. The expanding superfamily of phospholipase A(2) enzymes: classification and characterization. Biochim Biophys Acta. 2000;1488:1–19. doi: 10.1016/s1388-1981(00)00105-0. [DOI] [PubMed] [Google Scholar]
- 5.Leslie CC. Regulation of the specific release of arachidonic acid by cytosolic phospholipase A2. Prostaglandins Leukot Essent Fatty Acids. 2004;70:373–376. doi: 10.1016/j.plefa.2003.12.012. [DOI] [PubMed] [Google Scholar]
- 6.Winstead MV, Balsinde J, Dennis EA. Calcium-independent phospholipase A(2): structure and function. Biochim Biophys Acta. 2000;1488:28–39. doi: 10.1016/s1388-1981(00)00107-4. [DOI] [PubMed] [Google Scholar]
- 7.Balsinde J, Balboa MA. Cellular regulation and proposed biological functions of group VIA calcium-independent phospholipase A(2) in activated cells. Cellular Signalling. 2005;17:1052–1062. doi: 10.1016/j.cellsig.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 8.Berg OG, Gelb MH, Tsai MD, Jain MK. Interfacial enzymology: The secreted phospholipase A2-paradigm. Chem Rev. 2001;101:2613–2654. doi: 10.1021/cr990139w. [DOI] [PubMed] [Google Scholar]
- 9.Balestrieri B, Arm JP. Group V sPLA2: Classical and novel functions. Biochim Biophys Acta. 2006;1761:1280–1288. doi: 10.1016/j.bbalip.2006.07.008. [DOI] [PubMed] [Google Scholar]
- 10.Rosengren B, Jonsson-Rylander AC, Peilot H, Camejo G, Hurt-Camejo E. Distinctiveness of secretory phospholipase A2 group IIA and V suggesting unique roles in atherosclerosis. Biochim Biophys Acta. 2006;1761:1301–1308. doi: 10.1016/j.bbalip.2006.06.008. [DOI] [PubMed] [Google Scholar]
- 11.Mounier CM, Ghomashchi F, Lindsay MR, James S, Singer AG, Parton RG, Gelb MH. Arachidonic acid release from mammalian cells transfected with human groups IIA and X secreted phospholipase A2 occurs predominantly during the secretory process and with the involvement of cytosolic phospholipase A2-α. J Biol Chem. 2004;279:25024–25038. doi: 10.1074/jbc.M313019200. [DOI] [PubMed] [Google Scholar]
- 12.Shirai Y, Balsinde J, Dennis EA. Localization and functional interrelationships among cytosolic Group IV, secreted Group V, and Ca2+-independent Group VI phospholipase A2s in P388D1 macrophages using GFP/RFP constructs. Biochim Biophys Acta. 2005;1735:119–129. doi: 10.1016/j.bbalip.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 13.Magrioti V, Kokotos G. Synthetic inhibitors of Group IVA and Group VIA phospholipase A2. Anti-Inflamm Anti-Allergy Agents Med Chem. 2006;5:189–203. [Google Scholar]
- 14.Kokotos G, Kotsovolou S, Six DA, Constantinou-Kokotou V, Beltzner CC, Dennis EA. Novel 2-oxoamide inhibitors of human Group IVA phospholipase A2. J Med Chem. 2002;45:2891–2893. doi: 10.1021/jm025538p. [DOI] [PubMed] [Google Scholar]
- 15.Kokotos G, Six DA, Loukas V, Smith T, Constantinou-Kokotou V, Hadjipavlou-Litina D, Kotsovolou S, Chiou A, Beltzner CC, Dennis EA. Inhibition of Group IVA cytosolic phospholipase A2 by novel 2-oxoamides in vitro, in cells and in vivo. J Med Chem. 2004;47:3615–3628. doi: 10.1021/jm030485c. [DOI] [PubMed] [Google Scholar]
- 16.Constantinou-Kokotou V, Peristeraki A, Kokotos CG, Six DA, Dennis EA. Synthesis and activity of 2-oxoamides containing long chain beta-amino acids. J Pept Sci. 2005;11:431–435. doi: 10.1002/psc.628. [DOI] [PubMed] [Google Scholar]
- 17.Yaksh TL, Kokotos G, Svensson CI, Stephens D, Kokotos CG, Fitzsimmons B, Hadjipavlou-Litina D, Hua XY, Dennis EA. Systemic and intrathecal effects of a novel series of phospholipase A2 inhibitors on hyperalgesia and spinal prostaglandin E2 release. J Pharmacol Exp Ther. 2006;316:466–475. doi: 10.1124/jpet.105.091686. [DOI] [PubMed] [Google Scholar]
- 18.Stephens D, Barbayianni E, Constantinou-Kokotou V, Peristeraki A, Six DA, Cooper J, Harkewicz R, Deems RA, Dennis EA, Kokotos G. Differential inhibition of Group IVA and Group VIA phospholipases A(2) by 2-oxoamides. J Med Chem. 2006;49:2821–2828. doi: 10.1021/jm050993h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Six DA, Barbayianni E, Loukas V, Constantinou-Kokotou V, Hadjipavlou-Litina D, Stephens D, Wong AC, Magrioti V, Moutevelis-Minakakis P, Baker SF, Dennis EA, Kokotos G. Structure-activity relationship of 2-oxoamide inhibition of Group IVA cytosolic phospholipase A2 and Group V secreted phospholipase A2. J Med Chem. 2007;50:4222–4235. doi: 10.1021/jm0613673. [DOI] [PubMed] [Google Scholar]
- 20.Moutevelis-Minakakis P, Neokosmidi A, Filippakou M, Stephens D, Dennis EA, Kokotos G. Synthesis of lipophilic 2-oxoamides based on γ-aminobutyric and δ-aminovaleric analogues and their activity against phospholipase A2. J Pept Sci. 2007;13:634–641. doi: 10.1002/psc.889. [DOI] [PubMed] [Google Scholar]
- 21.Patani GA, LaVoie EJ. Bioisosterism: A Rational Approach in Drug Design. Chem Rev. 1996;96:3147–3176. doi: 10.1021/cr950066q. [DOI] [PubMed] [Google Scholar]
- 22.Schaaf TK, Hess HJ. Synthesis and biological activity of carboxyl-terminus modified prostaglandin analogues. J Med Chem. 1979;22:1340–1346. doi: 10.1021/jm00197a012. [DOI] [PubMed] [Google Scholar]
- 23.Johansson A, Poliakov A, Akerblom E, Wiklund K, Lindeberg G, Winiwarter S, Danielson UH, Samuelsson B, Hallberg A. Acyl sulfonamides as potent protease inhibitors of the hepatitis C virus full-length NS3 (Protease-Helicase/NTPase): A comparative study of different C-terminals. Bioorg Med Chem. 2003;11:2551–2568. doi: 10.1016/s0968-0896(03)00179-2. [DOI] [PubMed] [Google Scholar]
- 24.Liu DG, Gao Y, Voigt JH, Lee K, Nicklaus MC, Wu L, Zhang ZY, Burke TR. Acylsulfonamide-containing PTP1B inhibitors designed to mimic an enzyme-bound water of hydration. Bioorg Med Chem Lett. 2003;13:3005–3007. doi: 10.1016/s0960-894x(03)00635-8. [DOI] [PubMed] [Google Scholar]
- 25.Allegretti M, Bertini R, Cesta MC, Bizzarri C, Di Bitondo R, Di Cioccio V, Galliera E, Berdini V, Topai A, Zampella G, Russo V, Di Bello N, Nano G, Nicolini L, Locati M, Fantucci P, Florio S, Colotta F. 2-Arylpropionic CXC chemokine receptor 1 (CXCR1) ligands as novel noncometitive CXCL8 inhibitors. J Med Chem. 2005;48:4312–4331. doi: 10.1021/jm049082i. [DOI] [PubMed] [Google Scholar]
- 26.Bellis E, Vasilatou K, Kokotos G. 4-Substituted propyl sulfonamides as enantioselective organocatalysts for aldol reactions. Synthesis. 2005;14:2407–2413. [Google Scholar]
- 27.Cobb AJA, Shaw DM, Longbottom DA, Gold JB, Ley SV. Organocatalysis with proline derivatives: improved catalysts for the asymmetric Mannich, nitro-Michael and aldol reactions. Org Biomol Chem. 2005;3:84. doi: 10.1039/b414742a. [DOI] [PubMed] [Google Scholar]
- 28.Sheehan JC, Cruickshank PA, Boshart GL. A convenient synthesis of water-soluble carbodiimides. J Org Chem. 1961;26:2525–2528. [Google Scholar]
- 29.Kokotos G. A convenient one-pot conversion of N-protected amino acids and peptides into alcohols. Synthesis. 1990:299–301. [Google Scholar]
- 30.Kokotos G, Noula C. Selective one-pot conversion of carboxylic acids into alcohols. J Org Chem. 1996;61:6994–6996. doi: 10.1021/jo9520699. [DOI] [PubMed] [Google Scholar]
- 31.Loukas V, Noula C, Kokotos G. Efficient protocols for the synthesis of enantiopure γ-amino acids with proteinogenic side chains. J Pept Sci. 2003;9:312–319. doi: 10.1002/psc.458. [DOI] [PubMed] [Google Scholar]
- 32.Moutevelis-Minakakis P, Filippakou M, Sinanoglou C, Kokotos G. Synthesis of tetrazole analogs of γ- and δ–amino acids. J Pept Sci. 2006;12:377–382. doi: 10.1002/psc.737. [DOI] [PubMed] [Google Scholar]
- 33.Constantinou-Kokotou V, Kokotos G. Synthesis of 1,3-diamines. Org Prep Proced Int. 1994;26:599–602. [Google Scholar]