

Abstract
A highly enantioselective and straightforward route to trisaccharide natural product digoxose and digitoxin has been developed. Key to this approach is the iterative application of the palladium-catalyzed glycosylation reaction, reductive 1,3-transposition, diastereoselective dihydroxylation and regioselective protection. The first total synthesis of natural product digoxose was accomplished in 19 total steps from achiral 2-acylfuran and the digitoxin was fashioned in 15 steps starting from digitoxigenin 2 and pyranone 8β. This flexible synthetic strategy also allows for the preparation of mono- and disaccharide analogues of digoxose and digitoxin.
Introduction
The cardiac glycoside digitoxin (1) (Figure 1), the extract from the leaves of Digitalis purpurea (purple foxglove), has long been used to slow the heart rate while increase the contractility of the heart muscle (inotropic activity). It has been widely prescribed for congestive heart failure and cardiac arrhythmia for over 200 years. However, extensive care must be taken when treating patients with digitoxin, because the typical therapeutic dose (14–26 ng mL−1) is dangerously close to the toxic dose (>35 ng mL−1).2 Digitoxin has also been shown to possess potential anticancer activities.3 Structurally, digitoxin is the combination of two natural products, the aglycon digitoxigenin (2)4 and the trisaccharide digoxose (3).5 Oligosaccharides are known to play important roles in many pharmacologically important antibiotics, vaccines and antitumor agents.6 For instance, while the digitoxigenin is considered to be the pharmacophore portion of digitoxin 1, the aglycon 2 is inactive without the attachment of digoxose trisaccharide 3. Thus, it is reasonable to assume that by manipulating of the sugar portion, the resulting analogues may have improved activities. An excellent example of the interplay between carbohydrate structure and biological activity can be seen in the digitoxin neoglycoside library constructed by Thorson and co-workers.7 Several of these digitoxigenin monosaccharide analogues showed improved anticancer activity yet lower cardiotoxicity.7 In an effort to differentiate the effects the carbohydrate substitution versus the neoglycoside substitution on activity, we desired access to digitoxin and its bis- and mono-saccharide analogues as well as the free sugars digoxose and its mono- and disaccharide.
Figure 1.

Digitoxin, Digoxose and Digitoxigenin
The stereocontrolled synthesis of 2-deoxy-β-glycosides is difficult due to the missing control element at the C-2 position.8,9 The use of the participation of attached C-3 group to control the anomeric stereochemistry had been shown to be unreliable.10 To date, the most direct method involved an SN2 substitution of α-D-glycopyranosyl donor and alcohol acceptor.11 The selectivity of this method, however, is highly dependent on the protecting-group manipulation (activation) of the pyranose donor and the reactivity of the acceptor.12 Their use is also limited by the low availability of corresponding monosaccharide in nature. Other strategies utilized equatorial C-2 heteroatom neighboring group (−Br,13 −I,14 −SAr,15 −SePh,16 −OAC,17 and −NHCHO18) to control the anomeric stereochemistry, which post-glycosylation need to be reductively removed using radical tin-hydride chemistry. Similarly the Barton-type deoxygenation of C-2 hydroxyl groups has been used in combination with nucleophilic opening of anomeric epoxides.19 This challenge of stereocontrolled synthesis of 2-deoxy β-glycosides is evident in the two previous syntheses of the tris-1,4-linked 2,6-dideoxy-β-D-allose portion of digitoxin. While there has been no synthesis of the natural product digoxose (3), there have been two syntheses of digitoxin (1), a carbohydrate approach by Wiesner and co-workers and a de novo approach by McDonald and co-workers.20,21 Herein we describe the full account of our successful de novo approach to 2-deoxy-β-allo-glycosides and its application to syntheses of β-linked 1,4-oligosaccharide natural products digoxose (3) and digitoxin (1).22 This de novo methodology (Scheme 1) features the iterative use of a β-selective palladium catalyzed glycosylation reaction23 and subsequent chemo- and stereoselective transformations for the diastereoselective installation of the C-3/C-4 hydroxy groups and regioselective C-3 protection, which we believe resulted in a more efficient and flexible route in terms of number of steps and stereocontrol than the previous approaches.24
Scheme 1.

Digitoxin/Digoxose Retrosynthesis
Results and Discussion
Approach to β-pyranones
Previously, we have shown that α-glycosyl donors, like the α-Boc-pyranones 8α and 15α can be oligomerized and subsequently transformed into α-linked oligosaccharides.23d Because these oligosaccharides are prepared from achiral acylfurans, like 9 and 10, we call this a de novo approach. By employing an enantioselectively Noyori reduction,25,26 an Achmatowicz oxidation and diastereoselectively acylation the acylfurans can be converted into the α-Boc-pyranones 8α and 15α (Scheme 2)27 Alternatively, the Boc-protection can be preformed at elevated temperature ((Boc)2O/NaOAc in benzene at 80 °C) such that the β-pyranones 8β and 15β can be isolated in ~50% yields from the Achmatowicz products 13 and 14, with the ratio of β-pyranones to α-pyranones at these higher temperatures can be as high as 1.3:1 (Scheme 2). As with the α-pyranones 8α and 15α, the β-pyranones 8β and 15β can be converted via palladium (0) catalysis into their corresponding mixed acetal pyranones with complete retention of stereochemistry (i.e., 8 to 7 and 15β to 16, Scheme 1 and 3).
Scheme 2.

De Novo Pyranone Synthesis
Scheme 3.

Synthesis of 2-Deoxy-L-allose 19
Approach to 2-Deoxy-β-glycosides
With a practical route to the β-pyranones glycosyl donors, we next turned our attention toward the preparation of 2-deoxy-β-L-allose, which started with the palladium catalyzed glycosylation (5 mol% Pd(0)/10% PPh3) of BnOH with the β-pyranone 15β27 to form the β-benzyloxy pyranone 16 in 84% yield as a single diastereomer (Scheme 3). Ketone reduction of pyranone 16 with NaBH4 28 gave a mixture of allylic alcohols 17a and 17b in 88% yield with the diastereomeric ratio of ca. 1.5 to 1, respectively. Fortunately, both diastereomers 17a/b could be used in the next reaction. The diastereomeric ratio of alcohols could be improved with the use of DibalH (dr = 6:1); however, because of the slightly lower yields, we preferred to use the operational simpler Luche procedure (NaBH4/CeCl3, −78 °C; 88% 1.5:1). Applying the Myers’ reductive rearrangement conditions29 (NBSH, PPh3/DEAD, NMM, −30 °C to rt) to the mixture of allylic alcohols 17 cleanly provided olefin 18 in 71% yield. Finally exposing olefin 18 to the Upjohn conditions30 (OsO4/NMO) gave exclusively the diol 19 in 91% yield.31
With the successful synthesis of 2-deoxy-L-allose 19, we next investigated the synthesis of allo-disaccharides using the same strategy (Scheme 4). Our attempts at the regioselective glycosylation of diol 19 using pyranones 15β were not promising, giving a mixture of C-3 and C-4 glycosylated product 20 and 21 in a ratio of ca 1.3 to 1. The regiochemistry of glycosylation was assigned by coupling constant analysis of the benzoate 22 from the major isomer 20. Because of the inability of our palladium glycosylation methodology to differentiate the equatorial alcohol from the axial alcohol in 19, we decided to incorporate a selective protection step as applied this methodology toward oligosaccharides (vide infra).
Scheme 4.

Pd(0) Catalyzed Glycosylation of 2-Deoxy-L-allose 19
Synthesis of Digoxose and Digitoxin
Encouraged by these promising results towards the synthesis of 2-deoxy-L-allose 19, we next investigated the use of this approach for the synthesis of digitoxose 25a. Thus, palladium catalyzed glycosylation of pyranone (D)-8β with benzyl alcohol provided the pyranone 7a in 85% yield as a single diastereomer (Scheme 5). The Luche reduction of pyranone 7a gave a mixture of allylic alcohols 23a in 85% yield. Reductive rearrangement of allylic alcohols 23a provided olefin 24a in 84% yield. Dihydroxylation of 24a using the Upjohn conditions (OsO4/NMO)30 gave exclusively the diol 25a in 92% yield.31 Using the same strategy starting from the digitoxigenin and β-pyranone (D)-8β, the digitoxigenin monodigitoxoside 25b was prepared with similar efficiency.32,33 It is worth noting that both the tertiary alcohol and the butenolide ring of the aglycon were left untouched and thus compatible to the palladium catalyzed glycosylation and subsequent transformation.
Scheme 5.

Synthesis of Digitoxose and Digitoxin Monosaccharides 25a/b
To prepare the digitoxose disaccharide, a regioselective protection of diols 25a/b was needed. This was achieved by the formation of an orthoester and regioselective ring opening (Scheme 6).34 Specifically, the diol 25a was treated with trimethyl orthoacetate and catalytic p-toluenesulfonic acid to form an orthoester intermediate, which upon regiospecific acid hydrolysis (TsOH/H2O) opened to the kinetically preferred axial acetate 26a in excellent yield (95%). In the case of digitoxigenin monodigitoxoside 25b, the reaction works as well as 25a. The tertiary alcohol, the butenolide ring of the aglycon, and glycosidic bond were compatible with the acidic condition. The remaining equatorial alcohol was then ready to serve as a glycosyl acceptor for a second Pd(0) catalyzed glycosylation.
Scheme 6.

Regioselective Protection of Diol 25
We next explored the potential for the synthesis of β-linked 1,4-oligosaccharides via glycosylation of the C-4 secondary alcohol in 26a/b.35 Applying the same palladium catalyzed glycosylation conditions to 26a/b (2 equiv of pyranone (D)-8β, with 5% Pd(0)/10% PPh3) afforded the C-4 disaccharides 27a/b with complete stereocontrol at the anomeric center in 78% and 80% yields, respectively (Scheme 7). Once again, the 1,2-reduction of the keto-group in pyranone 27a/b under Luche conditions provided a mixture of allylic alcohols 28a/b (~1:1), which when exposed to the Myers’ reductive 1,3-allylic transposition conditions provided olefin 29a/b in 82% yield. Finally, applying the Upjohn conditions to 29a/b gave exclusively the dihydroxylated products 30a/b in 90% and 91% yield, respectively. 31 The digitoxose disaccharide and digitoxin disaccharide 31a/b were fashioned by deprotection of the acetate-protecting groups in 30a/b in 93% and 82% yield, respectively.
Scheme 7.

Synthesis of Digitoxin and Digitoxose Disaccharides
Gratifyingly, the preparation of trisaccharide occurred with the same efficiency and high degree of stereocontrol as with the disaccharides 31a/b (Scheme 7). Thus, regioselective protection of the C-3 axial alcohol in diol 30a/b provided the equatorial alcohol 32a/b which were subjected to the pyranone (D)-8β under Pd(0) catalyst to fashion the 1,4-linked trisaccharides 33a/b in 79% and 90% yield, respectively (Scheme 8). When enone 33a/b were subjected to Luche reduction, a mixture of allylic alcohols 34a/b was obtained in 93% and 98% yields, respectively. The alcohols 34a/b were then reductively rearranged to corresponding olefin 35a/b in 81% and 89% yield, respectively. The trisaccharides 4 and 1 were prepared with high yield and complete stereocontrol by dihydroxylation of olefins in 35a/b and followed by deprotection of acetate groups in 36a/b. The synthetic material 1 has identical physical and spectral data to that of the commercially available natural product 136,37 (1H NMR, 13C NMR, optical rotation and melting point).
Scheme 8.

Synthesis of Digitoxin and Digitoxose Trisaccharides 1 and 4
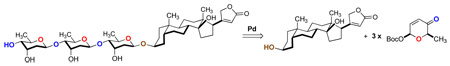
Finally the digoxose bisdigitoxoside 37 and natural product digoxose 3 was prepared by hydrogenolysis of the anomeric benzyl group (H2, Pd/C), which gave synthetic material with identical physical and spectral data to that of the isolated natural product 3 (1H NMR, 13C NMR, optical rotation and melting point). 37,38
Conclusions
In conclusion, a highly enantioselective route to digitoxin (1) and digoxose (3) as well as corresponding mono- and disaccharides (25a, 25b and 31b, 37) has been developed. Key to the success of this approach is the iterative use of the palladium-catalyzed glycosylation reaction, Myers’ reductive rearrangement, diastereoselective dihydroxylation and regioselective protection. Digoxose (3) was enantioselectively prepared in 16 steps and 12% overall yield from pyranone 8β (19 steps from achiral 2-acylfuran), which constitutes the first total synthesis of 3. The digitoxin (1) was achieved in 15 steps from digitoxigenin 2 and β-pyranone 8, which is not only the shortest but also the most stereocontrolled synthesis so far.39 This approach is equally amenable for the synthesis of the diastereomeric l-sugar digitoxin analogues. The uses of this strategy for the synthesis of these various analogues are ongoing and will be reported in due course.
Experimental Section40
(2R,6R)-2-Methyl-6-(phenylmethoxy)-2H-pyran-3(6H)-one (7a)
A CH2Cl2 (3 mL) solution of Boc pyranone 8β (716 mg, 3.14 mmol) and benzyl alcohol (678 mg, 6.28 mmol) was cooled to 0 °C. A CH2Cl2 (2 mL) solution of Pd2(DBA)3•CHCl3 (81 mg, 2.5 mol%) and PPh3 (82 mg, 10 mol%) was added to the reaction mixture at 0 °C. The reaction mixture was stirred at 0 °C for 2 hours and was quenched with 10 mL of saturated aqueous NaHCO3, extracted (3 × 10 mL) with Et2O, dried (Na2SO4), and concentrated under reduced pressure. The crude product was purified by silica gel flash chromatography eluting with 8% EtOAc/hexanes to give 7a (582 mg, 2.67 mmol, 85%) as a viscous oil: Rf (15% EtOAc/hexanes) = 0.23; ; IR (thin film, cm−1) 2933, 1698, 1453, 1373, 1163, 1057, 1023, 903, 800, 733, 697; 1H NMR (270 MHz, CDCl3) δ 7.37 (m, 5H), 6.92 (dd, J = 10.3, 2.0 Hz, 1H), 6.14 (dd, J = 10.3, 1.6 Hz, 1H), 5.40 (m, 1H), 4.95 (d, J = 11.7 Hz, 1H), 4.69 (d, J = 11.7 Hz, 1H), 4.24 (q, J = 6.9 Hz, 1H), 1.53 (d, J = 6.9 Hz, 3H); 13C NMR (67.5 MHz, CDCl3) δ 196.8, 146.4, 136.8, 128.5 (2C), 128.1(3C), 128.0, 94.3, 75.2, 70.1, 17.2; HRCIMS Calcd for [C13H14O3Na+]: 241.0835, Found 241.0843.
(2R,6R)-3,6-Dihydro-2-methyl-6-(phenylmethoxy)-2H-pyran-3-ol (23a)
A CH2Cl2 (2 mL) solution of enone 7a (435 mg, 2.0 mmol) and CeCl3 in MeOH solution (1.7 mL) was cooled to −78 °C. NaBH4 (75 mg, 2.0 mmol) was added and the reaction mixture was stirred at −78 °C for 3 hours. The reaction mixture was diluted with Et2O (5 mL) and was quenched with 5 mL of saturated aqueous NaHCO3, extracted (3 × 5 mL) with Et2O, dried (Na2SO4), and concentrated under reduced pressure. The crude product was purified by silica gel flash chromatography eluting with 20% EtOAc/hexanes to give allylic alcohols 23a (374 mg, 1.70 mmol, 85%) as a viscous oil (diastereomeric ratio I:II = 1.5:1, inseparable by silica gel chromatography): Rf (40% EtOAc/hexanes) = 0.30; IR (thin film, cm−1) 3397, 2978, 2933, 2869, 1498, 1455, 1378, 1053, 1010, 808, 738, 698; 1H NMR (600 MHz, CDCl3): isomer I: δ 7.35 (m, 5H), 6.16 (ddd, J = 10.2, 5.4, 1.2 Hz, 1H), 5.86 (d, J = 10.2 Hz, 1H), 5.14 (ddd, J = 1.8, 1.8, 1.2 Hz, 1H), 4.92 (d, J = 12.0 Hz, 1H), 4.66 (d, J = 12.0 Hz, 1H), 3.75 (qd, J = 6.0, 2.4 Hz, 1H), 3.68 (m, 1H), 2.0 (d, J = 10.2 Hz, 1H), 1.34 (d, J = 6.0 Hz, 3H); isomer II: δ 7.30 (m, 5H), 5.95 (ddd, J = 10.2, 2.4, 1.8 Hz, 1H), 5.79 (ddd, J = 10.2, 1.8, 1.2 Hz, 1H), 5.18 (ddd, J = 1.8, 1.8, 1.2 Hz, 1H), 4.86 (d, J = 12.0 Hz, 1H), 4.61 (d, J = 12.0 Hz, 1H), 3.90 (m, 1H), 3.64 (dq, J = 6.6, 6.0 Hz, 1H), 2.10 (d, J = 6.6 Hz, 1H), 1.38 (d, J = 6.0 Hz, 3H); 13C NMR (150 MHz, CDCl3) isomer I: δ137.5, 131.3, 130.6, 128.4 (2C), 127.9 (2C), 127.7, 97.0, 71.4, 10 69.9, 64.7, 16.6; isomer II: δ 137.7, 132.1, 128.7, 128.3 (2C ), 127.9 (2C), 127.6, 95.5, 74.4, 69.2, 68.3, 17.8; HRCIMS Calcd for [C13H16O3Na+]: 243.0992, Found 243.0983.
Cis-3,6-dihydro-6-methyl-2-(phenylmethoxy)-2H-pyran (24a)
A flask was charged with dry N-methyl morpholine (NMM) 3.0 mL, triphenyl phosphine (1.45 g, 5.54 mmol) and was cooled to −30 °C under Ar atmosphere. Diethylazodicarboxylate (0.8 mL, 5.05 mmol) was added and the reaction was stirred for 5 minutesutes, allylic alcohol 23a (370 mg, 1.68 mmol) was added in a 1M solution of NMM and the reaction mixture was stirred for 10 minutes, followed by addition of o-nitrobenzenesulfonyl hydrazide (NBSH) (1.02 g, 5.05 mmol). The reaction was stirred at −30 °C for 2h and was monitored by TLC, upon consumption of starting material, warm up to room temperature and stirred for another 2h. The reaction mixture was diluted with Et2O (10 mL) and was quenched with 5 mL of saturated aqueous NaHCO3, extracted (3 × 5 mL) with Et2O, dried (Na2SO4), and concentrated under reduced pressure. The crude product was purified using silica gel flash chromatography eluting with 2% Et2O/hexanes to give product 24a (293 mg, 1.44 mmol, 84%) as a viscous oil: Rf (15% EtOAc/hexanes) = 0.48; ; IR (thin film, cm−1) 2973, 2927, 1453, 1366, 1158, 1080, 1028, 880, 777, 733. 1H NMR (600 MHz, CDCl3) δ 7.35 (m, 5H), 5.69 (ddd, J = 10.2, 4.8, 2.4 Hz, 1H), 5.60 (ddd, J = 10.2, 1.2, 1.2 Hz, 1H), 4.95 (d, J = 12.0 Hz, 1H), 4.75 (dd, J = 9.0, 3.0 Hz, 1H), 4.63 (d, J = 12.0 Hz, 1H), 4.35 (m, 1H), 2.27 (dddd, J = 17.4, 8.4, 3.6, 2.4 Hz, 1H), 2.19 (dddd, J = 17.4, 6.6, 2.4, 1.2 Hz, 1H), 1.33 (d, J = 6.6 Hz, 3H); 13C NMR (150 MHz, CDCl3) δ 137.9, 130.9, 128.3 (2C), 127.9 (2C), 127.6, 122.5, 97.7, 70.6, 69.8, 30.9, 21.1; HRCIMS Calcd for [C13H16O2Na+]: 227.1042, Found 227.1045.
Phenylmethyl 2,6-dideoxy-β-D-ribo-hexopyranoside (25a)
To a CH2Cl2 (3 mL) solution of olefin 24a (291 mg, 1.43 mmol) at 0 °C was added a solution of (50% w/v) of N-methyl morpholine N-oxide / water (0.67 mL). Crystalline OsO4 (3.6 mg, 1 mol %) was added and the reaction was stirred for 3 h. The reaction was quenched by adding EtOAc and satd. NaHCO3. The organic layer was separated and concentrated. It was purified by a silica gel column using 35% EtOAc/hexanes. Pure fractions were combined and concentrated to afford diol 25a as a viscous oil (313 mg, 1.31 mmol, 92%): Rf (50% EtOAc/hexanes) = 0.23; ; IR (thin film, cm−1) 3426, 2883, 1496, 1454, 1364, 1164, 1137, 1072, 1007, 867, 731, 698; 1H NMR (600 MHz, CDCl3)δ 7.34 (m, 5H), 4.90 (dd, J = 9.0, 1.8 Hz, 1H), 4.88 (d, J = 11.4 Hz, 1H), 4.57 (d, J = 12.0 Hz, 1H), 4.09 (m, 1H), 3.74 (dq, J = 9.0, 6.0 Hz, 1H), 3.32 (m, 1H), 2.51(s, 1H), 2.35(s, 1H), 2.12 (ddd, J = 13.8, 3.6, 2.4 Hz, 1H), 1.78 (ddd, J = 13.8, 9.0, 3.0 Hz, 1H), 1.33(d, J = 6.0 Hz, 3H); 13C NMR (150 MHz, CDCl3) δ 137.7, 128.4 (2C), 127.9 (2C), 127.7, 96.9, 73.0, 70.5, 69.5, 67.9, 37.7, 18.1; HRCIMS Calcd for [C13H18O4Na+]: 261.1097, Found 261.1087.
Phenylmethyl 3-O-acetyl-2,6-dideoxy-β-D-ribo-hexopyranoside (26a)
A round bottom flask containing a 0.5 M solution of diol 25a (300 mg, 1.26 mmol) in benzene (2.5 mL) was stirring at room temperature. To this solution were added trimethylorthoacetate (0.8 mL, 6.29 mmol) and a catalytic amount of p-toluenesulfonic acid (12 mg, 63 µmol). The reaction was allowed to stir until starting material is gone. The solvent was removed under reduced pressure and the residue was dissolved in 3 mL THF/H2O (1:1,v/v) solution. Then p-toluenesulfonic acid (600 mg, 3.15 mmol) was added. Stirring was continued until hydrolysis was complete as seen by TLC. The reaction was quenched by adding EtOAc and satd NaHCO3. The organic layer was separated and concentrated. It was purified by a silica gel column using 30% EtOAc/hexanes. Pure fractions were combined and concentrated to afford compound 26a (335 mg, 1.20 mmol, 95%): Rf (50% EtOAc/hexanes) = 0.38; ; IR (thin film, cm−1) 3471, 2975, 2934, 1740, 1498, 1455, 1372, 1242, 1164, 1075, 1006, 698; 1H NMR (600 MHz, CDCl3) δ 7.34 (m, 5H), 5.29 (ddd, J = 3.6, 3.0, 3.0 Hz, 1H), 4.91 (d, J = 12.0 Hz, 1H), 4.83 (dd, J = 9.0, 2.4 Hz, 1H), 4.57 (d, J = 12.0 Hz, 1H), 3.73 (dq, J = 9.0, 6.0 Hz, 1H), 3.46 (dd, J = 9.0, 3.0 Hz, 1H), 2.14 (ddd, J = 14.4, 3.6, 2.4 Hz, 1H), 2.10 (s, 3H), 1.87 (ddd, J = 14.4, 9.0, 3.0 Hz, 1H), 1.36 (d, J = 6.0 Hz, 3H); 13C NMR (67.5 MHz, CDCl3) δ 171.2, 137.5, 128.3(2C), 127.8 (2C), 127.7, 97.0, 72.2, 71.0, 70.4, 70.3, 35.6, 21.1, 18.0; HRCIMS Calcd for [C15H20O5Na+]: 303.1203, Found 303.1201.
Phenylmethyl 3-O-acetyl-2,6-dideoxy-4-O-[(2R,6R)-5,6-dihydro-6-methyl-5-oxo-2H-pyran-2-yl]-β-D-ribo-hexopyranoside (27a)
A CH2Cl2 (0.8 mL) solution of Boc pyranone 8β (337 mg, 1.48 mmol) and alcohol 26a (207 mg, 0.74 mmol) was cooled to 0 °C. A CH2Cl2 (0.4 mL) solution of Pd2(DBA)3•CHCl3 (19 mg, 2.5 mol%) and PPh3 (20 mg, 10 mol%) was added to the reaction mixture at 0 °C. The reaction mixture was stirred at 0 °C for 2 hour. The reaction mixture was quenched with 5 mL of saturated aqueous NaHCO3, extracted (3 × 5 mL) with Et2O, dried (Na2SO4), and concentrated under reduced pressure. The crude product was purified using silica gel flash chromatography eluting with 22% EtOAc/hexanes to give enone 27a (228 mg, 0.58 mmol, 78%) as a viscous oil: Rf (30% EtOAc/hexanes) = 0.23; ; IR (thin film, cm−1) 2931, 1739, 1698, 1454, 1373, 1256, 1242, 1155, 1050, 1004, 787, 698; 1H NMR (600 MHz, CDCl3) δ 7.34 (m, 5H), 6.89 (dd, J = 10.2, 1.2 Hz, 1H), 6.13 (dd, J = 10.2, 1.2 Hz, 1H), 5.44 (ddd, J = 3.6, 3.0, 3.0 Hz, 1H), 5.42 (d, J = 1.2 Hz, 1H), 4.90 (d, J = 11.4 Hz, 1H), 4.83 (dd, J = 9.0, 2.4 Hz, 1H), 4.57 (d, J = 11.4 Hz, 1H), 4.16 (q, J = 6.6 Hz, 1H), 3.96 (dq, J = 9.0, 6.6 Hz, 1H), 3.55 (dd, J = 9.0, 3.0 Hz, 1H), 2.19 (ddd, J = 14.4, 3.6, 2.4 Hz, 1H), 2.06 (s, 3H), 1.85 (ddd, J = 14.4, 9.0, 3.0 Hz, 1H), 1.40 (d, J = 6.6 Hz, 3H), 1.37 (d, J = 6.6 Hz, 3H); 13C NMR (150 MHz, CDCl3) δ 196.2, 170.1, 146.3, 137.6, 128.8, 128.4 (2C), 127.8 (2C), 127.7, 97.1, 97.0, 79.4, 75.2, 70.5, 69.4, 69.3, 35.6, 21.2, 18.3, 16.3; HRCIMS Calcd for [C21H26O7Na+]: 413.1571, Found 413.1558.
Phenylmethyl 3-O-acetyl-2,6-dideoxy-4-O-[(2R,6R)-5,6-dihydro-5-hydroxy-6-methyl-2H-pyran-2-yl]-β-D-ribo-hexopyranoside (28a)
A CH2Cl2 (0.6 mL) solution of enone 27a (228 mg, 0.584 mmol) and CeCl3 in MeOH solution (0.6 mL) was cooled to −78 °C. NaBH4 (22 mg, 0.585 mmol) was added and the reaction mixture was stirred at −78 °C for 3 h. The reaction mixture was diluted with Et2O (5 mL) and was quenched with 5 mL of saturated aqueous NaHCO3, extracted (3 × 5 mL) with Et2O, dried (Na2SO4), and concentrated under reduced pressure. The crude product was purified using silica gel flash chromatography eluting with 35% EtOAc/hexanes to give allylic alcohols 28a (211 mg, 0.538 mmol, 92%) as a viscous oil (diastereometric ratio I:II = 1.6:1, inseparable by silica gel chromatography): Rf (40% EtOAc/hexanes) = 0.15; IR (thin film, cm−1) 3471, 2980, 2934, 2875, 1740, 1498, 1455, 1372, 1243, 1154, 1057, 1009, 736, 698; 1H NMR (600 MHz, CDCl3): isomer I: δ 7.28 (m, 5H), 6.17 (dd, J = 10.2, 5.4 Hz, 1H), 5.75 (d, J = 10.2 Hz, 1H), 5.57 (ddd, J = 3.0, 3.0, 3.0 Hz, 1H), 5.15 (m, 1H), 4.91 (d, J = 12.0 Hz, 1H), 4.84 (dd, J = 9.0, 1.8 Hz, 1H), 4.57 (d, J = 12.0 Hz, 1H), 3.91 (dq, J = 9.0, 6.0 Hz, 1H), 3.70 (dq, J = 1.8, 6.0 Hz, 1H), 3.60 (dd, J = 11.4, 6.0 Hz, 1H), 3.47 (dd, J = 10.2, 3.6 Hz, 1H), 2.27 (d, J = 14.4 Hz, 1H), 2.10 (ddd, J = 14.4, 4.8, 2.4 Hz, 1H), 2.05 (s, 3H), 1.87 (ddd, J = 14.4, 9.6, 2.4 Hz, 1H), 1.32 (d, J = 6.0 Hz, 3H), 1.27 (d, J = 6.6 Hz, 3H); isomer II: δ 7.34 (m, 5H), 5.96 (d, J = 10.2 Hz, 1H), 5.78 (d, J = 10.2 Hz, 1H), 5.42 (ddd, J = 3.0, 3.0, 3.0 Hz, 1H), 5.18 (m, 1H), 4.90 (d, J = 12.0 Hz, 1H), 4.80 (dd, J = 9.0, 1.8 Hz, 1H), 4.56 (d, J = 12.0 Hz, 1H), 3.86 (m, 2H), 3.64 (dq, J = 6.6, 6.0 Hz, 1H), 3.45 (dd, J = 9.6, 3.0 Hz, 1H), 2.16 (ddd, J = 14.4, 3.6, 2.4 Hz, 1H), 2.06 (s, 3H), 1.83 (ddd, J = 14.4, 9.0, 3.0 Hz, 1H), 1.63 (d, J = 7.8 Hz, 1H), 1.34 (d, J = 6.6 Hz, 3H), 1.29 (d, J = 6.6 Hz, 3H); 13C NMR (150 MHz, CDCl3) isomer I: δ 170.3, 137.67, 131.8, 128.4, 128.2 (2C), 127.7 (3C), 98.3, 97.21, 78.2, 71.5, 70.51, 70.1, 69.3, 64.4, 35.9, 21.28, 18.1, 16.7; isomer II: δ 170.2, 137.65, 132.9, 129.3, 128.37 (2C), 127.8 (3C), 97.5, 97.16, 78.1, 74.5, 70.49, 69.8, 69.4, 68.5, 35.8, 21.25, 18.3, 18.2; HRCIMS Calcd for [C21H28O7Na+]: 415.1727, Found 415.1726.
Phenylmethyl 3-O-acetyl--2,6-dideoxy-4-O-[(2S,6R)-3,6-dihydro-6-methyl-2H-pyran-2-yl]-β-D-ribo-hexopyranoside (29a)
A flask was charged with dry N-methyl morpholine (NMM) 0.9 mL, triphenyl phosphine (465 mg, 1.78 mmol) and was cooled to −30 °C under Ar atmosphere. Diethylazodicarboxylate (0.25 mL, 1.61 mmol) was added and the reaction was stirred for 5 minutes, allylic alcohols 28a (195 mg, 0.50 mmol) was added in a 1M solution of NMM and the reaction mixture was stirred for 10 minutes, followed by addition of o-nitrobenzenesulfonyl hydrazide (NBSH) (328 mg, 1.61 mmol). The reaction was stirred at −30 °C for 2 hours and was monitored by TLC. Upon consumption of starting material, the reaction was warmed up to room temperature and stirred for another 2 hours. The reaction mixture was diluted with Et2O (10 mL) and was quenched with 5 mL of saturated aqueous NaHCO3, extracted (3 × 5 mL) with Et2O, dried (Na2SO4), and concentrated under reduced pressure. The crude product was purified by silica gel flash chromatography eluting with 8% EtOAc/hexanes to give product 29a (156 mg, 0.41 mmol, 82%) as a viscous oil: Rf (15% EtOAc/hexanes) = 0.31; ; IR (thin film, cm−1) 2974, 2927, 1742, 1453, 1367, 1243, 1156, 1090, 1065, 1044, 781, 698. 1H NMR (600 MHz, CDCl3) δ 7.34 (m, 5H), 5.63 (dddd, J = 9.6, 4.8, 2.4, 2.4 Hz, 1H), 5.55 (ddd, J = 10.2, 2.4, 1.2 Hz, 1H), 5.55 (ddd, J = 3.6, 3.0, 3.0 Hz, 1H), 4.90 (d, J = 12.0 Hz, 1H), 4.81 (dd, J = 9.6, 1.8 Hz, 1H), 4.69 (dd, J = 8.4, 3.0 Hz, 1H), 4.56 (d, J = 12.0 Hz, 1H), 4.28 (m, 1H), 3.93 (dq, J = 9.0, 6.0 Hz, 1H), 3.38 (dd, J = 9.0, 3.0 Hz, 1H), 2.20 (ddd, J = 14.4, 3.6, 2.4 Hz, 1H), 2.18 (ddd, J = 17.4, 7.2, 4.2 Hz, 1H), 2.13 (ddd, J = 17.4, 6.6, 3.0 Hz, 1H), 2.07(s, 3H), 1.84 (ddd, J = 14.4, 9.0, 3.0 Hz, 1H), 1.33 (d, J = 6.0 Hz, 3H), 1.21 (d, J = 6.6 Hz, 3H); 13C NMR (150 MHz, CDCl3) δ 170.3, 137.7, 131.2, 128.4 (2C), 127.8 (2C), 127.7, 122.2, 100.3, 97.2, 79.2, 70.9, 70.5, 69.9, 69.4, 35.7, 30.9, 21.3, 20.8, 18.2; HRCIMS Calcd for [C21H28O6Na+]: 399.1778, Found 399.1773.
Phenylmethyl 3-O-acetyl-4-O-[2,6-dideoxy-β-D-ribo-hexopyranosyl]-2,6-dideoxy-β-D-ribo-hexopyranoside (30a)
To a CH2Cl2 (3 mL) solution of olefin 29a (148 mg, 0.39 mmol) at 0 °C was added a solution of (50% w/v) of N-methyl morpholine N-oxide / water (0.11 mL). Crystalline OsO4 (1.2 mg, 1 mol %) was added and the reaction was stirred for 3 hours. The reaction was quenched by adding EtOAc and saturated aqueous NaHCO3. The organic layer was separated and concentrated. It was purified by a silica gel column using 60% EtOAc/hexanes. Pure fractions were combined and concentrated to afford diol 30a (145 mg, 0.35 mmol, 90%): Rf(70% EtOAc/hexanes) = 0.18; ; IR (thin film, cm−1) 3436, 2972, 2932, 2879, 1741, 1370, 1247, 1165, 1066, 1012, 868, 740, 698; 1H NMR (600 MHz, CDCl3) δ 7.33 (m, 5H), 5.38 (ddd, J = 3.6, 3.0, 3.0 Hz, 1H), 4.89 (d, J = 12.0 Hz, 1H), 4.84 (dd, J = 9.6, 2.4 Hz, 1H), 4.79 (dd, J = 9.6, 2.4 Hz, 1H), 4.55 (d, J = 12.0 Hz, 1H), 4.05 (m, 1H), 3.88 (dq, J = 9.0, 6.0 Hz, 1H), 3.67 (dq, J = 9.0, 6.0 Hz, 1H), 3.35 (dd, J = 9.6, 3.0 Hz, 1H), 3.24 (ddd, J = 9.0, 6.6, 3.6 Hz, 1H), 2.54 (s, 1H), 2.33(d, J = 5.4 Hz, 1H), 2.16 (ddd, J = 14.4, 3.6, 2.4 Hz, 1H), 2.08 (ddd, J = 14.4, 3.0, 2.4 Hz, 1H), 2.06 (s, 3H), 1.82 (ddd, J = 14.4, 9.6, 3.0 Hz, 1H), 1.68 (ddd, J = 14.4, 9.6, 3.0 Hz, 1H), 1.31(d, J = 6.0 Hz, 3H), 1.22(d, J = 6.0 Hz, 3H); 13C NMR (150 MHz, CDCl3) δ 170.3, 137.6, 128.3 (2C), 127.8 (2C), 127.7, 98.6, 97.2, 79.4, 72.7, 70.5, 69.7, 69.3, 69.2, 68.0, 37.6, 35.6, 21.3, 18.2, 17.9; HRCIMS Calcd for [C21H30O8Na+]: 433.1833, Found 433.1826.
Phenylmethyl 2,6-dideoxy-4-O-[2,6-dideoxy-β-D-ribo-hexopyranosyl]-β-D-ribo-hexopyranoside (31a)
To a MeOH/H2O (0.1 mL, 1:1, 1M) solution of diol 30a (6 mg, 14.6 µmol) at room temperature was added LiOH (0.35 mg, 14.6 µmol) and the reaction was stirred for 3 hours. The reaction was quenched by adding EtOAc and saturated aquous NaHCO3. The organic layer was separated and concentrated. It was purified by a silica gel column using 65% EtOAc/hexanes. Pure fractions were combined and concentrated to afford triol 31a (5 mg, 13.6 µmol, 93%) as a white solid: Rf (80% EtOAc/hexanes) = 0.28; mp: 145–145.5 °C; ; IR (thin film, cm−1) 3437, 2962, 2931, 2886, 1454, 1405, 1368, 1164, 1068, 1011, 868, 735, 698; 1H NMR (600 MHz, CDCl3) δ 7.32 (m, 5H), 4.92 (m, 1H), 4.91 (m, 1H), 4.88 (d, J = 12.0 Hz, 1H), 4.56 (d, J = 12.0 Hz, 1H), 4.26 (dddd, J = 6.6, 3.6, 3.6, 1.8 Hz, 1H), 4.12 (dddd, J = 5.4, 4.2, 3.0, 2.4 Hz, 1H), 3.82 (dq, J = 9.0, 6.0 Hz, 1H), 3.67 (dq, J = 9.6, 6.0 Hz, 1H), 3.30 (ddd, J = 9.6, 6.6, 3.0 Hz, 1H), 3.26 (dd, J = 9.6, 3.0 Hz, 1H), 2.96 (m, J = 2.4, 1.8, 1.2 Hz, 1H), 2.28 (d, J = 1.2 Hz, 1H), 2.17 (ddd, J = 14.4, 4.2, 2.4 Hz, 1H), 2.13 (ddd, J = 14.4, 3.0, 2.4 Hz, 1H), 1.96 (d, J = 6.6 Hz, 1H), 1.79 (m, 1H), 1.75 (m, 1H), 1.29 (d, J = 6.0 Hz, 3H), 1.28(d, J = 6.0 Hz, 3H); 13C NMR (150 MHz, CDCl3) δ 137.8, 128.4 (2C), 127.9 (2C), 127.6, 98.3, 97.1, 82.7, 72.8, 70.6, 69.5, 68.3, 68.2, 66.3, 37.9, 36.6, 18.2, 18.1; HRCIMS Calcd for [C19H28O7Na+]: 391.1727, Found 391.1726.
Phenylmethyl 3-O-acetyl-4-O-[3-O-acetyl-2,6-dideoxy-β-D-ribo-hexopyranosyl]-2,6-dideoxy-β-D-ribo-hexopyranoside (32a)
A round bottom flask containing a 0.5 M solution of diol 30a (140 mg, 0.34 mmol) in benzene (0.6 mL) was stirring at room temperature. To this solution were added trimethylorthoacetate (0.13 mL, 1.02 mmol) and a catalytic amount of p-toluenesulfonic acid (3.2 mg, 17 µmol). The reaction was allowed to stir until starting material is gone. The solvent was removed under reduced pressure and the residue was dissolved in 0.8 mL THF/H2O (1:1,v/v) solution. Then p-toluenesulfonic acid (97 mg, 0.51 mmol) was added. Stirring was continued until hydrolysis was complete as seen by TLC. The reaction was quenched by adding EtOAc and saturated aqueous NaHCO3. The organic layer was separated and concentrated. It was purified by a silica gel column using 45% EtOAc/hexanes. Pure fractions were combined and concentrated to afford compound 32a (143 mg, 0.32 mmol, 93%) as a white solid: Rf (80% EtOAc/hexanes) = 0.48; mp: 105–106 °C; ; IR (thin film, cm−1) 3475, 2972, 2932, 2879, 1741, 1370, 1243, 1165, 1068, 1009, 947, 870, 704; 1H NMR (600 MHz, CDCl3) δ 7.33 (m, 5H), 5.39 (ddd, J = 3.6, 3.0, 3.0 Hz, 1H), 5.25 (ddd, J = 3.6, 3.0, 2.4 Hz, 1H), 4.89 (d, J = 12.0 Hz,1H), 4.79 (dd, J = 9.6, 1.8 Hz, 1H), 4.74 (dd, J = 9.6, 1.8 Hz, 1H), 4.55 (d, J = 12.0 Hz, 1H), 3.88 (dq, J = 9.0, 6.0 Hz, 1H), 3.65 (dq, J = 9.0, 6.0 Hz, 1H), 3.36 (dd, J = 9.0, 3.0 Hz, 1H), 3.35 (ddd, J = 9.0, 3.0, 3.0 Hz, 1H), 2.17 (ddd, J = 14.4, 3.6, 1.8 Hz, 1H), 2.13 (s, 3H), 2.08 (ddd, J = 14.4, 3.6, 2.4 Hz, 1H), 2.05 (s, 3H), 1.81 (ddd, J = 14.4, 9.6, 3.0 Hz, 1H), 1.78 (ddd, J = 14.4, 9.6, 3.0 Hz, 1H), 1.30 (d, J = 6.0 Hz, 3H), 1.24 (d, J = 6.0 Hz, 3H); 13C NMR (150 MHz, CDCl3) δ 171.5, 170.4, 137.8, 128.6 (2C), 128.2 (2C), 127.9, 98.8, 97.4, 79.8, 72.2, 71.3, 70.7, 70.4, 69.7, 69.5, 36.1, 35.8, 21.5, 21.4, 18.4, 18.1; HRCIMS Calcd for [C23H32O9Na+]: 475.1938, Found 475.1926.
Phenylmethyl 3-O-acetyl-2,6-dideoxy-4-O-[[3-O-acetyl-2,6-dideoxy-4-O-[(2R,6R)-5,6-dihydro-6-methyl-5-oxo-2H-pyran-2-yl]-β-D-ribo-hexopyranosyl]-β-D-ribo-hexopyranoside (33a)
A CH2Cl2 (0.3 mL) solution of Boc pyranone 8β (228 mg, 0.62 mmol) and alcohol 32a (141 mg, 0.31 mmol) was cooled to 0 °C. A CH2Cl2 (0.2 mL) solution of Pd2(DBA)3•CHCl3 (16 mg, 2.5 mol%) and PPh3 (16 mg, 10 mol%) was added to the reaction mixture at 0 °C. The reaction mixture was stirred at 0 °C for 2 hours and was quenched with 5 mL of saturated aqueous NaHCO3, extracted (3 × 5 mL) with Et2O, dried (Na2SO4), and concentrated under reduced pressure. The crude product was purified by silica gel flash chromatography eluting with 33% EtOAc/hexanes to give enone 33a (138 mg, 0.25 mmol, 79%) as a white solid: Rf (40% EtOAc/hexanes) = 0.18; mp: 95–96 °C; ; IR (thin film, cm−1)2980, 1740, 1702, 1454, 1372, 1243, 1158, 1055, 1006; 1H NMR (600 MHz, CDCl3) δ 7.33 (m, 5H), 6.87 (dd, J = 10.2, 1.2 Hz, 1H), 6.11 (dd, J = 10.2, 1.8 Hz, 1H), 5.40 (ddd, J = 3.6, 3.0, 3.0 Hz, 1H), 5.39 (m, 2H), 4.89 (d, J = 12.0 Hz, 1H), 4.79 (dd, J = 9.6, 1.8 Hz, 1H), 4.74 (dd, J = 9.6, 1.8 Hz, 1H), 4.55 (d, J = 12.0 Hz, 1H), 4.14 (q, J = 6.0 Hz, 1H), 3.88 (dq, J = 9.0, 6.0 Hz, 1H), 3.86 (dq, J = 9.0, 6.0 Hz, 1H), 3.45 (dd, J = 9.6, 3.0 Hz, 1H), 3.34 (dd, J = 9.6, 3.0 Hz, 1H), 2.15 (ddd, J = 14.4, 3.6, 2.4 Hz, 1H), 2.11 (ddd, J = 14.4, 3.6, 2.4 Hz, 1H), 2.09 (s, 3H), 2.06 (s, 3H), 1.81 (ddd, J = 14.4, 9.0, 3.0 Hz, 1H), 1.76 (ddd, J = 14.4, 9.0, 3.0 Hz, 1H), 1.38 (d, J = 6.6 Hz, 3H), 1.30 (d, J = 6.6 Hz, 3H), 1.26 (d, J = 6.0 Hz, 3H); 13C NMR (150 MHz, CDCl3) δ 196.1, 170.1, 170.07, 146.2, 137.6, 128.7, 128.3 (2C), 127.8 (2C), 127.7, 98.7, 97.1, 97.07, 79.6, 79.2, 75.1, 70.5, 69.64, 69.60, 69.2, 69.0, 35.9, 35.7, 21.24, 21.20, 18.2, 18.0, 16.3; HRCIMS Calcd for [C29H38O11Na+]: 585.2306, Found 585.2299.
Phenylmethyl 3-O-acetyl-2,6-dideoxy-4-O-[[3-O-acetyl-2,6-dideoxy-4-O-[(2R,6R)-5,6-dihydro-5-hydroxy-6-methyl-2H-pyran-2-yl]-β-D-ribo-hexopyranosyl]-β-D-ribo-hexopyranoside (34a)
A CH2Cl2 (0.3 mL) solution of enone 33a (138 mg, 0.245 mmol) and CeCl3 in MeOH solution (0.3 mL) was cooled to −78 °C. NaBH4 (10 mg, 0.25 mmol) was added and the reaction mixture was stirred at −78 °C for 3 hours. The reaction mixture was diluted with Et2O (5 mL) and was quenched with 5 mL of saturated aqueous NaHCO3, extracted (3 × 5 mL) with Et2O, dried (Na2SO4), and concentrated under reduced pressure. The crude product was purified using silica gel flash chromatography eluting with 50% EtOAc/hexanes to give allylic alcohols 34a (374 mg, 1.70 mmol, 85%) as a viscous oil (diastereometric ratio I:II = 1.4:1, inseparable in chromatography): Rf (60% EtOAc/hexanes) = 0.25; IR (thin film, cm−1) 3478, 2972, 2932, 2874, 1742, 1371, 1243, 1156, 1059, 1010; 1H NMR (600 MHz, CDCl3): isomer I: δ 7.33 (m, 5H), 6.15 (dd, J = 10.2, 6.0, Hz, 1H), 5.72 (d, J = 9.6 Hz, 1H), 5.54 (ddd, J = 3.0, 3.0, 3.0 Hz, 1H), 5.40 (ddd, J = 3.6, 3.0, 3.0 Hz, 1H), 5.12 (m, 1H), 4.89 (d, J = 12.0 Hz, 1H), 4.788 (dd, J = 9.6, 2.4 Hz, 1H), 4.75 (dd, J = 9.6, 1.8 Hz, 1H), 4.551 (d, J = 12.0 Hz, 1H), 3.88 (dq, J = 9.6, 6.0 Hz, 1H), 3.87 (m, 1H), 3.77 (dq, J = 9.6, 6.0 Hz, 1H), 3.68 (qd, J = 6.0, 1.8 Hz, 1H), 3.58 (dd, J = 11.4, 5.4 Hz, 1H), 3.39 (dd, J = 9.6, 3.0 Hz, 1H), 3.34 (dd, J = 9.6, 3.6 Hz, 1H), 2.16 (ddd, J = 14.4, 3.6, 2.4 Hz, 1H), 2.08 (s, 3H), 2.069 (s, 3H), 2.02 (ddd, J = 14.4, 3.0, 2.4 Hz, 1H), 1.79 (ddd, J = 14.4, 9.0, 3.0 Hz, 1H), 1.72 (ddd, J = 14.4, 9.0, 3.0 Hz, 1H), 1.70 (s, 1H), 1.31 (d, J = 6.6 Hz, 3H), 1.25 (d, J = 6.6 Hz, 3H), 1.21 (d, J = 6.6 Hz, 3H); isomer II: δ 7.33 (m, 5H), 5.94 (ddd, J = 10.8, 1.8, 1.8 Hz, 1H), 5.86 (d, J = 10.2 Hz, 1H), 5.75 (ddd, J = 10.2, 1.8, 1.2 Hz, 1H), 5.39 (ddd, J = 3.6, 3.0, 3.0Hz, 1H), 5.38 (ddd, J = 3.0, 3.0, 2.4 Hz, 1H), 5.13 (m, 1H), 4.88 (d, J = 12.0 Hz, 1H), 4.783 (dd, J = 9.6, 2.4 Hz, 1H), 4.71 (dd, J = 9.6, 1.8 Hz, 1H), 4.548 (d, J = 12.0 Hz, 1H), 3.87 (m, 1H), 3.83 (dq, J = 9.6, 6.0 Hz, 1H), 3.54 (dq, J = 6.6, 6.0 Hz, 1H), 3.35 (m, 2H), 2.32 (d, J = 11.4 Hz, 1H), 2.16 (ddd, J = 14.4, 3.6, 2.4 Hz, 1H), 2.09 (s, 3H), 2.056 (s, 3H), 2.02 (ddd, J = 14.4, 3.0, 2.4 Hz, 1H), 1.81 (ddd, J = 14.4, 9.0, 3.0 Hz, 1H), 1.76 (ddd, J = 14.4, 9.0, 3.0 Hz, 1H), 1.29 (d, J = 6.6 Hz, 3H), 1.27 (d, J = 6.6 Hz, 3H), 1.23 (d, J = 6.6 Hz, 3H); 13C NMR (150 MHz, CDCl3) isomer I: δ 170.1 (2C), 133.0 (2C), 131.7, 128.1 (2C ), 127.7 (3C), 98.75 (2C), 98.24, 98.22, 79.5, 77.8, 71.4, 69.9, 69.7, 69.2, 69.1, 64.4, 36.0, 35.7, 21.30, 21.28, 18.18, 18.03, 17.9; isomer II: δ 170.3 (2C), 137.6 (2C), 129.2, 128.3 (2C), 127.8 (3C), 98.72, 97.6, 97.2 (2C), 79.6, 78.0, 74.5, 70.5, 70.2, 69.6, 69.3, 68.4, 35.9, 35.6, 21.28, 21.26, 18.17, 18.16, 16.7; HRCIMS Calcd for [C29H40O11Na+]: 587.2463, Found 587.2453.
Phenylmethyl 3-O-acetyl-2,6-dideoxy-4-O-[[3-O-acetyl-2,6-dideoxy-4-O-[(2S,6R)-3,6-dihydro-6-methyl-2H-pyran-2-yl]-β-D-ribo-hexopyranosyl]-β-D-ribo-hexopyranoside (35a)
A flask was charged with dry N-methyl morpholine (NMM) 0.4 mL, triphenyl phosphine (191 mg, 0.73 mmol) and was cooled to −30 °C under Ar atmosphere. Diethylazodicarboxylate (0.1 mL, 0.66 mmol) was added and the reaction was stirred for 5 minutes, allylic alcohols 34a (125 mg, 0.22 mmol) was added in a 1M solution of NMM and the reaction mixture was stirred for 10 minutes, followed by addition of o-nitrobenzenesulfonyl hydrazide (NBSH) (135 mg, 0.66 mmol). The reaction was stirred at −30 °C for 4 hours and was monitored by TLC. Upon consumption of starting material, the reaction was warmed up to room temperature and stirred for another 2 hours. The reaction mixture was diluted with Et2O (10 mL) and was quenched with 5 mL of saturated aqueous NaHCO3, extracted (3 × 5 mL) with Et2O, dried (Na2SO4), and concentrated under reduced pressure. The crude product was purified using silica gel flash chromatography eluting with 20% EtOAc/hexanes to give product 35a (97 mg, 0.18 mmol, 81%) as a white solid: Rf (50% EtOAc/hexanes) = 0.44; mp: 101–101.5 °C; ; IR (thin film, cm−1) 2981, 2932, 2871, 1742, 1368, 1243, 1157, 1090, 1067, 1008, 704; 1H NMR (600 MHz, CDCl3) δ 7.33 (m, 5H), 5.62 (dddd, J = 10.2, 4.8, 2.4, 2.4 Hz, 1H), 5.53 (ddd, J = 9.6, 1.2, 1.2 Hz, 1H), 5.41 (ddd, J = 3.6, 3.0, 3.0 Hz, 1H), 5.37 (ddd, J = 3.6, 3.0, 3.0 Hz, 1H), 4.89 (d, J = 12.0 Hz, 1H), 4.78 (dd, J = 9.6, 1.8 Hz, 1H), 4.71 (dd, J = 9.6, 1.8 Hz, 1H), 4.65 (dd, J = 8.4, 3.6 Hz, 1H), 4.55 (d, J = 12.0 Hz, 1H), 4.25 (m, 1H), 3.87 (dq, J = 9.0, 6.0 Hz, 1H), 3.84 (dq, J = 9.6, 6.0 Hz, 1H), 3.34 (dd, J = 9.6, 3.6 Hz, 1H), 3.28 (dd, J = 9.6, 3.6Hz, 1H), 2.14 (m, 4H), 2.11 (s, 3H), 2.06 (s, 3H), 1.81 (dddd, J = 14.4, 9.0, 3.0 Hz, 1H), 1.74 (dddd, J = 14.4, 9.6, 3.0 Hz, 1H), 1.30 (d, J = 6.0 Hz, 3H), 1.23 (d, J = 6.6 Hz, 3H), 1.20 (d, J = 6.6 Hz, 3H); 13C NMR (150 MHz, CDCl3) δ 170.3, 170.1, 137.6, 131.1, 128.3 (2C), 127.8 (2C), 127.7, 122.2, 100.3, 98.8, 97.2, 79.5, 78.9, 70.9, 70.5, 70.1, 69.7, 69.3, 69.2, 35.9, 35.7, 30.9, 21.33, 21.26, 20.8, 18.2, 18.0; HRCIMS Calcd for [C29H40O10Na+]: 571.2514, Found 571.2525.
Phenylmethyl 3-O-acetyl-2,6-dideoxy-4-O-[[3-O-acetyl-4-O-[2,6-dideoxy-β-D-ribo-hexopyranosyl]-2,6-dideoxy-β-D-ribo-hexopyranosyl]-β-D-ribo-hexopyranoside (36a)
To a CH2Cl2 (0.6 mL) solution of olefin 35a (94 mg, 0.17 mmol) at 0 °C was added a solution of (50% w/v) of N-methyl morpholine N-oxide / water (80 µL). Crystalline OsO4 (0.4 mg, 1 mol %) was added and the reaction was stirred for 3 hours. The reaction was quenched by adding EtOAc and satd NaHCO3. The organic layer was separated and concentrated. It was purified by a silica gel column using 60% EtOAc/hexanes. Pure fractions were combined and concentrated to afford alcohol 36a (92 mg, 0.16 mmol, 92%) as a white solid: Rf (80% EtOAc/hexanes) = 0.25; mp: 167–167.5 °C; ; IR (thin film, cm−1) 3455, 2972, 2932, 2880, 1741, 1370, 1244, 1162, 1065, 1011, 869, 704; 1H NMR (600 MHz, CDCl3) δ 7.33 (m, 5H), 5.39 (ddd, J = 3.6, 3.0, 3.0 Hz, 1H), 5.34 (ddd, J = 3.0, 3.0, 3.0 Hz, 1H), 4.88 (d, J = 12.0 Hz, 1H), 4.81 (dd, J = 9.6, 1.8 Hz, 1H), 4.78 (dd, J = 9.6, 1.8 Hz, 1H), 4.70 (dd, J = 9.6, 1.8 Hz, 1H), 4.54 (d, J = 12.0 Hz, 1H), 4.05 (m, 1H), 3.86 (dq, J = 9.0, 6.0 Hz, 1H), 3.80 (dq, J = 9.0, 6.0 Hz, 1H), 3.65 (dq, J = 9.0, 6.0 Hz, 1H), 3.33 (dd, J = 9.6, 3.0 Hz, 1H), 3.27 (dd, J = 9.6, 3.6 Hz, 1H), 3.23 (ddd, J = 9.6, 6.0, 3.6 Hz, 1H), 2.47 (s, 1H), 2.25 (d, J = 6.6 Hz, 1H), 2.15 (ddd, J = 14.4, 3.6, 1.8 Hz, 1H), 2.09 (s, 3H), 2.08 (ddd, J = 14.4, 3.0, 1.8 Hz, 1H), 2.06 (ddd, J = 14.4, 3.0, 1.8 Hz, 1H), 2.05 (s, 3H), 1.80 (ddd, J = 14.4, 9.6, 2.4 Hz, 1H), 1.72 (ddd, J = 14.4, 9.6, 3.0 Hz, 1H), 1.66 (ddd, J = 13.8, 9.6, 3.0 Hz, 1H), 1.29 (d, J = 6.6 Hz, 3H), 1.22 (d, J = 6.0 Hz, 3H), 1.20 (d, J = 6.0 Hz, 3H); 13C NMR (150 MHz, CDCl3) δ 170.3, 170.2, 137.6, 128.3(2C), 127.8 (2C), 127.7, 98.8, 98.7, 97.2, 79.5, 79.2, 72.7, 70.5, 69.9, 69.7, 69.3 (2C), 69.1, 68.0, 37.6, 35.9, 35.6, 21.3, 21.2, 18.2, 17.9 (2C); HRCIMS Calcd for [C29H42O12Na+]: 605.2568, Found 605.2580.
Phenylmethyl 2,6-dideoxy-4-O-[[2,6-dideoxy-β-D-ribo-hexopyranosyl]-2,6-dideoxy-β-D-ribo-hexopyranosyl]-β-D-ribo-hexopyranoside (4)
To a MeOH/H2O (0.3 mL, 1:1, 1M) solution of alcohol 36a (14 mg, 24 µmol) at room temperature was added LiOH (2.5 mg, 60 µmol) and the reaction was stirred for 3 hours. The reaction was quenched by adding EtOAc and saturated aqueous NaHCO3. The organic layer was separated and concentrated. It was purified by a silica gel column using 75% EtOAc/hexanes. Pure fractions were combined and concentrated to afford 4 (11.5 mg, 23 µmol, 96%) as a white solid: Rf (80% EtOAc/hexanes) = 0.18; mp: 120–121 °C; ; IR (thin film, cm−1) 3424, 2927, 2886, 1455, 1369, 1318, 1163, 1130, 1068, 1012, 869, 733, 699; 1H NMR (600 MHz, CDCl3) δ 7.33 (m, 5H), 4.91 (dd, J = 9.6, 3.0 Hz, 1H), 4.90 (m, 2H), 4.88 (d, J = 12.0 Hz, 1H), 4.56 (d, J = 12.0 Hz, 1H), 4.25 (m, 2H), 4.12 (ddd, J = 3.6, 3.6, 2.4 Hz, 1H), 3.83 (dq, J = 9.6, 6.0 Hz, 1H), 3.81(dq, J = 9.0, 6.0 Hz, 1H), 3.76 (dq, J = 9.6, 6.0 Hz, 1H), 3.30 (m, 1H), 3.26 (dd, J = 9.6, 3.0 Hz, 1H), 3.20 (dd, J = 9.6, 3.0 Hz, 1H), 2.99 (s, 1H), 2.95 (s, 1H), 2.33 (s, 1H), 2.16 (ddd, J = 13.8, 3.6, 2.4 Hz, 1H), 2.14 (ddd, J = 14.4, 3.0, 2.4 Hz, 1H), 2.11 (ddd, J = 13.8, 3.0, 2.4 Hz, 1H), 2.03 (s, 1H), 1.78 (ddd, J = 14.4, 9.0, 3.0 Hz, 1H), 1.74 (m, 2H), 1.28 (d, J = 6.6 Hz, 6H), 1.22 (d, J = 6.0 Hz, 3H); 13C NMR (150 MHz, CDCl3) δ 137.8, 128.3 (2C), 127.9 (2C), 127.6, 98.30, 98.26, 97.1, 82.6, 82.2, 72.7, 70.6, 69.5, 68.32, 68.26, 68.1, 66.4, 66.2, 37.8, 36.7, 36.6, 18.2 (2C), 18.1; HRCIMS Calcd for [C25H38O10Na+]: 521.2357, Found 521.2360.
O-2,6-dideoxy-β-D-ribo-hexopyranosyl-(1→4)-2,6-dideoxy-D-ribo-hexose (37)
To an EtOH (2 mL) solution of 31a (15.6 mg, 42 µmol) under H2 atmosphere at room temperature was added Pd/C (8 mg) and the reaction was stirred for 6 hours. The reaction mixture was filtered through a pad of Celite using MeOH. The filtrate was concentrated and purified by a silica gel column using 1% MeOH/EtOAc. Pure fractions were combined and concentrated to afford digoxose bisdigitoxide 37 (11 mg, 39.5 µmol, 94%) as a white solid: Rf (10% MeOH/EtOAc) = 0.18; mp: 132–135 °C; ; IR (thin film, cm−1) 3426, 2930, 1376, 1319, 1165, 1132, 1068, 1014, 992, 869, 729; 1H NMR (600 MHz, CD3OD/CDCl3) β: δ 5.03 (dd, J = 9.6, 1.8 Hz, 1H), 4.84 (dd, J = 9.6, 2.4 Hz, 1H), 4.17 (ddd, J = 3.6, 3.0, 2.4 Hz, 1H), 3.98 (m, 1H), 3.78 (dq, J = 9.0, 6.0 Hz, 1H), 3.69 (dq, J = 9.0, 6.0 Hz, 1H), 3.15 (dd, J = 9.0, 3.0 Hz, 1H), 3.13 (dd, J = 9.0, 3.0 Hz, 1H), 2.04 (m, 2H), 1.61 (ddd, J = 13.8, 9.6, 3.0 Hz, 1H), 1.67 (m, 1H), 1.20 (d, J = 6.0 Hz, 3H), 1.17 (d, J = 6.6 Hz, 3H); α: 5.01 (d, J = 3.0 Hz, 1H), 4.87 (dd, J = 9.6, 1.8 Hz, 1H), 4.26 (ddd, J = 3.6, 3.0, 3.0 Hz, 1H), 4.07 (dq, J = 9.6, 6.0 Hz, 1H), 3.97 (m, 1H), 3.68 (dq, J = 9.6, 6.0 Hz, 1H), 3.18 (dd, J = 9.0, 3.0 Hz, 1H), 3.14 (dd, J = 9.0, 3.0 Hz, 1H), 2.06 (m, 2H), 1.80 (ddd, J = 14.4, 3.0, 3.0 Hz, 1H), 1.68 (m, 1H), 1.19 (d, J = 6.0 Hz, 3H), 1.16 (d, J = 6.0 Hz, 3H); 13C NMR (150 MHz, CD3OD/CDCl3) β: δ 98.56, 91.4, 82.4, 72.46, 69.59, 68.0, 67.62, 66.3, 37.76, 37.69, 17.92, 17.86; α: δ 98.58, 91.5, 82.2, 72.44, 69.60, 67.6, 66.9, 61.6, 37.74, 34.3, 17.93, 17.7; HRCIMS Calcd for [C12H22O7Na+]: 301.1263, Found 301.1255.
O-2,6-dideoxy-β-D-ribo-hexopyranosyl-(1→4)-O-2,6-dideoxy-β-D-ribo-hexopyranosyl-(1→4)-2,6-dideoxy-D-ribo-hexose (3)
To an EtOH (0.3 mL) solution of 4 (11 mg, 22 µmol) under H2 atmosphere at room temperature was added Pd/C (6 mg) and the reaction was stirred for 6 hours. The reaction mixture was filtered through a pad of Celite using MeOH. The eluent was concentrated and purified by a silica gel column using 2% MeOH/EtOAc. Pure fractions were combined and concentrated to afford digoxose 3 (8 mg, 20 µmol, 92%) as a white solid: Rf (10% MeOH/EtOAc) = 0.29; mp: 210–212 °C; ; IR (thin film, cm−1) 3425, 2929, 1376, 1319, 1231, 1165, 1133, 1067, 1013, 992, 869, 729; 1H NMR (600 MHz, CD3OD/CDCl3) β: δ 5.04 (dd, J = 9.6, 2.4 Hz, 1H), 4.845 (dd, J = 9.6, 1.8 Hz, 1H), 4.841 (dd, J = 9.6, 1.8 Hz, 1H), 4.18 (ddd, J = 3.6, 3.0, 3.0 Hz, 1H), 4.17 (ddd, J = 3.6, 3.0, 3.0 Hz, 1H), 3.98 (ddd, J = 3.6, 3.0, 3.0 Hz, 1H), 3.78 (m, 2H), 3.69 (dq, J = 9.0, 6.0 Hz, 1H), 3.16 (dd, J = 9.6, 3.0 Hz, 1H), 3.147 (dd, J = 9.6, 3.0 Hz, 1H), 3.144 (dd, J = 9.6, 3.0 Hz, 1H), 2.16 (m, 3H), 1.65 (m, 3H), 1.213 (d, J = 6.6 Hz, 3H), 1.175 (d, J = 6.6 Hz, 3H), 1.165 (d, J = 6.6 Hz, 3H); α: 5.01 (d, J = 3.0 Hz, 1H), 4.87 (dd, J = 10.2, 2.4 Hz, 1H), 4.848 (dd, J = 9.6, 2.4 Hz, 1H), 4.26 (ddd, J = 3.6, 3.0, 3.0 Hz, 1H), 4.06 (m, 2H), 3.77 (m, 2H), 3.70 (dq, J = 9.0, 6.0 Hz, 1H), 3.19 (dd, J = 9.6, 3.0 Hz, 1H), 3.137 (dd, J = 9.6, 3.0 Hz, 1H), 3.140 (m, 1H), 2.06 (m, 3H), 1.64 (m, 3H), 1.216 (d, J = 6.6 Hz, 3H), 1.19 (d, J = 6.6 Hz, 3H), 1.160 (d, J = 6.6 Hz, 3H); 13C NMR (150 MHz, CD3OD/CDCl3) β: δ 98.6, 98.5, 91.4, 82.4, 82.19, 82.17, 72.5, 69.64, 68.2, 67.6, 66.9, 66.15, 37.75 (2C), 36.61, 17.95 (2C), 17.75; α: δ 98.6, 98.5, 91.5, 82.21, 82.19, 82.17, 69.63, 68.0, 66.4, 66.18, 66.16, 61.50, 37.71, 36.62, 34.3, 17.95, 17.93, 17.90; HRCIMS Calcd for [C18H32O10Na+]: 431.1889, Found 431.1888.
Scheme 9.

Synthesis of Digoxose Bisdigitoxoside 37 and Digoxose 3
Acknowledgment
We are grateful to NIH (GM63150) and NSF (CHE-0415469) for the support of our research program and NSF-EPSCoR (0314742) for a 600 MHz NMR at WVU.
Footnotes
Supporting Information Available: Complete experimental procedures and spectral data for all new compounds can be found in the Supporting Information. This material is available free of charge via the Internet at http://pubs.acs.org.
Reference Section
- 1.This paper is dedicated to a dear friend and former colleague, Professor Wayland Noland, on the occasion of his 80th birthday.
- 2.Greeff K. Handbook of Experimental Pharmacology. Vol. 56. Berlin; New York: Springer-Verlag; 1981. Cardiac Glycosides, Part 1: Experimental Pharmacology. [Google Scholar]
- 3.(a) Haux J. Med. Hyp. 1999;53:543–548. doi: 10.1054/mehy.1999.0985. [DOI] [PubMed] [Google Scholar]; (b) Kometiani P, Liu L, Askari A. Mol. Pharmacol. 2005;67:929–936. doi: 10.1124/mol.104.007302. [DOI] [PubMed] [Google Scholar]; (c) Lopez-Lazaro M, Pastor N, Azrak SS, Ayuso MJ, Austin CA, Cortes F. J. Nat. Prod. 2005;68:1642–1645. doi: 10.1021/np050226l. [DOI] [PubMed] [Google Scholar]; (d) Winnicka K, Bielawski K, Bielawska A. Acta Poloniae Pharmaceutica. 2006;63:109–115. [PubMed] [Google Scholar]
- 4.For the synthesis of digitoxigenin see: Danieli N, Mazur Y, Sondheimer F. J. Am. Chem. Soc. 1962;84:875–876. Donovan Stephen F, Avery Mitchell A, McMurry John E. Tetrahedron Lett. 1979;35:3287–3290. Tsai TYR, Minta A, Wiesner K. Heterocycles. 1979;12:1397–1402. Marini-Bettolo R, Flecker P, Tsai TYR, Wiesner K. Can. J. Chem. 1981;59:1403–1410. Marek M Kabat. J. Org. Chem. 1995;60:1823–1827. Stork G, West F, Lee HY, Isaacs RCA, Manabe S. J. Am. Chem. Soc. 1996;118:10660–10661. Almirante N, Cerri A. J. Org. Chem. 1997;62:3402–3404. doi: 10.1021/jo962237c. Bocknack BM, Wang L-C, Krische MJ. Proc. Natl. Acad. Sci. 2004;101:5421–5424. doi: 10.1073/pnas.0307120101.
- 5.The attempts at the selective hydrolysis of digitoxin to form digoxose (3) has been futile, only the monosaccharide digitoxose was isolated. Surprisingly, 3 can be isolated from the dried twigs of Orthenthera viminea, see: Tiwari KN, Khare NK, Khare A, Khare MP. Carbohydr. Res. 1984;129:179–187.
- 6.(a) Toshima K, Tatsuta K. Chem. Rev. 1993;93:1503–1531. [Google Scholar]; (b) Danishefsky SJ, Bilodeau MT. Angew. Chem., Int. Ed. Engl. 1996;35:1380–1419. [Google Scholar]; (c) Kirschning A, Bechthold AF-W, Rohr J. Top. Curr. Chem. 1997;188:1–84. [Google Scholar]; (d) Marzabadi CH, Frank RW. Tetrahedron. 2000;56:8385–8417. [Google Scholar]; (e) Kirschning A, Jesberger M, Schoning KU. Synthesis. 2001:507–540. [Google Scholar]; (f) Nicolaou KC, Mitchell HJ. Angew. Chem. Int. Ed. 2001;40:1576–1624. [PubMed] [Google Scholar]; (g) Sears P, Wong C-H. Science. 2001;291:2344–2350. doi: 10.1126/science.1058899. [DOI] [PubMed] [Google Scholar]; (h) He X, Liu H-W. Annu. Rev. Biochem. 2002;71:701–754. doi: 10.1146/annurev.biochem.71.110601.135339. [DOI] [PubMed] [Google Scholar]
- 7.Langenhan JM, Peters NR, Guzei IA, Hoffmann FM, Thorson JS. Proc. Natl. Acad. Sci. U.S.A. 2005;102:12305–12310. doi: 10.1073/pnas.0503270102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) Roush WR, Bennett CE. J. Am. Chem. Soc. 1999;121:3541–3542. [Google Scholar]; (b) Sherry BD, Loy RN, Toste FD. J. Am. Chem. Soc. 2004;126:4510–4511. doi: 10.1021/ja031895t. [DOI] [PubMed] [Google Scholar]
- 9.Toshima K, Tatsuta K. Chem. Rev. 1993;93:1503–1531. [Google Scholar]
- 10.(a) Binkley RW, Koholic DJ. J. Carbohydr. Chem. 1988;7:487–499. [Google Scholar]; (b) Hashimoto S, Sano A, Sakamoto H, Nakajima M, Yanagiya Y, Ikegami S. Synlett. 1995:1271–1273. [Google Scholar]; (c) Guo Y, Sulikowski GA. J. Am. Chem. Soc. 1998;120:1392–1397. [Google Scholar]; (d) Pongdee R, Wu B, Sulikowski GA. Org. Lett. 2001;3:3523–3525. doi: 10.1021/ol016593f. [DOI] [PubMed] [Google Scholar]
- 11.(a) Paulsen H, Kutschker W, Lockhoff O. Chem. Ber. 1981;114:3233–3241. [Google Scholar]; (b) Paulsen H, Lockhoff O. Chem. Ber. 1981;114:3102–3214. [Google Scholar]
- 12.(a) van Boeckel CAA, Beetz T, van Aelst SF. Tetrahedron. 1984;40:4097–4107. [Google Scholar]; (b) van Boeckel CAA, Beetz T. Recl. Trav. Chim. Pays-Bas. 1985;104:171–173. [Google Scholar]
- 13.(a) Thiem J, Karl H. Chem. Ber. 1980;113:3039–3048. [Google Scholar]; (b) Thiem J, Gerken M. J. Carbohydr. Chem. 19823;1:229–249. [Google Scholar]; (c) Thiem J, Gerken M, Bock K. Liebigs Ann. Chem. 1983:462–470. [Google Scholar]; (d) Bock K, Lundt I, Pedersen C. Carbohydr. Res. 1984;130:125–134. [Google Scholar]; (e) Lundt I, Thiem J, Prahst A. J. Org. Chem. 1984;49:3063–3069. [Google Scholar]; (f) Thiem J, Gerken M. J. Org. Chem. 1985;50:954–958. [Google Scholar]
- 14.Roush WR, Lin X-F. J. Am. Chem. Soc. 1995;117:2236–2250. [Google Scholar]
- 15.(a) Ramesh S, Kaila N, Grewal G, Franck RW. J. Org. Chem. 1990;55:5–7. [Google Scholar]; (b) Nicolaou KC, Ladduwahetty T, Randall JL, Chucholowski A. J. Am Chem. Soc. 1986;108:2466–2467. doi: 10.1021/ja00269a066. [DOI] [PubMed] [Google Scholar]; (c) Ito Y, Ogawa T. Tetrahedron Lett. 1987;28:2723–2726. [Google Scholar]; (d) Preuss R, Schmidt RR. Synthesis. 1988:694–697. [Google Scholar]; (e) Hashimoto S, Yanagiya Y, Honda T, Ikegami S. Chem. Lett. 1992:1511–1514. [Google Scholar]
- 16.Perez M, Beau J-M. Tetrahedron Lett. 1989;30:75–78. [Google Scholar]
- 17.(a) Trumtel M, Veyribres A, Sinay P. Tetrahedron Lett. 1989;30:2529–2532. [Google Scholar]; (b) Trumtel M, Tavecchia P, Veyribres A, Sinay P. Carbohydr. Res. 1989;191:29–52. doi: 10.1016/0008-6215(90)84084-8. [DOI] [PubMed] [Google Scholar]
- 18.Tavecchia P, Trumtel M, VeyriBres A, Sinay P. Tetrahedron Lett. 1989;30:2533–2536. [Google Scholar]
- 19.Gervay J, Danishefsky S. J. Org. Chem. 1991;56:5448–5451. [Google Scholar]
- 20. Wiesner K, Tsai TYR, Jin H. Helv. Chim. Acta. 1985;68:300–314. Wiesner K, Tsai TYR. Pure Appl. Chem. 1986;58:799–810. and McDonald FE, Reddy KS, Diaz Y. J. Am. Chem. Soc. 2000;122:4304–4309. McDonald FE, Reddy KS. Angew. Chem. Int. Ed. 2001;40:3653–3655. doi: 10.1002/1521-3773(20011001)40:19<3653::aid-anie3653>3.0.co;2-w.
- 21.In contrast, McDonald and coworkers are able to use his methodology to prepare an all-alpha analogue of digitoxin with high stereocontrol, see: McDonald FE, Wu M. Org. Lett. 2002;4:3979–3981. doi: 10.1021/ol026886o.
- 22.For the communication of our initial approach to 1 see: Zhou M, O’Doherty GA. Org. Lett. 2006;8:4339–4342. doi: 10.1021/ol061683b.
- 23. Babu RS, O’Doherty GA. J. Am. Chem. Soc. 2003;125:12406–12407. doi: 10.1021/ja037097k. Comely AC, Eelkema R, Minnaard AJ, Feringa BL. J. Am. Chem. Soc. 2003;125:8714–8715. doi: 10.1021/ja0347538. Kim H, Men H, Lee C. J. Am. Chem. Soc. 2004;126:1336–1337. doi: 10.1021/ja039746y. For its application in the de novo synthesis of α-linked 1,4- and 1,6-oligosaccharides, see: Babu RS, Zhou M, O’Doherty GA. J. Am. Chem. Soc. 2004;126:3428–3429. doi: 10.1021/ja039400n.
- 24.In both the Wiesner and McDonald syntheses of digitoxin at least one of the three glycosidic bonds was assembled with poor stereoselectivity, see: ref. 20.
- 25.Fujii A, Hashiguchi S, Uematsu N, Ikariya T, Noyori R. J. Am. Chem. Soc. 1996;118:2521–2522. [Google Scholar]
- 26.(a) Li M, Scott JG, O'Doherty GA. Tetrahedron Lett. 2004;45:1005–1009. [Google Scholar]; (b) Li M, O'Doherty GA. Tetrahedron Lett. 2004;45:6407–6411. [Google Scholar]
- 27.For the preparation of Boc-protected pyranones, see: Ref. 26 Babu RS, O'Doherty GA. J. Carb. Chem. 2005;24:169–177. Guo H, O’Doherty GA. Org. Lett. 2005;7:3921–3924. doi: 10.1021/ol051383e.
- 28.For reduction of β-pyanones, the CeCl3 is necessary to avoid 1,4-reduction products. For Luche reduction, see: Luche JL. J. Am. Chem. Soc. 1978;100:2226–2227. Haukaas MH, O’Doherty GA. Org. Lett. 2001;3:401–404. doi: 10.1021/ol006907j.
- 29.(a) Myers AG, Zheng B. J. Am. Chem. Soc. 1996;118:4492–4493. [Google Scholar]; (b) Myers AG, Zheng B. Tetrahedron Lett. 1996;37:4841–4844. [Google Scholar]; (c) Myers AG, Zheng B, Movassaghi M. J. Org. Chem. 1997;62:7507. doi: 10.1021/jo9710137. [DOI] [PubMed] [Google Scholar]; (d) Haukaas MH, O’Doherty GA. Org. Lett. 2002;4:1771–1774. doi: 10.1021/ol025844x. [DOI] [PubMed] [Google Scholar]
- 30.VanRheenen V, Kelly RC, Cha DY. Tetrahedron Lett. 1976;17:1973–1976. [Google Scholar]
- 31.No minor diastereomer was detected by 1H NMR.
- 32.Mono- and bis-digtoxosides has been prepared by gradual degradation of digitoxin, see: Satoh D, Aoyama K. Chem.Pharm. Bull. 1970;18:94–98. Templeton FJ, Setiloane P, Kumar VPS, Yan Y, Zeglam HT, LaBella FS. J. Med. Chem. 1991;34:2778–2782. doi: 10.1021/jm00113a015. Our synthetic material 25b and 31b have identical physical and spectral data with degraded products in the terms of 1H NMR, 13C NMR, optical rotation and melting point.
- 33.The nomenclature of compounds 25b and 31b was used according to ref. 32.
- 34.(a) King JF, Allbutt AD. Can. J. Chem. 1970;48:1754–1769. [Google Scholar]; (b) Lowary TL, Hindsgaul O. Carbohydr. Res. 1994;251:33–67. doi: 10.1016/0008-6215(94)84275-2. [DOI] [PubMed] [Google Scholar]
- 35.For glycosylation at the C-4 position, we found that the best yields were obtained when a 2:1 ratio of glycosyl donor to acceptor was used.
- 36.Digitoxin purchased from Acros was used to do the comparison.
- 37.Stereochemical assignments were made by coupling constant analysis and verified by the synthesis of known natural products 1 and 3.
- 38.We found our synthetic digoxose had higher optical rotation and melting points , mp: 210–212 °C) than the literature values; mp: 171–174 °C), which is probably j ust an indication of purity.
- 39.The synthesis By McDonald and coworkers prepares digitoxin in 18-steps and experiences poor stereocontrol in the installation of the final glycosidic bond, see: ref. 20d.
- 40.Presented in this experimental section are the procedures and spectral data for all new compounds. Complete experimental procedures and spectral data for all compounds are presented in Supporting Information.