

Abstract
Human cancers arise from an imbalance of cell growth and cell death. Key proteins that govern this balance are those that mediate the cell cycle. Several different molecular effectors have been identified that tightly regulate specific phases of the cell cycle, including cyclins, cyclin-dependent kinases (CDKs) and CDK inhibitors. Notably, loss of expression or function of two G1-checkpoint CDK inhibitors - p21 (CDKN1A) and p27 (CDKN1B) - has been implicated in the genesis or progression of many human malignancies. Additionally, there is a growing body of evidence suggesting that functional loss of p21 or p27 can mediate a drug-resistance phenotype. However, reports in the literature have also suggested p21 and p27 can promote tumours, indicating a paradoxical effect. Here, we review historic and recent studies of these two CDK inhibitors, including their identification, function, importance to carcinogenesis and finally their roles in drug resistance.
The importance of cell cycle mediators to human carcinogenesis is now well established. In particular, critical genes that regulate cell cycle checkpoints have been demonstrated to be lost and/or have loss of function in many different human cancers. However, it has only recently been appreciated that dysfunction or loss of many of these same genes can also mediate resistance versus sensitivity to currently used cancer therapies. In this review, we focus on two important cell cycle regulators - the cyclin-dependent kinase (CDK) inhibitors p21 (CDKN1A) and p27 (CDKN1B) - by discussing their early discovery as mediators of cell cycle checkpoints and subsequent work showing how their loss or dysfunction leads to carcinogenesis and, more recently, resistance to therapeutic drugs. We also briefly discuss their paradoxical effects as cell cycle and tumour promoters.
Cell cycle, cyclins, CDKs and CDK inhibitors
The cell cycle comprises a very carefully orchestrated set of events that can lead to cell proliferation, senescence or apoptosis. Cells progress through the various phases of the cell cycle via the interactions of different cyclins with their respective CDK subunits (Fig. 1). The name cyclin comes from the fact that these proteins were first identified as molecules that accumulated and were then degraded at distinct points during the cell cycle of embryonic sea urchin eggs (Ref. 1). There are now several recognised classes or types of cyclins, active in different stages of the cell cycle. The D- and E-type cyclins are associated with the G1-S phase transition of the cell cycle (Refs 2, 3). Human cyclin D1 (CCND1; also called PRAD1 and BCL-1) and E were identified from mRNAs able to restore cyclin function in cyclin-deficient yeast (Refs 4, 5). Cyclin D1 quickly generated interest when it was mapped to chromosomal locus 11q13 close to a region known to be a breakpoint for certain chromosomal inversions that lead to parathyroid adenomas (Ref. 6). This ultimately led to further studies that uncovered additional cyclins in human and other mammalian systems.
Figure 1. Cell cycle progression is regulated by cyclin-CDK complexes.

Following a mitogenic stimulus, the decision to enter the cell cycle is regulated by complexes of cyclin-B-CDK1 and cyclin-C-CDK3 complexes (not shown). Next, during G1, D- and E-type cyclins are upregulated and assemble with their respective CDK binding partners. The cyclin-D-CDK4/6 and cyclin-E-CDK2 complexes then phosphorylate the retinoblastoma protein (pRB), thus inactivating it and releasing the transcription factor E2F from its inhibition. E2F then activates a variety of growth-promoting genes, which sends the cell cycle into the synthesis (S) phase. The cyclin-B-CDK1 complex continues to inhibit pRB at the S-G2 and G2-mitosis (M) checkpoints via phosphorylation to ensure completion of the cell cycle. Finally, following dephosphorylation of pRB, cells exit the M phase and pRB once again inhibits E2F. The CDK inhibitors p21 and p27 regulate the cell cycle by inhibiting the functions of the various CDK proteins as indicated. Abbreviations: CDK, cyclin-dependent kinase; P, phosphorylation.
CDKs were identified by virtue of the fact that mutations of these proteins in yeast affected their growth rates (Refs 7, 8). Because many of the early CDKs were discovered independently before their functions were well understood, the nomenclature of these proteins was nonuniform and often confusing. In 1991, researchers met at Cold Spring Harbor for the Symposium on Cell Cycle to agree on the following designations: cdc2 would become CDK1, PSK-J3 would become CDK4, PSSALRE was renamed CDK5, and PLSTIRE was renamed CDK6. Subsequently discovered CDKs were sequentially named (Ref. 9). CDKs were classified as kinases based on the observation that the total amount of phosphorylated proteins increased following injection of CDKs into the oocytes of a variety of different organisms (Refs 10, 11). p34cdc2 (CDK1) was the first CDK that was found to affect the cell cycle, as it was rate limiting for entry into mitosis (Refs 7, 12, 13, 14). Levels of this protein are constant throughout the cell cycle yet it is only catalytically active during M phase, implying that it is post-translationally modified (Ref. 15). Later, a human variant of p34cdc2 was discovered (Ref. 16), and was found to be regulated via phosphorylation (Refs 17, 18) and a CDK inhibitor (Ref. 19).
Among the most important CDKs that regulated the cell cycle are CDK4 and CDK6 (which are structurally related) and CDK2. Following mitogenic stimuli, quiescent cells enter the cell cycle and upregulate the D- and then E-type cyclins during G1 (Refs 4, 20, 21). The D-type cyclins then associate with CDK4 and CDK6 (Refs 22, 23, 24), while cyclin E associates with CDK2 (Refs 25, 26). Once assembled, the cyclin-CDK complexes enter the nucleus, where they are phosphorylated by a CDK-activating kinase (Refs 27, 28, 29). These activated complexes then phosphorylate additional proteins including various members of the retinoblastoma family, such as pRB (RB1), p107 (RBL1) and p130 (RBL2). Phosphorylation of pRB prevents its binding to E2F transcription factors, enabling them to activate the expression of genes that regulate entry into S phase (Ref. 30). Additionally, cyclin-CDK complexes have been shown to control centrosome duplication (Refs 31, 32, 33, 34) and histone gene transcription (Ref. 35), which are also key mediators of cell cycle progression.
Inhibitors of cyclin-CDKs can modulate the cell cycle by preventing or limiting cyclin-CDKs from phosphorylating their normal substrates. There are two classes of CDK inhibitors. Proteins in the first class were originally grouped as inhibitors of CDK4 (also known as INK4), but eventually included inhibitors of CDK6 as well. INK4 class proteins include the molecules p15INK4B (CDKN2B) (Ref. 36), p16INK4A (CDNK2A) (Ref. 37), p18INK4C (CDKN2C) (Ref. 38) and p19INK4D (CDKN2D) (Refs 39, 40); these specifically bind CDK4 and CDK6 and inhibit their association with D-type cyclins. Proteins in the second class of inhibitors are classified as kinase inhibitor proteins (KIPs), and include p21CIP1/WAF1 (CDKN1A; hereafter referred to as p21) (Refs 41, 42), p27KIP1 (CDKN1B; hereafter referred to as p27) (Refs 43, 44) and p57KIP2 (CDKN1C) (Refs 45, 46). As a more general classification, proteins of the KIP class are recognised as inhibitors of cyclin-E-CDK and cyclin-A-CDK complexes, although there are many reports that demonstrate that KIP class proteins can also interact with cyclin-D-associated CDKs (Refs 27, 41, 47, 48). When complexed with their respective cyclin binding partner, p21 and p27 are able to block the kinase activity of CDKs.
Additionally, cyclin-CDK complexes are further regulated by the molecules WEE1 and PKMYT1, which phosphorylate specific threonine and tyrosine residues on CDKs that inhibit their kinase activity; by contrast, CDC25 phosphatases can dephosphorylate these same amino acid residues on CDKs, thus reactivating their kinase function within the cyclin-CDK complex (Ref. 9).
CDK inhibitors act as vital checkpoints during various stages of the cell cycle. An important function of these checkpoints is to prevent the replication and propagation of damaged DNA to future daughter cells; they act as a fail-safe mechanism to allow for damaged DNA to be repaired or, if repair is not possible, the initiation of cell death. Since most cancer cells harbour genetically altered or ‘damaged’ DNA, it logically follows that these cancer cells must have circumvented these checkpoints to allow for uninhibited cell proliferation. Thus, the search for and identification of these checkpoint mediators naturally led to the recognition of their potential roles in mediating carcinogenesis.
Discovery of p21 and p27
p21
During the period 1992-1993, several independent groups separately identified p21 using various techniques. In 1991, the discovery of cyclin D1 and its association with the cell cycle led to further work to identify its mechanism of action (Refs 6, 49). Thus, Xiong et al. first identified p21 in 1992 through the detection of a 21 kDa protein of unknown function, but identified as a binding partner of cyclin D1 and D3 through an effort to find regulators of cyclin-CDK complexes (Ref. 47). This group used immunoprecipitation experiments with cyclin D1 and CDK2 in parallel and ultimately found p21 in both sets of lysates. They also noted that p21 was associated with another protein, called proliferating cell nuclear antigen (PCNA), and hypothesised that PCNA, cyclin D1, CDK2 and p21 formed a quaternary structure. The following year, this same group then demonstrated that p21 functioned as a universal inhibitor of cyclin-CDKs (Ref. 50).
Concurrent with the identification of p21 as a cyclin-CDK inhibitor, two independent groups simultaneously published the cloning of its gene in 1993. el-Deiry et al. sought to find proteins that were upregulated by the tumour suppressor p53 (TP53) (Ref. 42). Using a subtractive hybridisation technique, this group cloned the gene for p21 and found it to be a downstream target of p53 that was upregulated following p53 activation. They subsequently mapped the gene to the chromosomal region 6p21.2 and demonstrated that it suppressed tumour growth in experimental models. They named this gene wild-type p53 activated fragment 1 (WAF1). At the same time, Harper et al. identified a human gene encoding p21, which they named CDK-interacting protein 1 (CIP1), using a yeast two-hybrid system to identify proteins that interacted with CDK2 complexes (Ref. 41). Because of the concurrent yet independent discovery of p21 by these two groups, p21 is often referred to as p21CIP1/WAF1.
Other reports by numerous investigators soon followed. David Morgan’s group purified a 20 kDa protein that blocked the in vitro kinase activity of cyclin-CDK complexes, which they termed CAP20 (Ref. 51). James Smith’s group then published the results of their genetic screen to identify mediators of senescence and isolated a gene they dubbed SDI1 (Ref. 52). Finally, Steven Reed’s group identified p21 through a screen to find mediators of radiation-induced cell cycle arrest (Ref. 53). Comparison of protein and DNA sequences led to the realisation that all of these groups had independently found p21, and that it was an important mediator of cell cycle arrest through its role as a cyclin-CDK inhibitor.
p27
Almost concurrent with the discovery of p21, other investigators were analysing a protein with similar size and function. Initial work led to the hypothesis of a protein that mediated the assembly and inhibition of cyclin-E-CDK2 complexes (Ref. 54). This protein, p27, was subsequently isolated by biochemical analysis, thus confirming the original supposition (Ref. 55). Similar to the identification of the gene for p21, two independent groups concurrently cloned the gene encoding p27, resulting in the simultaneous publication of their reports (Refs 43, 44). The group led by Joan Massague identified the gene for p27 by applying lysates of contact-inhibited cells (which were known to prevent cyclin-E-CDK2 activation) to cyclin-E-CDK2 affinity columns, subjecting the eluents to automated Edman degradation, and designing degenerate PCR primers for cDNA amplification. They subsequently found that p27 inhibited cyclin-CDK complexes from phosphorylating histone H1. Furthermore, overexpression of p27 was found to prevent CDK activation and entry into the S phase of the cell cycle (Ref. 43). At the same time, Tony Hunter’s group cloned the gene for p27 using a yeast two-hybrid system consisting of a cyclin-D1-GAL4 fusion peptide with CDK4 to find peptides that were able to bind to this complex. Ultimately their work revealed a 91 amino acid fragment that was structurally similar to p21 and was identical to the product of the gene cloned by the Massague group (Ref. 44).
Function and regulation
CDK regulation
Overexpression of p21 inhibits proliferation in mammalian cells, and has been found to inhibit all cyclin-CDK complexes, indicating that it is a universal cyclin-CDK inhibitor (Ref. 50). Nuclear p21 is the form of this protein that mediates CDK inhibition (Ref. 56). Functional mapping of p21 has demonstrated that this inhibitory control on cyclin-CDK complexes is mediated through its N-terminal domain and is distinct from its ability to bind PCNA (Ref. 57). Further studies have revealed variable substrate specificity of p21, depending on the system and cells used. For example, p21 has been demonstrated to inhibit cyclin A or cyclin E bound to CDK2 (Ref. 51), as well as CDK4 bound to cyclin D1 or cyclin D2 (Ref. 41). Similar to p21, p27 inhibits cyclin-CDK complexes in G0/G1 and is functional in the nucleus (Ref. 19). However, p27 is dynamic, and can appear briefly in the cytoplasm during the G1-S transition prior to its degradation (Ref. 58).
Curiously, in certain cell systems p21 and p27 have also been shown to have paradoxical roles in the regulation of the cell cycle. Some investigators have demonstrated that p21 or p27 can facilitate the association of cyclin D to CDK4/6, with subsequent entry into the nucleus (Refs 59, 60). Others have shown that cyclin-D-CDK4/6 complexes act to sequester p21 away from cyclin-E-CDK2 complexes, which leads to their activation (Ref. 61). These opposing events are thought to be mediated by phosphorylation sites on the CDK inhibitors. For example, G1 arrest induced by TGF-β was associated with different patterns of p27 phosphorylation compared with cells resistant to the growth-inhibitory effects of TGF-β, and this correlated with the ability of p27 to bind cyclin-D-CDK4 versus cyclin-E-CDK2 complexes (Ref. 62). Further work by others subsequently demonstrated that differential tyrosine phosphorylation of p27 could dictate its inhibitory versus noninhibitory function when bound to cyclin-D-CDK4 complexes in mink lung epithelial cells (Ref. 63).
In sum, the above studies reveal that p21 and p27 are multifaceted proteins, potentially with several important roles in cell cycle regulation.
Regulation of p21
p21 is known to be directly regulated by the tumour suppressor p53, and is considered to be one of the most important and potent effector molecules of p53. The p53 protein directly activates p21 expression by binding its promoter (Ref. 64). Hence, in human cancers, inactivation of p53 will also lead to decreased levels of p21.
Despite its early recognised association with p53, it has now become clear that p21 can be regulated via many p53-independent pathways. For example, p21 levels can be transcriptionally regulated by MYC, as microarray analysis has demonstrated that p21 is a target of this oncogene (Ref. 65). In addition, post-translational modification can also affect the function of p21. Notably, phosphorylation of Thr145 (termed the nuclear localisation signal of p21) leads to cytoplasmic localisation of p21, which functionally results in its inactivation (Ref. 56). Indeed, it has been suggested that mislocalisation of p21 is a common method of its inactivation (Refs 56, 66) (Fig. 2).
Figure 2. CDK inhibitors are regulated by phosphorylation within the nucleus.
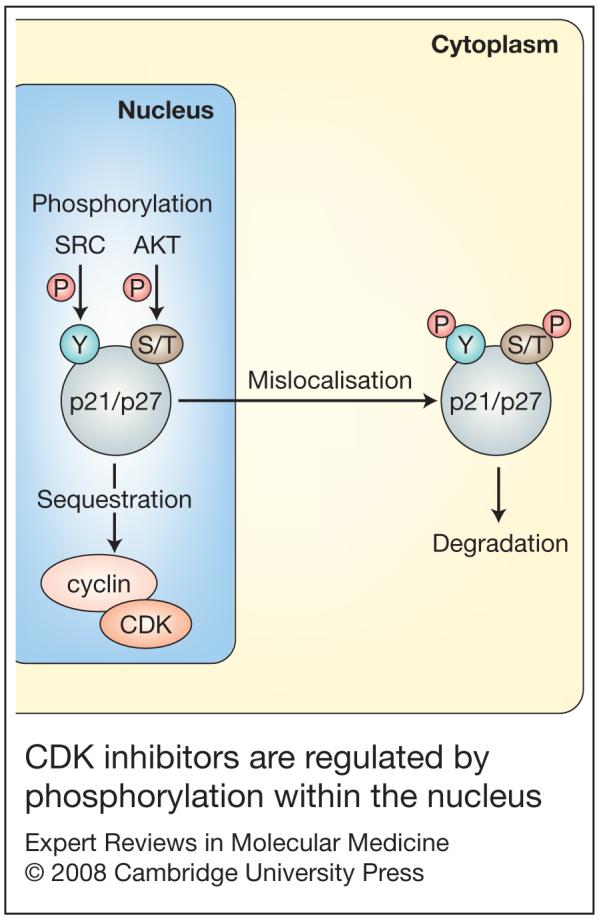
p21 or p27 protein is able to interact with cyclin-CDK complexes within the nucleus and hence regulate the cell cycle. p21 and p27 are themselves regulated either by sequestration via cyclin-CDK complexes, or by phosphorylation on serine/threonine (S/T) residues by enzymes such as AKT. In the case of p27, the tyrosine kinase SRC can also phosphorylate this protein. Following phosphorylation, p21 or p27 exit the nucleus and enter the cytoplasm, where they are not able to bind nuclear cyclin-CDK complexes and can then be degraded. Abbreviations: P, phosphorylation; S/T, serine/threonine; Y, tyrosine.
Still other mechanisms of p21 regulation have been reported. Similar to p53, E-box-binding proteins have also been shown to activate p21 (Ref. 67). Interestingly, the product of the breast cancer susceptibility gene BRCA1 has also been associated with p21 and p27 regulation (Refs 68, 69, 70, 71, 72). Since BRCA1 acts as a tumour suppressor by initiating the DNA-repair pathway, it can indirectly stimulate p21 by activating its upstream partner, p53. Additionally, there may be a direct association between BRCA1 and the expression of p21 or p27: BRCA1 binds to the transcription factor ZBRK1 via the DNA sequence GGGXXXCAGXXXTTT, and this same binding motif is present on the promoter of p21 (Refs 69, 71, 72). Further evidence of this lies in the fact that unlike wild-type BRCA1, mutant BRCA1 was unable to activate the p21 promoter. It is thought that BRCA1 activates p21 in order to block S-phase entry into the cell cycle following DNA damage (Ref. 71).
Regulation of p27
As is the case with p21, p27 can be regulated by several different independent mechanisms, and tumour cells can switch between different modes of p27 inactivation as progression of the tumour ensues (Ref. 73). p27 mRNA levels are generally constant throughout the cell cycle; however, p27 protein levels are under tight translational control, as they are highest in cells undergoing senescence and are reduced during the G1 and S phases of the cell cycle (Refs 73, 74, 75). Proteolysis is considered an important mechanism for regulating p27 levels, and the protein can be marked for degradation through a series of phosphorylation events (Refs 76, 77) (Fig. 2).
One pathway leading to p27 proteolytic degradation is mediated by cyclin-E-CDK2 via phosphorylation of Thr187 (Refs 19, 78). This phosphorylation during late G1 and early S phase facilitates the interaction of p27 with the SKP2-dependent E3 ligase complex, which ultimately leads to its degradation (Ref. 79). More recently, researchers have demonstrated that phosphorylation of Tyr88 by oncogenic tyrosine kinases can facilitate Thr187 phosphorylation, leading to its degradation at the G1-S transition (Ref. 80). Another site of p27 phosphorylation is Ser10; following mitogenic stimuli, phosphorylation of this residue during G1 leads to direct nuclear export of p27 (Ref. 81), which renders p27 nonfunctional, similar to the situation with p21. Furthermore, phosphorylation of p27 at Thr157 has been shown to regulate its localisation to the cytoplasm: mutation of Thr157 to alanine resulted in this mutant p27 being located exclusively in the nucleus (Ref. 82). Finally, SRC was shown to phosphorylate p27 at Tyr74 and Tyr88, resulting in less nuclear p27 due to an accumulation of cytoplasmic p27 (Ref. 83); pharmacological inhibition of SRC subsequently restored p27 function (see also section ‘Drug resistance’)
As mentioned previously, p21 and p27 inactivation can also occur through binding and subsequent sequestration by cyclin-D-CDK4/6 (Refs 84, 85).
It should be noted that absence of nuclear localisation of p21 or p27 may not be equivalent to loss of protein, as some groups have reported other functions of cytoplasmic p27 such as inhibition of GRB2, which can alter RAS function (Refs 86, 87). In addition, p27 can also associate with Rho (ARHGAP1), and thus may affect cell migration independently of its ability to bind cyclin-CDK complexes (Ref. 88).
Role in cancer
Given the fact that p21 was found to be a major effector of the tumour suppressor p53, it was initially thought that p21 would be of similar importance to human cancers. However, p21 somatic mutations are extremely rare, although epigenetic silencing has been reported in select cases (Refs 89, 90, 91). Furthermore, although p21 is capable of inhibiting cell growth, it does not fit the classic definition of a tumour suppressor, since germline mutations of p21 that lead to an increased susceptibility to tumour formation have not been observed. However, loss of p27 results in a more pronounced phenotype, and strong evidence suggests that p27 does indeed lead to an increased incidence of certain cancers. Below we discuss critical studies that implicate p21 and p27 as important mediators of carcinogenesis.
Knockout mouse studies
Early studies on the significance of p21 in tumourigenesis using mouse models also cast doubt on the notion of p21 as an important tumour suppressor. Researchers used mouse p21-knockouts to demonstrate that lack of p21 abrogated the ability of cells to undergo G1 arrest following damage to DNA, but unlike p53-knockout mice, it was initially thought that these animals did not develop spontaneous tumours (Refs 92, 93). Mice lacking p21 were found to develop normally, and this same study demonstrated that p21 was not needed for contact growth inhibition or for terminal differentiation of p21-null embryonic fibroblasts (Ref. 93). However, p21-null mice were found to be more susceptible to chemically induced tumours of the skin (Ref. 94) and colon (Ref. 95), and following irradiation they displayed increased tumourigenesis and metastases (Ref. 96). In addition, using different mouse strains, others have found that p21-null mice exhibit spontaneous tumour formation in the background of other genetic knockouts, such as Muc2-/- (lacking mucin 2) (Ref. 97) and Apc1638+/- (carrying a mutant allele of the adenomatosis polyposis coli gene) (Ref. 98). Furthermore, subsequent to the initial description of p21-null mice, investigators have found that p21-null mice bred on a 129Sv/C57BL6 50:50 background did in fact develop spontaneous tumours at an average age of 16 months (Ref. 99). Collectively, these mouse studies demonstrated the importance of p21 in mediating the G1 checkpoint, and its ability to function as a tumour suppressor.
Similar to p21, p27 has been implicated in increased tumour susceptibility. However, based on the fact that heterozygous p27+/- mice can develop pituitary tumours late in life and have accelerated and increased tumours when exposed to carcinogens, p27 has been suggested to be a tumour suppressor with a haploinsufficiency phenotype (Ref. 100). Moreover, there appears to be a dose-dependency effect, as p27 homozygous null mice have a more severe phenotype. Several groups have created p27-null mice and demonstrated that these mice grew to be 25-40% larger than their littermate controls (Refs 100, 101, 102). Mice lacking p27 also displayed greater CDK2 activity and exhibited enlarged pituitaries, thymii, adrenals and gonads. Similar to the situation for p21-null mice, p27 knockout also works cooperatively with other genetic modifiers of tumour susceptibility. Thus, p27-null mice crossed on the background of mice homozygous or hemizygous for gene knockout of p53, PTEN, pRB, p16, APC and others display varying degrees of increased tumour formation in different tissue types (Refs 103, 104, 105, 106, 107). In total, these studies present strong evidence that the lack of functional p27 has a profound effect on tumourigenesis and is a major factor in the development of human tumours.
Consistent with the notion that p21 and p27 may have paradoxical cell-cycle-promoting roles (see section ‘CDK regulation’), several investigators have shown either reduced tumour susceptibility with p21/p27 loss or a carcinogenic role of these proteins using various knockout mouse models. For example, loss of p21 resulted in decreased tumour incidence in certain tissues depending on additional crosses with other knockout mice, and/or in conjunction with ionising radiation (Refs 99, 108, 109). Additional mouse studies have supported a direct oncogenic role for p21 and p27. In one report, mutational analysis via an elegant system of somatic cell gene transfer in p21-null mice allowed the investigators to demonstrate that the cyclin-binding motifs of p21 may have a tumourigenic effect in a mouse model of oligodendroglioma (Ref. 110). In a similar vein, other investigators selectively mutated the cyclin- and CDK-binding domain of p27 and then demonstrated that in the absence of CDK-inhibitory activity, these knock-in mice developed hyperplastic lesions and tumours in various organs due to increased numbers of stem cells (Ref. 111). Thus, it would appear that in addition to their well-known property of CDK inhibition, this family of proteins may have a secondary role in promoting carcinogenesis.
Clinical implications/applications
Familial cancer syndromes
Although p21 does not meet the strictest definition of a human tumour suppressor gene as outlined above, one group has identified a germline nonsense mutation in p27 that leads to multiple endocrine neoplasia (MEN) syndrome (Ref. 112). In this study, the authors identified a family with a strong history of MEN, but without a germline mutation in the MEN1 gene, which is the most commonly mutated gene in MEN type 1. Based on a rodent model of p27 mutation yielding a MEN phenotype, this group discovered a truncating nonsense mutation in two affected family members. Functional studies demonstrated that this truncated mutant p27 was not capable of nuclear entry. Although this analysis was limited, it is in concordance with mouse knockout studies and suggests that p27 is indeed a bona fide tumour suppressor.
Expression and prognosis
Although the significance of CDK inhibitors has been investigated in several different cancers, including lung (Refs 113, 114), renal (Ref. 115), gallbladder (Ref. 116), gastric (Ref. 117), oesophageal (Ref. 118), endometrial (Ref. 119), head and neck (Ref. 120), ovarian (Refs 121, 122), and colorectal (Ref. 123) carcinomas, clinically the association between CDK inhibitors and cancer has been controversial. Using breast cancer as an example, multiple studies have yielded conflicting results. One group of investigators showed that p21 overexpression as detected by immunohistochemistry (IHC) was associated with larger tumour size, positive nodal status and a worse prognosis. Their analysis further demonstrated that p21 expression or lack thereof, could be used as a prognostic marker, but varied depending on the patient’s p53 status (using IHC) and whether the patient received adjuvant hormonal and/or chemotherapy (Ref. 124). Along these same lines, another group sought to study the relationship between p53 mutational status and p21 expression and showed that overexpression of p53 due to mutations led to decreased levels of p21 in 37 of 38 patient samples (Ref. 125). Others have shown that p21 status was not correlated with overall survival of breast cancer patients with up to 5 years of follow up (Ref. 126). However, similar to the first example, in the adjuvant hormonal and chemotherapy setting, patients with p21-/p53+ IHC-labelled tumours had a worse 5-year survival rate than p21+/p53+-tumour-bearing patients. Collectively, these studies suggest that the absence of p21 may be a negative predictor for response to systemic adjuvant therapies and could play a role in mediating drug resistance versus sensitivity in human cancers.
In addition to loss of p21 expression, there is a growing body of evidence suggesting that the presence of cytoplasmic p21 or p27 within a patient’s tumour is an indicator of poor prognosis (Refs 55, 73, 127, 128, 129, 130, 131). It is thought that this is due to the inability of CDK inhibitors to interact with their cyclin-CDK targets within the nucleus (Refs 2, 132). In the case of breast cancer, cytoplasmic localisation of p27 was found in up to 40% of primary breast malignancies (Refs 82, 132, 133, 134). For p27, the underlying mechanism found in the majority of these studies was thought to be phosphorylation of Thr157. Furthermore, when staining primary tumours with an antibody specific for p27 phosphorylated at Thr157, only cytoplasmic staining was observed. Similarly, p21 phosphorylation at Thr145 also excludes it from the nucleus as described above. The main enzyme thought to be responsible for regulating p21 and p27 phosphorylation at these residues is AKT [AKT1; also known as protein kinase B (PKB)]. AKT is a serine/threonine kinase that is central in mediating cell proliferation and cell death through several downstream pathways. AKT can be activated through various genetic alterations, including phosphoinositide 3-kinase activation from oncogenic mutations of PIK3CA, PTEN loss or HER2/neu (ERBB2) amplification. Interestingly, AKT activation has been shown to correlate with resistance to hormonal therapies used in breast cancers (Refs 135, 136, 137).
Drug resistance
Absence or cytoplasmic localisation of p21 and p27 has been linked to drug resistance in many in vitro cellular models (Refs 61, 138, 139, 140). However, to date, clinical evidence that these CDK inhibitors truly mediate a drug-resistance phenotype in human cancers has been incomplete. The study of drug resistance in human cancer patients has been understandably limited, because procurement of tissues from recurrent or metastatic disease is difficult, since these lesions are often not biopsied. Thus, as in the analyses described above, multiyear follow up of correlative marker studies using initial biopsy specimens in the adjuvant setting have only recently been described. Despite this, researchers have of late shed light on an important potential role for p21 and p27 in mediating drug resistance in human breast cancers.
The most effective drugs used to treat breast cancers are hormonal therapies that target breast cancers overexpressing the nuclear hormone receptors oestrogen (estrogen) receptor alpha (ESR1) and/or progesterone receptor (PGR), hereafter designated ER and PR, respectively. The prototypical drug in this class is the selective estrogen-receptor modulator (SERM) tamoxifen. Tamoxifen is an unusual drug in that it has mixed agonist and antagonist properties when bound to the ER. Thus, some tissues such as endometrial epithelium are growth stimulated by this drug, while breast tissue is normally growth inhibited. Some of the molecular mediators of the selective effects of tamoxifen have been elucidated (Ref. 141), but mechanisms of tamoxifen drug resistance have only recently been described.
When administered to women who have undergone mastectomy or lumpectomy with radiation therapy for local treatment, this drug can reduce a woman’s chance of developing recurrent breast cancer by 47% (Ref. 142). Similarly, this drug can be highly effective even in women who initially present with metastatic disease. Tamoxifen is also used as a preventative agent in women at high risk for developing breast cancers. Although newer classes of hormonal agents called aromatase inhibitors have recently supplanted tamoxifen as first-line therapy in postmenopausal women with ER- and/or PR-positive breast cancers (Ref. 141), tamoxifen remains as first-line therapy for premenopausal women with ER- and/or PR-expressing breast cancers and for primary prevention of breast cancer in women at increased risk. However, resistance to this drug is still a considerable clinical problem. Many breast cancers are initially not sensitive to tamoxifen while others acquire drug resistance, as seen in women who originally respond but then develop recurrent and/or metastatic disease. An additional observation has been that some breast cancers are able to use tamoxifen as an ER agonist, as is evidenced by tumour regression upon tamoxifen withdrawal (Ref. 143). This is a poorly understood process, but it is thought that tamoxifen is able to cause differential phosphorylation of the ER, which leads to the recruitment of coactivators rather than corepressors (Refs 144, 145, 146).
A recent study has implicated loss of p21 function as one possible cause of a tamoxifen-resistant/agonistic phenotype (Ref. 147). Withdrawal of tamoxifen in a woman whose primary ER-positive breast cancer had sequentially recurred in her colon and skin induced a clinical regression at the skin site, indicating that tamoxifen was acting as an ER agonist on her metastatic skin lesions. IHC labelling for p21 in serial biopsies from all three sites (breast, colon, skin) showed that loss of p21 expression correlated with tamoxifen resistance/agonism. Molecular analysis in isogenic p21-knockout cell lines revealed that the absence of p21 enabled cyclin-CDK complexes to aberrantly phosphorylate ER when bound to tamoxifen, resulting in a growth-stimulatory phenotype. These data correlated with the patient’s tissue specimens: the presence of phosphorylated ER in the patient’s skin lesion (p21-), but not her colonic lesion (p21+), strongly suggesting that loss of p21 led to tamoxifen resistance and a growth-promoting response in this patient.
Similar to p21, p27 inhibition via SRC, an oncogenic kinase expressed at high levels in many cancers, has recently been associated with a tamoxifen-resistance phenotype (Ref. 83). As mentioned above, SRC was shown to regulate p27 through its phosphorylation at Tyr74 and Tyr88 in tamoxifen-resistant breast cancer cell lines with high SRC activity; furthermore, the SRC-family inhibitor AZD0530 could restore tamoxifen sensitivity by preventing SRC-mediated phosphorylation of p27. In addition, using a panel of primary human breast cancers, the authors showed a correlation between SRC activation and low levels of nuclear p27, suggesting that SRC inhibition in patients may aid in preventing or restoring sensitivity to tamoxifen.
Research in progress and outstanding research questions
Since the early 1990s, it has been demonstrated that both p21 and p27 are unequivocally significant players in cell cycle regulation. Logically, this has led to strong implications for tumour growth and survival. Functional inactivation of these proteins can lead to the propagation of damaged DNA. Subsequently, damaged DNA may include genetic aberrations that confer a growth phenotype, drug resistance, and/or ablation of other cell cycle checkpoints. A more thorough understanding of these mechanisms will lead to a better appreciation of how cancers evade these checkpoints, and ultimately aid in the development of more-effective targeted therapies. Since loss or inactivation of p21 and p27 is seen clinically in primary solid tumours, it is important to target these mechanisms prior to the development of metastatic drug-resistant disease. As stated previously, studies associating loss of p21 expression with clinical outcome have been conflicting (Ref. 126), and there remains much uncertainty on correlations between sensitivity and resistance to chemotherapeutic agents and the levels of p21 and p27 in various cancers (Refs 148, 149, 150). One reason for this may be that loss of p21 and p27 is a predominant mechanism of acquired, but not de novo, drug resistance, which has largely not been evaluated. However, even with better therapies targeted toward specific genetic alterations, further genetic changes due to the genomic instability inherent in human cancers may confer a drug-resistance phenotype. Therefore, in order to truly eradicate this disease, it will be necessary to define and target new mechanisms of drug resistance, possibly involving other cell cycle checkpoint regulators. In this way, a multipronged approach that targets the numerous genetic anomalies present in cancerous but not normal cells will enable effective therapies that prevent the emergence of drug-resistant malignancies.
Acknowledgements and funding
We thank members of the Park laboratory for critical review of the manuscript. We apologise to authors whose work could not be included due to space constraints. We thank Andrew Koff and the anonymous peer reviewers for their careful reading of this manuscript and thoughtful comments and suggestions. This work was supported by The Flight Attendant’s Medical Research Institute (FAMRI), the Maryland Cigarette Restitution Fund, NIH/NCI CA109274, the Stewart Trust Fund, The Mary Kay Ash Charitable Foundation and the Avon Foundation. A.M.A. was supported by the Susan G. Komen for the Cure Foundation (PDF0707944).
References
- 1.Evans T, et al. Cyclin: a protein specified by maternal mRNA in sea urchin eggs that is destroyed at each cleavage division. Cell. 1983;33:389–396. doi: 10.1016/0092-8674(83)90420-8. [DOI] [PubMed] [Google Scholar]
- 2.Slingerland J, Pagano M. Regulation of the cdk inhibitor p27 and its deregulation in cancer. J Cell Physiol. 2000;183:10–17. doi: 10.1002/(SICI)1097-4652(200004)183:1<10::AID-JCP2>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 3.Sherr CJ. G1 phase progression: cycling on cue. Cell. 1994;79:551–555. doi: 10.1016/0092-8674(94)90540-1. [DOI] [PubMed] [Google Scholar]
- 4.Lew DJ, Dulic V, Reed SI. Isolation of three novel human cyclins by rescue of G1 cyclin (Cln) function in yeast. Cell. 1991;66:1197–1206. doi: 10.1016/0092-8674(91)90042-w. [DOI] [PubMed] [Google Scholar]
- 5.Xiong Y, et al. Human D-type cyclin. Cell. 1991;65:691–699. doi: 10.1016/0092-8674(91)90100-d. [DOI] [PubMed] [Google Scholar]
- 6.Motokura T, et al. A novel cyclin encoded by a bcl1-linked candidate oncogene. Nature. 1991;350:512–515. doi: 10.1038/350512a0. [DOI] [PubMed] [Google Scholar]
- 7.Nurse P, Thuriaux P. Regulatory genes controlling mitosis in the fission yeast Schizosaccharomyces pombe. Genetics. 1980;96:627–637. doi: 10.1093/genetics/96.3.627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nurse P. Genetic control of cell size at cell division in yeast. Nature. 1975;256:547–551. doi: 10.1038/256547a0. [DOI] [PubMed] [Google Scholar]
- 9.Malumbres M, Barbacid M. Mammalian cyclin-dependent kinases. Trends Biochem Sci. 2005;30:630–641. doi: 10.1016/j.tibs.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 10.Doree M, Peaucellier G, Picard A. Activity of the maturation-promoting factor and the extent of protein phosphorylation oscillate simultaneously during meiotic maturation of starfish oocytes. Dev Biol. 1983;99:489–501. doi: 10.1016/0012-1606(83)90298-1. [DOI] [PubMed] [Google Scholar]
- 11.Maller J, Wu M, Gerhart JC. Changes in protein phosphorylation accompanying maturation of Xenopus laevis oocytes. Dev Biol. 1977;58:295–312. doi: 10.1016/0012-1606(77)90093-8. [DOI] [PubMed] [Google Scholar]
- 12.Gautier J, et al. Purified maturation-promoting factor contains the product of a Xenopus homolog of the fission yeast cell cycle control gene cdc2+ Cell. 1988;54:433–439. doi: 10.1016/0092-8674(88)90206-1. [DOI] [PubMed] [Google Scholar]
- 13.Dunphy WG, et al. The Xenopus cdc2 protein is a component of MPF, a cytoplasmic regulator of mitosis. Cell. 1988;54:423–431. doi: 10.1016/0092-8674(88)90205-x. [DOI] [PubMed] [Google Scholar]
- 14.Norbury C, Nurse P. Animal cell cycles and their control. Annu Rev Biochem. 1992;61:441–470. doi: 10.1146/annurev.bi.61.070192.002301. [DOI] [PubMed] [Google Scholar]
- 15.Sherr CJ. Mammalian G1 cyclins. Cell. 1993;73:1059–1065. doi: 10.1016/0092-8674(93)90636-5. [DOI] [PubMed] [Google Scholar]
- 16.Lee MG, Nurse P. Complementation used to clone a human homologue of the fission yeast cell cycle control gene cdc2. Nature. 1987;327:31–35. doi: 10.1038/327031a0. [DOI] [PubMed] [Google Scholar]
- 17.Gould KL, et al. Phosphorylation at Thr167 is required for Schizosaccharomyces pombe p34cdc2 function. EMBO J. 1991;10:3297–3309. doi: 10.1002/j.1460-2075.1991.tb04894.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gould KL, Nurse P. Tyrosine phosphorylation of the fission yeast cdc2+ protein kinase regulates entry into mitosis. Nature. 1989;342:39–45. doi: 10.1038/342039a0. [DOI] [PubMed] [Google Scholar]
- 19.Alkarain A, Jordan R, Slingerland J. p27 deregulation in breast cancer: prognostic significance and implications for therapy. J Mammary Gland Biol Neoplasia. 2004;9:67–80. doi: 10.1023/B:JOMG.0000023589.00994.5e. [DOI] [PubMed] [Google Scholar]
- 20.Koff A, et al. Human cyclin E, a new cyclin that interacts with two members of the CDC2 gene family. Cell. 1991;66:1217–1228. doi: 10.1016/0092-8674(91)90044-y. [DOI] [PubMed] [Google Scholar]
- 21.Matsushime H, et al. Colony-stimulating factor 1 regulates novel cyclins during the G1 phase of the cell cycle. Cell. 1991;65:701–713. doi: 10.1016/0092-8674(91)90101-4. [DOI] [PubMed] [Google Scholar]
- 22.Meyerson M, Harlow E. Identification of G1 kinase activity for cdk6, a novel cyclin D partner. Mol Cell Biol. 1994;14:2077–2086. doi: 10.1128/mcb.14.3.2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bates S, et al. CDK6 (PLSTIRE) and CDK4 (PSK-J3) are a distinct subset of the cyclin-dependent kinases that associate with cyclin D1. Oncogene. 1994;9:71–79. [PubMed] [Google Scholar]
- 24.Matsushime H, et al. Identification and properties of an atypical catalytic subunit (p34PSK-J3/cdk4) for mammalian D type G1 cyclins. Cell. 1992;71:323–334. doi: 10.1016/0092-8674(92)90360-o. [DOI] [PubMed] [Google Scholar]
- 25.Dulic V, Lees E, Reed SI. Association of human cyclin E with a periodic G1-S phase protein kinase. Science. 1992;257:1958–1961. doi: 10.1126/science.1329201. [DOI] [PubMed] [Google Scholar]
- 26.Koff A, et al. Formation and activation of a cyclin E-cdk2 complex during the G1 phase of the human cell cycle. Science. 1992;257:1689–1694. doi: 10.1126/science.1388288. [DOI] [PubMed] [Google Scholar]
- 27.Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 1999;13:1501–1512. doi: 10.1101/gad.13.12.1501. [DOI] [PubMed] [Google Scholar]
- 28.Kato JY, et al. Regulation of cyclin D-dependent kinase 4 (cdk4) by cdk4-activating kinase. Mol Cell Biol. 1994;14:2713–2721. doi: 10.1128/mcb.14.4.2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Matsuoka M, et al. Activation of cyclin-dependent kinase 4 (cdk4) by mouse MO15-associated kinase. Mol Cell Biol. 1994;14:7265–7275. doi: 10.1128/mcb.14.11.7265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Malumbres M, Barbacid M. To cycle or not to cycle: a critical decision in cancer. Nat Rev Cancer. 2001;1:222–231. doi: 10.1038/35106065. [DOI] [PubMed] [Google Scholar]
- 31.Fisk HA, Winey M. The mouse Mps1p-like kinase regulates centrosome duplication. Cell. 2001;106:95–104. doi: 10.1016/s0092-8674(01)00411-1. [DOI] [PubMed] [Google Scholar]
- 32.Okuda M, et al. Nucleophosmin/B23 is a target of CDK2/cyclin E in centrosome duplication. Cell. 2000;103:127–140. doi: 10.1016/s0092-8674(00)00093-3. [DOI] [PubMed] [Google Scholar]
- 33.Meraldi P, et al. Centrosome duplication in mammalian somatic cells requires E2F and Cdk2-cyclin A. Nat Cell Biol. 1999;1:88–93. doi: 10.1038/10054. [DOI] [PubMed] [Google Scholar]
- 34.Lacey KR, Jackson PK, Stearns T. Cyclin-dependent kinase control of centrosome duplication. Proc Natl Acad Sci U S A. 1999;96:2817–2822. doi: 10.1073/pnas.96.6.2817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ewen ME. Where the cell cycle and histones meet. Genes Dev. 2000;14:2265–2270. doi: 10.1101/gad.842100. [DOI] [PubMed] [Google Scholar]
- 36.Hannon GJ, Beach D. p15INK4B is a potential effector of TGF-beta-induced cell cycle arrest. Nature. 1994;371:257–261. doi: 10.1038/371257a0. [DOI] [PubMed] [Google Scholar]
- 37.Serrano M, Hannon GJ, Beach D. A new regulatory motif in cell-cycle control causing specific inhibition of cyclin D/CDK4. Nature. 1993;366:704–707. doi: 10.1038/366704a0. [DOI] [PubMed] [Google Scholar]
- 38.Guan KL, et al. Growth suppression by p18, a p16INK4/MTS1- and p14INK4B/MTS2-related CDK6 inhibitor, correlates with wild-type pRb function. Genes Dev. 1994;8:2939–2952. doi: 10.1101/gad.8.24.2939. [DOI] [PubMed] [Google Scholar]
- 39.Hirai H, et al. Novel INK4 proteins, p19 and p18, are specific inhibitors of the cyclin D-dependent kinases CDK4 and CDK6. Mol Cell Biol. 1995;15:2672–2681. doi: 10.1128/mcb.15.5.2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chan FK, et al. Identification of human and mouse p19, a novel CDK4 and CDK6 inhibitor with homology to p16ink4. Mol Cell Biol. 1995;15:2682–2688. doi: 10.1128/mcb.15.5.2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Harper JW, et al. The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell. 1993;75:805–816. doi: 10.1016/0092-8674(93)90499-g. [DOI] [PubMed] [Google Scholar]
- 42.el-Deiry WS, et al. WAF1, a potential mediator of p53 tumor suppression. Cell. 1993;75:817–825. doi: 10.1016/0092-8674(93)90500-p. [DOI] [PubMed] [Google Scholar]
- 43.Polyak K, et al. Cloning of p27Kip1, a cyclin-dependent kinase inhibitor and a potential mediator of extracellular antimitogenic signals. Cell. 1994;78:59–66. doi: 10.1016/0092-8674(94)90572-x. [DOI] [PubMed] [Google Scholar]
- 44.Toyoshima H, Hunter T. p27, a novel inhibitor of G1 cyclin-Cdk protein kinase activity, is related to p21. Cell. 1994;78:67–74. doi: 10.1016/0092-8674(94)90573-8. [DOI] [PubMed] [Google Scholar]
- 45.Matsuoka S, et al. p57KIP2, a structurally distinct member of the p21CIP1 Cdk inhibitor family, is a candidate tumor suppressor gene. Genes Dev. 1995;9:650–662. doi: 10.1101/gad.9.6.650. [DOI] [PubMed] [Google Scholar]
- 46.Lee MH, Reynisdottir I, Massague J. Cloning of p57KIP2, a cyclin-dependent kinase inhibitor with unique domain structure and tissue distribution. Genes Dev. 1995;9:639–649. doi: 10.1101/gad.9.6.639. [DOI] [PubMed] [Google Scholar]
- 47.Xiong Y, Zhang H, Beach D. D type cyclins associate with multiple protein kinases and the DNA replication and repair factor PCNA. Cell. 1992;71:505–514. doi: 10.1016/0092-8674(92)90518-h. [DOI] [PubMed] [Google Scholar]
- 48.Yu ZK, Gervais JL, Zhang H. Human CUL-1 associates with the SKP1/SKP2 complex and regulates p21(CIP1/WAF1) and cyclin D proteins. Proc Natl Acad Sci U S A. 1998;95:11324–11329. doi: 10.1073/pnas.95.19.11324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Withers DA, et al. Characterization of a candidate bcl-1 gene. Mol Cell Biol. 1991;11:4846–4853. doi: 10.1128/mcb.11.10.4846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xiong Y, et al. p21 is a universal inhibitor of cyclin kinases. Nature. 1993;366:701–704. doi: 10.1038/366701a0. [DOI] [PubMed] [Google Scholar]
- 51.Gu Y, Turck CW, Morgan DO. Inhibition of CDK2 activity in vivo by an associated 20K regulatory subunit. Nature. 1993;366:707–710. doi: 10.1038/366707a0. [DOI] [PubMed] [Google Scholar]
- 52.Noda A, et al. Cloning of senescent cell-derived inhibitors of DNA synthesis using an expression screen. Exp Cell Res. 1994;211:90–98. doi: 10.1006/excr.1994.1063. [DOI] [PubMed] [Google Scholar]
- 53.Dulic V, et al. p53-dependent inhibition of cyclin-dependent kinase activities in human fibroblasts during radiation-induced G1 arrest. Cell. 1994;76:1013–1023. doi: 10.1016/0092-8674(94)90379-4. [DOI] [PubMed] [Google Scholar]
- 54.Koff A, et al. Negative regulation of G1 in mammalian cells: inhibition of cyclin E-dependent kinase by TGF-beta. Science. 1993;260:536–539. doi: 10.1126/science.8475385. [DOI] [PubMed] [Google Scholar]
- 55.Polyak K, et al. p27Kip1, a cyclin-Cdk inhibitor, links transforming growth factor-beta and contact inhibition to cell cycle arrest. Genes Dev. 1994;8:9–22. doi: 10.1101/gad.8.1.9. [DOI] [PubMed] [Google Scholar]
- 56.Zhou BP, et al. Cytoplasmic localization of p21Cip1/WAF1 by Akt-induced phosphorylation in HER-2/neu-overexpressing cells. Nat Cell Biol. 2001;3:245–252. doi: 10.1038/35060032. [DOI] [PubMed] [Google Scholar]
- 57.Goubin F, Ducommun B. Identification of binding domains on the p21Cip1 cyclin-dependent kinase inhibitor. Oncogene. 1995;10:2281–2287. [PubMed] [Google Scholar]
- 58.Connor MK, et al. CRM1/Ran-mediated nuclear export of p27(Kip1) involves a nuclear export signal and links p27 export and proteolysis. Mol Biol Cell. 2003;14:201–213. doi: 10.1091/mbc.E02-06-0319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cheng M, et al. The p21(Cip1) and p27(Kip1) CDK ‘inhibitors’ are essential activators of cyclin D-dependent kinases in murine fibroblasts. EMBO J. 1999;18:1571–1583. doi: 10.1093/emboj/18.6.1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.LaBaer J, et al. New functional activities for the p21 family of CDK inhibitors. Genes Dev. 1997;11:847–862. doi: 10.1101/gad.11.7.847. [DOI] [PubMed] [Google Scholar]
- 61.Planas-Silva MD, Weinberg RA. Estrogen-dependent cyclin E-cdk2 activation through p21 redistribution. Mol Cell Biol. 1997;17:4059–4069. doi: 10.1128/mcb.17.7.4059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ciarallo S, et al. Altered p27(Kip1) phosphorylation, localization, and function in human epithelial cells resistant to transforming growth factor beta-mediated G(1) arrest. Mol Cell Biol. 2002;22:2993–3002. doi: 10.1128/MCB.22.9.2993-3002.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.James MK, et al. Differential modification of p27Kip1 controls its cyclin D-cdk4 inhibitory activity. Mol Cell Biol. 2008;28:498–510. doi: 10.1128/MCB.02171-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Westfall MD, et al. The Delta Np63 alpha phosphoprotein binds the p21 and 14-3-3 sigma promoters in vivo and has transcriptional repressor activity that is reduced by Hay-Wells syndrome-derived mutations. Mol Cell Biol. 2003;23:2264–2276. doi: 10.1128/MCB.23.7.2264-2276.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Coller HA, et al. Expression analysis with oligonucleotide microarrays reveals that MYC regulates genes involved in growth, cell cycle, signaling, and adhesion. Proc Natl Acad Sci U S A. 2000;97:3260–3265. doi: 10.1073/pnas.97.7.3260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Westbrook TF, et al. E7 abolishes raf-induced arrest via mislocalization of p21(Cip1) Mol Cell Biol. 2002;22:7041–7052. doi: 10.1128/MCB.22.20.7041-7052.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Prabhu S, et al. Regulation of the expression of cyclin-dependent kinase inhibitor p21 by E2A and Id proteins. Mol Cell Biol. 1997;17:5888–5896. doi: 10.1128/mcb.17.10.5888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.MacLachlan TK, Takimoto R, El-Deiry WS. BRCA1 directs a selective p53-dependent transcriptional response towards growth arrest and DNA repair targets. Mol Cell Biol. 2002;22:4280–4292. doi: 10.1128/MCB.22.12.4280-4292.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zheng L, et al. Sequence-specific transcriptional corepressor function for BRCA1 through a novel zinc finger protein, ZBRK1. Mol Cell. 2000;6:757–768. doi: 10.1016/s1097-2765(00)00075-7. [DOI] [PubMed] [Google Scholar]
- 70.Ouchi T, et al. Collaboration of signal transducer and activator of transcription 1 (STAT1) and BRCA1 in differential regulation of IFN-gamma target genes. Proc Natl Acad Sci U S A. 2000;97:5208–5213. doi: 10.1073/pnas.080469697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Somasundaram K, et al. Arrest of the cell cycle by the tumour-suppressor BRCA1 requires the CDK-inhibitor p21WAF1/CiP1. Nature. 1997;389:187–190. doi: 10.1038/38291. [DOI] [PubMed] [Google Scholar]
- 72.Williamson EA, Dadmanesh F, Koeffler HP. BRCA1 transactivates the cyclin-dependent kinase inhibitor p27(Kip1) Oncogene. 2002;21:3199–3206. doi: 10.1038/sj.onc.1205461. [DOI] [PubMed] [Google Scholar]
- 73.Timmerbeul I, et al. Testing the importance of p27 degradation by the SCFskp2 pathway in murine models of lung and colon cancer. Proc Natl Acad Sci U S A. 2006;103:14009–14014. doi: 10.1073/pnas.0606316103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hengst L, Reed SI. Translational control of p27Kip1 accumulation during the cell cycle. Science. 1996;271:1861–1864. doi: 10.1126/science.271.5257.1861. [DOI] [PubMed] [Google Scholar]
- 75.Millard SS, et al. Enhanced ribosomal association of p27(Kip1) mRNA is a mechanism contributing to accumulation during growth arrest. J Biol Chem. 1997;272:7093–7098. doi: 10.1074/jbc.272.11.7093. [DOI] [PubMed] [Google Scholar]
- 76.Pagano M, et al. Role of the ubiquitin-proteasome pathway in regulating abundance of the cyclin-dependent kinase inhibitor p27. Science. 1995;269:682–685. doi: 10.1126/science.7624798. [DOI] [PubMed] [Google Scholar]
- 77.Malek NP, et al. A mouse knock-in model exposes sequential proteolytic pathways that regulate p27Kip1 in G1 and S phase. Nature. 2001;413:323–327. doi: 10.1038/35095083. [DOI] [PubMed] [Google Scholar]
- 78.Boehm M, et al. A growth factor-dependent nuclear kinase phosphorylates p27(Kip1) and regulates cell cycle progression. EMBO J. 2002;21:3390–3401. doi: 10.1093/emboj/cdf343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sabile A, et al. Regulation of p27 degradation and S-phase progression by Ro52 RING finger protein. Mol Cell Biol. 2006;26:5994–6004. doi: 10.1128/MCB.01630-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Grimmler M, et al. Cdk-inhibitory activity and stability of p27Kip1 are directly regulated by oncogenic tyrosine kinases. Cell. 2007;128:269–280. doi: 10.1016/j.cell.2006.11.047. [DOI] [PubMed] [Google Scholar]
- 81.Rodier G, et al. p27 cytoplasmic localization is regulated by phosphorylation on Ser10 and is not a prerequisite for its proteolysis. Embo J. 2001;20:6672–6682. doi: 10.1093/emboj/20.23.6672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Viglietto G, et al. Cytoplasmic relocalization and inhibition of the cyclin-dependent kinase inhibitor p27(Kip1) by PKB/Akt-mediated phosphorylation in breast cancer. Nat Med. 2002;8:1136–1144. doi: 10.1038/nm762. [DOI] [PubMed] [Google Scholar]
- 83.Chu I, et al. p27 phosphorylation by Src regulates inhibition of cyclin E-Cdk2. Cell. 2007;128:281–294. doi: 10.1016/j.cell.2006.11.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Perez-Roger I, et al. Cyclins D1 and D2 mediate myc-induced proliferation via sequestration of p27(Kip1) and p21(Cip1) EMBO J. 1999;18:5310–5320. doi: 10.1093/emboj/18.19.5310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sherr CJ, Roberts JM. Inhibitors of mammalian G1 cyclin-dependent kinases. Genes Dev. 1995;9:1149–1163. doi: 10.1101/gad.9.10.1149. [DOI] [PubMed] [Google Scholar]
- 86.Moeller SJ, Head ED, Sheaff RJ. p27Kip1 inhibition of GRB2-SOS formation can regulate Ras activation. Mol Cell Biol. 2003;23:3735–3752. doi: 10.1128/MCB.23.11.3735-3752.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.McAllister SS, et al. Novel p27(kip1) C-terminal scatter domain mediates Rac-dependent cell migration independent of cell cycle arrest functions. Mol Cell Biol. 2003;23:216–228. doi: 10.1128/MCB.23.1.216-228.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Besson A, et al. p27Kip1 modulates cell migration through the regulation of RhoA activation. Genes Dev. 2004;18:862–876. doi: 10.1101/gad.1185504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Allan LA, et al. The p21(WAF1/CIP1) promoter is methylated in Rat-1 cells: stable restoration of p53-dependent p21(WAF1/CIP1) expression after transfection of a genomic clone containing the p21(WAF1/CIP1) gene. Mol Cell Biol. 2000;20:1291–1298. doi: 10.1128/mcb.20.4.1291-1298.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Roman-Gomez J, et al. 5′ CpG island hypermethylation is associated with transcriptional silencing of the p21(CIP1/WAF1/SDI1) gene and confers poor prognosis in acute lymphoblastic leukemia. Blood. 2002;99:2291–2296. doi: 10.1182/blood.v99.7.2291. [DOI] [PubMed] [Google Scholar]
- 91.Balbin M, et al. Functional analysis of a p21WAF1,CIP1,SDI1 mutant (Arg94 -> Trp) identified in a human breast carcinoma. Evidence that the mutation impairs the ability of p21 to inhibit cyclin-dependent kinases. J Biol Chem. 1996;271:15782–15786. doi: 10.1074/jbc.271.26.15782. [DOI] [PubMed] [Google Scholar]
- 92.Brugarolas J, et al. Radiation-induced cell cycle arrest compromised by p21 deficiency. Nature. 1995;377:552–557. doi: 10.1038/377552a0. [DOI] [PubMed] [Google Scholar]
- 93.Deng C, et al. Mice lacking p21CIP1/WAF1 undergo normal development, but are defective in G1 checkpoint control. Cell. 1995;82:675–684. doi: 10.1016/0092-8674(95)90039-x. [DOI] [PubMed] [Google Scholar]
- 94.Topley GI, et al. p21(WAF1/Cip1) functions as a suppressor of malignant skin tumor formation and a determinant of keratinocyte stem-cell potential. Proc Natl Acad Sci U S A. 1999;96:9089–9094. doi: 10.1073/pnas.96.16.9089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Poole AJ, et al. Tumor suppressor functions for the Cdk inhibitor p21 in the mouse colon. Oncogene. 2004;23:8128–8134. doi: 10.1038/sj.onc.1207994. [DOI] [PubMed] [Google Scholar]
- 96.Jackson RJ, et al. p21Cip1 nullizygosity increases tumor metastasis in irradiated mice. Cancer Res. 2003;63:3021–3025. [PubMed] [Google Scholar]
- 97.Yang W, et al. Inactivation of p21WAF1/cip1 enhances intestinal tumor formation in Muc2-/- mice. Am J Pathol. 2005;166:1239–1246. doi: 10.1016/S0002-9440(10)62342-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Yang WC, et al. Targeted inactivation of the p21(WAF1/cip1) gene enhances Apc-initiated tumor formation and the tumor-promoting activity of a Western-style high-risk diet by altering cell maturation in the intestinal mucosal. Cancer Res. 2001;61:565–569. [PubMed] [Google Scholar]
- 99.Martin-Caballero J, et al. Tumor susceptibility of p21(Waf1/Cip1)-deficient mice. Cancer Res. 2001;61:6234–6238. [PubMed] [Google Scholar]
- 100.Fero ML, et al. The murine gene p27Kip1 is haplo-insufficient for tumour suppression. Nature. 1998;396:177–180. doi: 10.1038/24179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kiyokawa H, et al. Enhanced growth of mice lacking the cyclin-dependent kinase inhibitor function of p27(Kip1) Cell. 1996;85:721–732. doi: 10.1016/s0092-8674(00)81238-6. [DOI] [PubMed] [Google Scholar]
- 102.Nakayama K, et al. Mice lacking p27(Kip1) display increased body size, multiple organ hyperplasia, retinal dysplasia, and pituitary tumors. Cell. 1996;85:707–720. doi: 10.1016/s0092-8674(00)81237-4. [DOI] [PubMed] [Google Scholar]
- 103.Philipp-Staheli J, et al. Distinct roles for p53, p27Kip1, and p21Cip1 during tumor development. Oncogene. 2004;23:905–913. doi: 10.1038/sj.onc.1207220. [DOI] [PubMed] [Google Scholar]
- 104.Di Cristofano A, et al. Pten and p27KIP1 cooperate in prostate cancer tumor suppression in the mouse. Nat Genet. 2001;27:222–224. doi: 10.1038/84879. [DOI] [PubMed] [Google Scholar]
- 105.Park MS, et al. p27 and Rb are on overlapping pathways suppressing tumorigenesis in mice. Proc Natl Acad Sci U S A. 1999;96:6382–6387. doi: 10.1073/pnas.96.11.6382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Martin-Caballero J, et al. Different cooperating effect of p21 or p27 deficiency in combination with INK4a/ARF deletion in mice. Oncogene. 2004;23:8231–8237. doi: 10.1038/sj.onc.1207863. [DOI] [PubMed] [Google Scholar]
- 107.Philipp-Staheli J, et al. Pathway-specific tumor suppression. Reduction of p27 accelerates gastrointestinal tumorigenesis in Apc mutant mice, but not in Smad3 mutant mice. Cancer Cell. 2002;1:355–368. doi: 10.1016/s1535-6108(02)00054-5. [DOI] [PubMed] [Google Scholar]
- 108.De la Cueva E, et al. Tumorigenic activity of p21Waf1/Cip1 in thymic lymphoma. Oncogene. 2006;25:4128–4132. doi: 10.1038/sj.onc.1209432. [DOI] [PubMed] [Google Scholar]
- 109.Wang YA, Elson A, Leder P. Loss of p21 increases sensitivity to ionizing radiation and delays the onset of lymphoma in atm-deficient mice. Proc Natl Acad Sci U S A. 1997;94:14590–14595. doi: 10.1073/pnas.94.26.14590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Liu Y, et al. Somatic cell type specific gene transfer reveals a tumor-promoting function for p21(Waf1/Cip1) EMBO J. 2007;26:4683–4693. doi: 10.1038/sj.emboj.7601886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Besson A, et al. Discovery of an oncogenic activity in p27Kip1 that causes stem cell expansion and a multiple tumor phenotype. Genes Dev. 2007;21:1731–1746. doi: 10.1101/gad.1556607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Pellegata NS, et al. Germ-line mutations in p27Kip1 cause a multiple endocrine neoplasia syndrome in rats and humans. Proc Natl Acad Sci U S A. 2006;103:15558–15563. doi: 10.1073/pnas.0603877103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hotta K, et al. Gefitinib induces premature senescence in non-small cell lung cancer cells with or without EGFR gene mutation. Oncol Rep. 2007;17:313–317. [PubMed] [Google Scholar]
- 114.Tsai MF, et al. A new tumor suppressor DnaJ-like heat shock protein, HLJ1, and survival of patients with non-small-cell lung carcinoma. J Natl Cancer Inst. 2006;98:825–838. doi: 10.1093/jnci/djj229. [DOI] [PubMed] [Google Scholar]
- 115.Delahunt B, Bethwaite PB, Nacey JN. Outcome prediction for renal cell carcinoma: evaluation of prognostic factors for tumours divided according to histological subtype. Pathology. 2007;39:459–465. doi: 10.1080/00313020701570061. [DOI] [PubMed] [Google Scholar]
- 116.Puhalla H, et al. Expression of p21(Wafl/Cip1), p57(Kip2) and HER2/neu in patients with gallbladder cancer. Anticancer Res. 2007;27:1679–1684. [PubMed] [Google Scholar]
- 117.Mizokami K, et al. Relationship of hypoxia-inducible factor 1alpha and p21WAF1/CIP1 expression to cell apoptosis and clinical outcome in patients with gastric cancer. World J Surg Oncol. 2006;4:94. doi: 10.1186/1477-7819-4-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Villwock Mde M, et al. Prevalence of p21 immunohistochemical expression in esophageal adenocarcinoma. Arq Gastroenterol. 2006;43:212–218. doi: 10.1590/s0004-28032006000300011. [DOI] [PubMed] [Google Scholar]
- 119.Pijnenborg JM, et al. TP53 overexpression in recurrent endometrial carcinoma. Gynecol Oncol. 2006;100:397–404. doi: 10.1016/j.ygyno.2005.09.056. [DOI] [PubMed] [Google Scholar]
- 120.Li G, et al. Genetic polymorphisms of p21 are associated with risk of squamous cell carcinoma of the head and neck. Carcinogenesis. 2005;26:1596–1602. doi: 10.1093/carcin/bgi105. [DOI] [PubMed] [Google Scholar]
- 121.Bali A, et al. Cyclin D1, p53, and p21Waf1/Cip1 expression is predictive of poor clinical outcome in serous epithelial ovarian cancer. Clin Cancer Res. 2004;10:5168–5177. doi: 10.1158/1078-0432.CCR-03-0751. [DOI] [PubMed] [Google Scholar]
- 122.Anttila MA, et al. p21/WAF1 expression as related to p53, cell proliferation and prognosis in epithelial ovarian cancer. Br J Cancer. 1999;79:1870–1878. doi: 10.1038/sj.bjc.6690298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Viale G, et al. p21WAF1/CIP1 expression in colorectal carcinoma correlates with advanced disease stage and p53 mutations. J Pathol. 1999;187:302–307. doi: 10.1002/(SICI)1096-9896(199902)187:3<302::AID-PATH243>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 124.Caffo O, et al. Prognostic value of p21(WAF1) and p53 expression in breast carcinoma: an immunohistochemical study in 261 patients with long-term follow-up. Clin Cancer Res. 1996;2:1591–1599. [PubMed] [Google Scholar]
- 125.Bukholm IK, et al. Relationship between abnormal p53 protein and failure to express p21 protein in human breast carcinomas. J Pathol. 1997;181:140–145. doi: 10.1002/(SICI)1096-9896(199702)181:2<140::AID-PATH745>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 126.Domagala W, et al. p21/WAF1/Cip1 expression in invasive ductal breast carcinoma: relationship to p53, proliferation rate, and survival at 5 years. Virchows Arch. 2001;439:132–140. doi: 10.1007/s004280100410. [DOI] [PubMed] [Google Scholar]
- 127.Hemmati PG, et al. Loss of p21 disrupts p14 ARF-induced G1 cell cycle arrest but augments p14 ARF-induced apoptosis in human carcinoma cells. Oncogene. 2005;24:4114–4128. doi: 10.1038/sj.onc.1208579. [DOI] [PubMed] [Google Scholar]
- 128.McBride SR, Leonard N, Reynolds NJ. Loss of p21(WAF1) compartmentalisation in sebaceous carcinoma compared with sebaceous hyperplasia and sebaceous adenoma. J Clin Pathol. 2002;55:763–766. doi: 10.1136/jcp.55.10.763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Porter PL, et al. Expression of cell-cycle regulators p27Kip1 and cyclin E, alone and in combination, correlate with survival in young breast cancer patients. Nat Med. 1997;3:222–225. doi: 10.1038/nm0297-222. [DOI] [PubMed] [Google Scholar]
- 130.Xia W, et al. Phosphorylation/cytoplasmic localization of p21Cip1/WAF1 is associated with HER2/neu overexpression and provides a novel combination predictor for poor prognosis in breast cancer patients. Clin Cancer Res. 2004;10:3815–3824. doi: 10.1158/1078-0432.CCR-03-0527. [DOI] [PubMed] [Google Scholar]
- 131.Bachman KE, et al. 21(WAF1/CIP1) mediates the growth response to TGF-beta in human epithelial cells. Cancer Biol Ther. 2004;3:221–225. doi: 10.4161/cbt.3.2.666. [DOI] [PubMed] [Google Scholar]
- 132.Blain SW, Massague J. Breast cancer banishes p27 from nucleus. Nat Med. 2002;8:1076–1078. doi: 10.1038/nm1002-1076. [DOI] [PubMed] [Google Scholar]
- 133.Liang J, et al. PKB/Akt phosphorylates p27, impairs nuclear import of p27 and opposes p27-mediated G1 arrest. Nat Med. 2002;8:1153–1160. doi: 10.1038/nm761. [DOI] [PubMed] [Google Scholar]
- 134.Shin I, et al. PKB/Akt mediates cell-cycle progression by phosphorylation of p27(Kip1) at threonine 157 and modulation of its cellular localization. Nat Med. 2002;8:1145–1152. doi: 10.1038/nm759. [DOI] [PubMed] [Google Scholar]
- 135.Silva J, et al. Akt-Induced Tamoxifen Resistance is Associated with Altered FKHR Regulation. Cancer Invest. 2007:1–5. doi: 10.1080/07357900701513538. [DOI] [PubMed] [Google Scholar]
- 136.deGraffenried LA, et al. Inhibition of mTOR activity restores tamoxifen response in breast cancer cells with aberrant Akt Activity. Clin Cancer Res. 2004;10:8059–8067. doi: 10.1158/1078-0432.CCR-04-0035. [DOI] [PubMed] [Google Scholar]
- 137.Campbell RA, et al. Phosphatidylinositol 3-kinase/AKT-mediated activation of estrogen receptor alpha: a new model for anti-estrogen resistance. J Biol Chem. 2001;276:9817–9824. doi: 10.1074/jbc.M010840200. [DOI] [PubMed] [Google Scholar]
- 138.Cariou S, et al. Down-regulation of p21WAF1/CIP1 or p27Kip1 abrogates antiestrogen-mediated cell cycle arrest in human breast cancer cells. Proc Natl Acad Sci U S A. 2000;97:9042–9046. doi: 10.1073/pnas.160016897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Giannakakou P, et al. Low concentrations of paclitaxel induce cell type-dependent p53, p21 and G1/G2 arrest instead of mitotic arrest: molecular determinants of paclitaxel-induced cytotoxicity. Oncogene. 2001;20:3806–3813. doi: 10.1038/sj.onc.1204487. [DOI] [PubMed] [Google Scholar]
- 140.Schmidt M, Fan Z. Protection against chemotherapy-induced cytotoxicity by cyclin-dependent kinase inhibitors (CKI) in CKI-responsive cells compared with CKI-unresponsive cells. Oncogene. 2001;20:6164–6171. doi: 10.1038/sj.onc.1204814. [DOI] [PubMed] [Google Scholar]
- 141.Shang Y, Brown M. Molecular determinants for the tissue specificity of SERMs. Science. 2002;295:2465–2468. doi: 10.1126/science.1068537. [DOI] [PubMed] [Google Scholar]
- 142.Anonymous, Early Breast Cancer Trialists’ Collaborative Group Tamoxifen for early breast cancer: an overview of the randomised trials. Lancet. 1998;351:1451–1467. [PubMed] [Google Scholar]
- 143.Canney PA, et al. Clinical significance of tamoxifen withdrawal response. Lancet. 1987;1:36. doi: 10.1016/s0140-6736(87)90717-3. [DOI] [PubMed] [Google Scholar]
- 144.Michalides R, et al. Tamoxifen resistance by a conformational arrest of the estrogen receptor alpha after PKA activation in breast cancer. Cancer Cell. 2004;5:597–605. doi: 10.1016/j.ccr.2004.05.016. [DOI] [PubMed] [Google Scholar]
- 145.Rayala SK, Molli PR, Kumar R. Nuclear p21-activated kinase 1 in breast cancer packs off tamoxifen sensitivity. Cancer Res. 2006;66:5985–5988. doi: 10.1158/0008-5472.CAN-06-0978. [DOI] [PubMed] [Google Scholar]
- 146.Shou J, et al. Mechanisms of tamoxifen resistance: increased estrogen receptor-HER2/ neu cross-talk in ER/HER2-positive breast cancer. J Natl Cancer Inst. 2004;96:926–935. doi: 10.1093/jnci/djh166. [DOI] [PubMed] [Google Scholar]
- 147.Abukhdeir AM, et al. Tamoxifen-stimulated growth of breast cancer due to p21 loss. Proc Natl Acad Sci U S A. 2008;105:288–293. doi: 10.1073/pnas.0710887105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Jankevicius F, et al. p21 and p53 Immunostaining and survival following systemic chemotherapy for urothelial cancer. Urol Int. 2002;69:174–180. doi: 10.1159/000063949. [DOI] [PubMed] [Google Scholar]
- 149.Kuwahara M, et al. p53, p21(Waf1/Cip1) and cyclin D1 protein expression and prognosis in esophageal cancer. Dis Esophagus. 1999;12:116–119. doi: 10.1046/j.1442-2050.1999.00034.x. [DOI] [PubMed] [Google Scholar]
- 150.Cheng JD, et al. Paradoxical correlations of cyclin-dependent kinase inhibitors p21waf1/cip1 and p27kip1 in metastatic colorectal carcinoma. Clin Cancer Res. 1999;5:1057–1062. [PubMed] [Google Scholar]