

Abstract
Hamartomatous polyposis syndromes are a diverse group of inherited conditions grouped together because they exhibit hamartomatous rather than epithelial polyp histology. Each syndrome exhibits characteristic polyp histology, gastrointestinal polyp distribution, gastrointestinal cancer risks, extra-intestinal benign findings and often extra-intestinal cancer risks. Identifying individuals at risk for these syndromes and accurately defining the precise diagnosis is necessary for planning surveillance and management in order to prevent the benign and malignant complications. Characteristic syndrome features including gastrointestinal findings, pathology, genetics, and management options for the three most common hamartomatous polyposis syndromes, Peutz-Jeghers syndrome, PTEN hamartoma tumor syndrome, and juvenile polyposis will be presented in this review.
Keywords: hamartomatous polyps, Peutz-Jeghers syndrome, Cowden syndrome, juvenile polyposis, PTEN hamartoma tumor syndrome, STK11, SMAD4, BMPR1A, genetic counseling, genetic testing
Introduction
Hamartomatous polyps account for a small percentage of all colon polyps. These polyps arise from an over proliferation of subepithelial cells native to the tissue of origin, and can contain cellular components from any of the three germinal layers forming the intestines. The progression of hamartomas to cancer progression has not yet been fully delineated. It is clear however, that individuals with inherited hamartomatous polyposis syndromes have a greatly increased risk for malignancy. Kinzler and Vogelstein hypothesize that the risk for malignancy is due to defective “landscape” in which the environment created by the abnormal stromal tissue promotes the malignant progression of the adjacent epithelium [1].
Inherited syndromes predisposing to hamartomatous polyps occur less commonly than hereditary conditions leading to adenomatous polyposis, and account for <1% of all colorectal cancers [2]. While accounting for only a small portion of cancer, appropriate identification of families affected with these syndromes is crucial for planning screening and management. Hereditary hamartomatous polyposis syndromes are associated with high rates of malignancy and intensive cancer screening is necessary to maximize the opportunity for early detection. With this review we will present the clinical features, pathology, genetics, and management recommendations for Peutz-Jeghers syndrome (PJS), PTEN hamartoma tumor syndrome (PTHS), and juvenile polyposis syndrome (JPS).
Peutz-Jeghers Syndrome
Clinical Features
Peutz-Jeghers syndrome (PJS) occurs with an estimated frequency of 1/8,300 to 1/280,000 individuals [3], and is characterized by the development of mucocutaneous pigmentation, hamartomatous polyps throughout the digestive system, and an increased risk for malignancy of several types.
The characteristic pigmented macules present most commonly on the lips, buccal mucosa, and periorbital area, but they can also occur on the fingers, soles, palms, perianal area, labia, and intestinal mucosa [2-4]. These pigmented lesions are benign and not thought to have malignant potential. The pigmentation presents in infancy, but tends to fade during adolescence. However, pigmented areas inside the mouth or on the gums tend to persist into adulthood [4]. While greater than 90% of affected individuals will have characteristic pigmentation, the extent of the phenotype varies considerably from diffuse, dark freckling over the face to light pigmentation in characteristic areas (Figure 1) [5]. Reviewing childhood pictures and having patients remove cosmetics may be necessary to accurately assess the presence or absence of mucocutaneous pigmentation.
Figure 1.
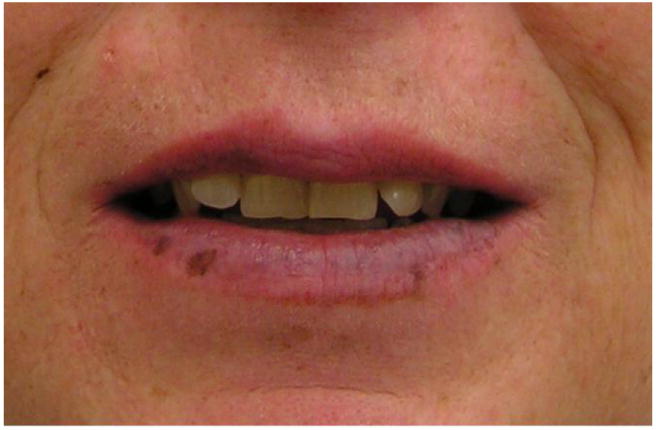
Peutz-Jeghers associated pigmented macules
The median age for onset of gastrointestinal (GI) symptoms is 13 years of age, and approximately 50% will have experienced symptoms by age 20 [4, 6]. Common presenting symptoms include small bowel intussusception and obstruction, rectal bleeding, and anemia. A small fraction of affected individuals will not develop symptoms until later in life or may only have vague symptoms such as abdominal pain [7, 8]. In contrast to the other hamartomatous syndromes in which polyps occur most commonly in the colon, PJS related polyps occur most frequently in the small intestine. Over 90% of affected individuals will develop polyps in the small intestine during their lifetime. The incidence within the small intestine is greatest in the jejunum and progressively decreases in the ileum and duodenum. Up to 30% will develop polyps in the colon, and approximately 25% will develop gastric polyps [4]. Hamartomatous polyps may also develop outside of the digestive tract in the uterus, nasal cavity, bladder, and lungs [2].
A clinical diagnosis of PJS can be made when an individual has two or more of the following features [7]:
2 or more PJS polyps of the small intestine
Characteristic pigmentation of the mouth, lips, nose, eyes, genitalia, or fingers
Family history of PJS
Individuals with PJS have an increased risk for numerous malignancies. The cumulative risk for developing any type of cancer has been estimated to be between 81%-93% [9-11]. Specific associated cancer risks are summarized in Table 1. The greatest specific cancer risk is female breast cancer (45%-54%) [9, 10]. Approximately 57%-68% of affected individuals will develop some type of GI cancer [10, 11]. While polyps occur most commonly in the small intestine, the colon is the most frequent site for GI malignancy. Sex cord tumors with annular tubules (SCTAT) of the ovary are a classic feature of PJS. Up to 36% of women who develop SCTAT have PJS [12]. Adenoma malignum, also called minimal deviation adenocarcinoma, is also characteristic PJS-related gynecological cancer [13]. Sertoli cell testicular tumors occur in males. These tumors pathologically resemble SCTAT tumors and often present prior to adolescence together with gynecomastia and accelerated physical growth [4].
Table 1.
Cancer risks associated with hamartomatous polyposis syndromes and management recommendations.
| Syndrome | Cancer Risks | Cancer Specific Management Recommendations | |
|---|---|---|---|
| Peutz-Jeghers Syndrome[4, 9, 10] | Breast | 45%-50% |
|
| Colon | 39% |
|
|
| Pancreas | 11%-36% |
|
|
| Stomach | 29% |
|
|
| Ovary | 18%-21% |
|
|
| Lung | 15%-17% |
|
|
| Small intestine | 13% |
|
|
| Cervix | 10% |
|
|
| Uterus | 9% |
|
|
| Testes | 9% |
|
|
| Juvenile Polyposis | Colon | 40%-50% |
|
| Stomach | 21% (if multiple polyps) |
|
|
| Small intestine | Rare, undefined |
|
|
| Pancreas | Rare, undefined |
|
|
| PTEN Hamartoma Tumor Syndrome | Breast | 25%-50% |
|
| Thyroid (non-medullary) | 10% |
|
|
| Endometrium | 5%-10% |
|
|
| Skin | Undefined |
|
|
| Colon + Kidney | Undefined |
|
|
magnetic resonance cholangiopancreatography
Pathology
PJS polyps of the small intestine are characterized by a distinctive pattern of arborization of the muscularis mucosa. This feature is less prominent or absent from polyps occurring in other areas of the digestive system. During polyp development, some epithelial tissue may be involuted into the polyps, and these ectopic islands of epithelial tissue may suggest pseudocarcinoma invasion [4, 14].
Genetics
PJS is caused by mutations in the STK11 (also called LKB1) gene located at 19p13.3. STK11 encodes a serine/threonine kinase which functions as a tumor suppressor. Mutations in this gene are detected in 50%-90% of individuals with PJS [6, 15, 16]. Variability in detection rates is likely due to differences in selection criteria and testing methodologies. The majority of mutations are truncating or missense mutations which eliminate the kinase function of the protein. However, up to 30% of mutations may be large deletions which would not be detected by sequencing alone [16]. Therefore the optimal approach for genetic testing of a proband would include both sequencing of the coding region of the gene and analysis for large deletions.
The likelihood of detecting a mutation is higher in individuals who meet the clinical diagnostic criteria compared to those who only have an isolated feature of PJS. Aretz et al. identified mutations in 53/56 (95%) of individuals meeting clinical criteria for PJS compared to 0/12 (0%) of those with only an isolated haratoma or pigmentation of the oral mucosa [16]. Other studies have likewise failed to find mutations in individuals exhibiting solitary features of PJS [6, 17].
The addition of large deletion analysis to STK11 testing has greatly increased the mutation detection rate. However, there is still a very small portion of individuals and families meeting the clinical diagnostic criteria in which a deleterious mutation cannot be identified. It is possible that current methods are still missing mutations, and there are reports of families with a clinical diagnosis of PJS who do not link to 19p13.3 suggesting that there may be another genetic locus causing PJS in rare families [18]. At this time STK11 is the only genetic cause of PJS for which clinical testing is available.
In 2004 Amos et al. identified a significantly later age of onset of polyps and symptoms in individuals with missense mutations when compared to those with truncating mutations or no identifiable mutation (23, 13, and 15 years respectively) [6]. In the largest published series of individuals with PJS, Hearle et al. did not detect a significant difference in cancer risk based on mutation status or type [10]. Hearle et al. also recently looked at the relationship between mutation type and risk for intussusception, and found a trend towards earlier onset with truncating mutations, but the difference was not statistically significant [19]. At this time further studies are needed to determine if there is a relationship between genotype and phenotype. No distinctions in management should be made on the basis of mutation type or gene location. Mutations in the STK11 gene are inherited in an autosomal dominant manner. However, 25% of cases appear to be the first person affected in their family. Due to the variability of presentation, it is important to carefully evaluate first degree relatives for features of PJS before assuming an individual is affected due to a de novo mutation.
Management
Screening recommendation for PJS are outlined in Table 1. Managing polyps in the distal small intestine is particularly challenging. Capsule endoscopy (CE) has been found to detect more polyps than small bowel radiographic studies [20, 21]. However, other studies have shown that CE still misses a significant portion of polyps. Soares et al. evaluated five patients having enteroscopy due to the finding of one or more large polyps with CE. During enteroscopy all of these patients had at least 20% or more additional polyps identified than had been seen with the CE screening alone [22]. Imaging with abdominal CT scan is another option for screening. A retrospective review of 165 Australian cases of small intestine tumors found that abdominal CT scans with oral contrast demonstrated greater sensitivity than small bowel radiography [23]. At this time there has not been a systematic trial comparing all these different methods. However, our center has been successfully using abdominal CT scan with oral contrast to screen PJS patients. Double-balloon enteroscopy unlikely has a role for routine screening, but may be used to remove polyps and reduce surgical procedures. Colonoscopy and upper endoscopy are recommended for screening the colon, stomach, and duodenum.
Approaches for pancreatic cancer screening include imaging with CT scan or magnetic resonance cholangiopancreatography (MRCP) and endoscopic ultrasound (EUS). EUS may be the most sensitive, but it is highly operator dependent [24]. Canto et al. have found intraductal papillary mucinous neoplasms (IMPNs) are a common finding in individuals at risk for familial pancreatic cancer or with a personal history of PJS. MRCP provides good visualization of the pancreatic ducts and cystic lesions and may be preferable to CT scan for screening these patients [25].
PTEN Hamartoma Tumor Syndrome
Germline mutations in the PTEN gene are responsible for a group of phenotypically diverse conditions, which have collectively been called the PTEN hamartoma tumor syndrome (PHTS). These rare autosomal dominant conditions include both Cowden syndrome (CS) and Bannayan-Riley Ruvalcaba syndrome (BRRS), which will be the focus here, in addition to possibly other seemingly unrelated disorders [26]. As the name implies, hamartomatous tumors, which can affect any organ, are the hallmark feature of the PHTS. CS is associated with a predisposition to benign lesions of the skin, mucousal membranes, GI tract and other organs, in addition to an increased risk of breast, thyroid and endometrial cancer. BRRS is a congenital disorder characterized by macrocephaly, lipomatosis, GI polyposis, and pigmented macules of the glans penis [27].
Cowden Syndrome
Cowden syndrome is the quintessential condition of the PHTS. It was first described by Lloyd and Dennis in 1963 and was named after their patient Rachel Cowden who presented with assorted benign findings and later died of breast cancer at age 30 years [28]. Although the prevalence of CS is currently unknown, it was once estimated to be as low as one in 1,000,000 [29]. The identification of the PTEN tumor suppressor gene in 1997 resulted in improved recognition of CS, which was made evident by updated prevalence estimates of 1 in 250,000 to 200,000 [29-31]. CS is likely still under diagnosed due to difficulties in recognition [32].
Diagnostic Criteria
Consensus criteria have been developed to aid in the operational diagnosis of CS, which often presents with assorted manifestations [33, 34]. The National Comprehensive Cancer Network (NCCN) Genetic/Familial High-Risk Assessment Panel (www.nccn.org) reviews the criteria on an annual basis and revisions have recently been published (Table 2). A clinical diagnosis occurs when individuals have any of the pathognomonic features or when specific combinations of the major and minor criteria are met (see Table 2).
Table 2.
Diagnostic Criteria for Cowden Syndrome*
Pathognomonic Criteria
|
Minor Criteria
|
Major Criterai
|
|
Operational Diagnosis in an Individual
| |
Operational Diagnosis for Individuals with CS in the Family
| |
Adapted from www.nccn.org
The most important indicators of CS are mucocutaneous features, as they are found in nearly all individuals and are invariably present by the third decade of life [33, 35]. Pathognomonic mucocutaneous features include trichilemmomas (flesh colored facial papules involving a hair follicle) which tend to occur around the mouth, nose and ears, papillomatous papules (histologically these are benign fibromas) which can result in a cobblestone appearance in the mucosal cavity, and acral keratoses (flesh colored or slightly pigmented smooth or verrucoid papules of the hands and feet) [33, 36]. Adult onset Lhermitte-Duclos disease, also referred to as dysplastic gangliocytoma of the cerebellum, was recently added to the pathognomonic criteria for CS (www.nccn.org) [33]. Lhermitte-Duclos disease presenting in childhood is less often associated with CS compared to those presenting in adults, although young onset cases have been reported [37].
Additional Benign Findings
Other cutaneous features commonly seen in CS include lipomas, neuromas, hemangiomas and scrotal tongue [36]. Macrocephaly is present in approximately 40% of affected individuals [26]. Fibrocystic breast disease and breast adenomas (up to 75%), thyroid lesions including mulitnodular goiter and thyroid adenomas (up to 75%), and uterine leiomyomas (~50%) are additional benign lesions associated with CS, however, these features are also common in the general population and are therefore included in the minor criteria.
Gastrointestinal polyps
Hamartomatous polyps occur throughout the GI tract and were originally estimated to occur in 60% of affected individuals [35]. This is likely an underestimate, since GI polyps in CS are often asymptomatic and as few as 25% of reported cases have had their GI tract examined [38]. In a review of CS cases in Japan, the most common sites for GI polyps included stomach (75%), colon (66%), esophagus (66%) and duodenum (37%) [38]. Juvenile polyps are the most frequent lesion in the colon, stomach and small bowel, although multiple other types do occur, including lipomas, inflammatory polyps, ganglioneuromas, lymphoid hyperplasia, and adenomas [38, 39]. The polyps in the esophagus are glycogenic acanthosis. These typically appear as white flat elevations, are numerous, and are spread throughout the esophagus (Figure 2) [39]. Although colon cancer has been reported in CS, it is still not clear whether this is part of the syndrome.
Figure 2.
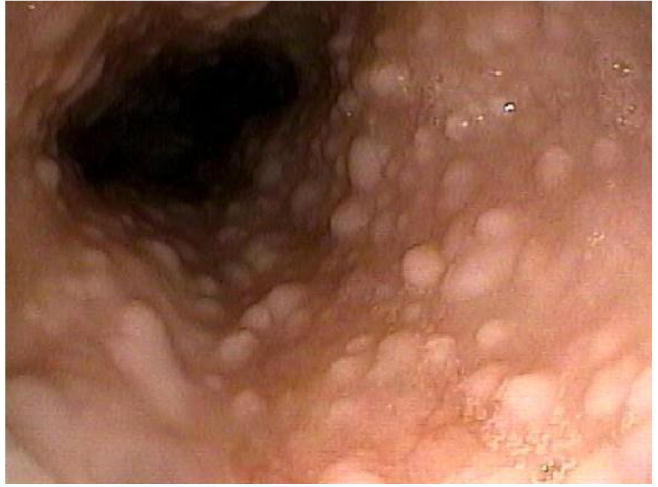
Glyocogenic acanthosis associated with Cowden syndrome
Associated malignancies
Although benign lesions are what usually prompt the identification of CS, breast, thyroid and endometrial cancers are included in the major criteria due to the increased risk associated with PTEN mutations (see Table 1 for associated malignancies). Women with CS have a 25%-50% lifetime risk for breast cancer, compared to a 10%-12% in the general population, and a 5%-10% risk for endometrial cancer, compare to less than a 3% risk in the general population [26]. Early onset malignancies are characteristic of CS with the average age of breast cancer onset occurring in the 40s [26]. Both men and women with CS have up to a 10% risk for non-medullary thyroid cancer; a female preponderance is seen. Renal cell carcinoma, melanoma, colon cancer and other cancers have been reported, however their association with CS is currently not known.
Bannayan-Riley-Ruvalcaba syndrome
In 1960 Riley and Smith described a mother and four children with macrocephaly and pseudopapilledema; two of these individuals also had hemangiomatas [24]. Bannayan in 1971 described a three year old with macrocephaly, lipomatosis, and angiomatosis [40]. A decade later Ruvalcaba and colleagues reported on two cases with macrocephaly, intestinal hamartomatous polypsosis, and pigmented macules of the glans penis [41]. Although originally believed to be distinct conditions, the cases described by Riley, Bannayan, Ruvalcaba, and colleagues are now believed to be part of the condition coined by Gorlin et al. in 1992 as Bannayan-Riley-Ruvalcaba syndrome (BRRS) [27]. BRRS is also known to be allelic to CS and is part of the phenotypic spectrum resulting from germline mutations in the PTEN gene [42, 43]. Kindreds with both CS and BRRS have been reported [42-44].
No consensus criteria have been developed for the diagnosis of BRRS. It has been suggested that individuals having various combinations of macrocephaly, lipomatosis, hemangiomas, GI polyposis, and pigmented macules of the glans penis be considered clinically affected [34]. Mental retardation and assorted congenital abnormalities are also seen in BRRS [27]. In contrast to CS, GI polyps are frequently symptomatic in BRRS [45]. Breast, thyroid, and endometrial cancers are now also believed to be a component feature of BRRS [42, 45].
Genetics
PTEN, also known as phosphatase and tensin homolog deleted on chromosome ten, is a tumor suppressor gene located on 10q22-q23. Germline mutations in PTEN are responsible for the PHTS. Mutations are detected in up to 80% of individuals with CS and 60% of individuals with BRRS [42, 46]. The highest mutation detection rate has been found in families with both BRRS and CS [42]. It is estimated that 10%-50% of CS cases are familial [45]. Recently, it has been shown that approximately 20% of individuals with autism spectrum disorders and macrocephaly have germline PTEN mutations [47]. Previous studies have suggested possible genotype-phenotype correlations, however data supporting these relationships are lacking [44].
Genetic testing of PTEN is commercially available and is necessary in order to confirm the diagnosis of the PHTS. Predictive genetic testing should be offered to all at-risk relatives when a mutation has been identified in the family, even if a de novo mutation is suspected in the individual. A comprehensive evaluation for associated features in these at-risk relatives is imperative due to the heterogenous nature and variability in age of onset associated with PTEN mutations.
Management
Practice guidelines for the management of individuals with PTEN mutations have been proposed (www.nccn.org) and are reviewed on an annual basis at that site. Currently, screening recommendations are targeted to associated cancer risks (see Table 1). Due to the young average age of onset, earlier and more frequent breast cancer screening is recommended for affected women. For men and women, annual thyroid ultrasound starting at age 18 years should be considered. The optimal screening approach for endometrial cancer, renal cell carcinoma or other potentially associated cancers has not been resolved and participation in a clinical trial is recommended. Due to the high penetrance of multiple mucocutaneous lesions and previous reports of skin cancer (including melanoma) in CS, annual dermatologic exams should be considered for affected individuals. Since most individuals with PHTS will have GI polyps, baseline colonoscopy by at least age 40 and regularly thereafter, is recommended at some centers. However, further studies are needed to address the efficacy of earlier and more frequent colonoscopies, given that data are lacking supporting an association between germline PTEN mutations and increased risk for colorectal cancer.
Juvenile Polyposis
Clinical Features
Juvenile polyposis syndrome (JPS) is characterized by the development of multiple juvenile polyps in the GI tract [48, 49]. An individual often presents with symptoms of JPS prior to age 20 [49].
Incidence
JPS occurs in approximately 1 in 100,000 individuals [2, 50]. Of note, the appearance of singular juvenile polyps in children is not uncommon. This finding is present in approximately 2% of children and is believed to be fundamentally different from true JPS which involves multiple polyps [49, 51]. Approximately 25%-50% of JPS cases are believed to be de novo – meaning the underlying genetic cause was not inherited from a parent, but could be passed on to children [14, 51].
Clinical criteria
A clinical diagnosis of JPS is considered in anyone who meets at least one of the following criteria [52, 53]:
At least 3-5 juvenile polyps of the colon
Multiple juvenile polyps found throughout the GI tract
Any number of juvenile polyps in an individual with a family history of JPS
Pathology
Juvenile polyps are often reported out as inflammatory or hyperplastic, sometimes delaying the diagnosis of JPS [54]. True juvenile polyps are distinguished by inflammatory stromal tissue with mucus-filled cystic glands [48]. Smooth-muscle proliferation is not seen in juvenile polyps [49].
Genetics
Two primary genes have been associated with JPS to date: SMAD4 and BMPR1A. Both genes are part of the TGF-β signaling pathway. This pathway is highly involved in regulating the cell cycle, particularly in colonic cells [49]. Recent studies have shown that deletions in SMAD4 and BMPR1A, while less frequent than point mutations, account for a significant proportion of JPS-causing mutations [54-56]. Based on their data, adding deletion testing to SMAD4 and BMPR1A gene analysis will increase current standard mutation detection rates in individuals with clinical JPS.
Mutations in ENG and PTEN have been postulated to also account for a few cases of JPS. Due to the GI phenotype overlap between CS and JPS, individuals originally classified as having JPS with a PTEN mutation may truly have CS instead [49, 55]. Out of a sample of 14 individuals clinically diagnosed with JPS and without identified SMAD4 or BMPR1A mutations, two mutations in ENG were identified [50]. ENG mutations are generally associated with a vascular genetic condition, hereditary hemorrhagic telangiectasia (HHT). Subsequent studies have questioned whether or not ENG mutations are causative of JPS. The contribution of ENG to JPS is still uncertain.
Mutations in SMAD4 and BMPR1A are believed to account for approximately equal proportions of individuals with JPS (~20% each) [49]. However, some distinct genotype/phenotype correlations have been identified between these two mutation groups. Thus the phenotype of some individuals can assist with prioritizing genetic testing of one gene over the other. Given the polyp pathology overlap with CS, individuals with suspected JPS should be closely scrutinized for the additional extra-GI features of CS. PTEN testing could then be pursued in individuals who appear to have features suggestive of the PHTS [54].
Genotype/phenotype correlations
Genotype correlations have been found that can give assistance in predicting the timing and location of polyp development in JPS, as well as the likelihood of extra-GI manifestations in some instances. Since juvenile polyps can be seen in JPS and PHTS, phenotypic overlap occurs between the two [48]. Of note, BMPR1A is contiguous with PTEN on chromosome 10. Some individuals with partial or full deletions of both BMPR1A and PTEN have been described in the literature with varying phenotypic presentations [48]. Some of these individuals had phenotypes consistent with juvenile polyposis of infancy (JPI).
JPS Subtypes
JPI has been classified as a rare subtype of JPS typically presenting before age two [57]. JPI has a severe presentation and is frequently fatal. Infants with JPI can present with rectal bleeding, intussusception and/or diarrhea [48]. To date, JPI appears to be caused by de novo genetic mutations – no confirmed familial cases have been reported [57]. A 2006 study reported four unrelated individuals who all presented with JPI and were subsequently found to have contiguous deletions of BMPR1A and PTEN [57]. All four individuals had rectal bleeding prior to age two and were found to have colonic juvenile polyposis. One patient was found to have adenomatous polyps and foci of grade 3 dysplasia in the small intestine at age three, while another had adenocarcinoma in a duodenal polyp at age 14. Of interest, this latter patient was found to be mosaic for the BMPR1A/PTEN deletion. Three of the patients underwent colectomies at ages 10 months, 17 months, and 8 years. One of the children passed away at age three due to continued severe GI symptoms. All four of the children also had physical and cutaneous features consistent with PHTS, including macrocephaly.
A subsequent case report described a female patient with a de novo 12 MB interstitial deletion of the paternal chromosome 10, including BMPR1A and PTEN [58]. She did not present with symptomatic colonic juvenile polyposis until age five [58]. Interestingly, the macrocephaly and cutaneous features commonly associated with CS were not seen in this patient [58]. This phenotypic variability is further supported by a study of four additional individuals with BMPR1A/PTEN contiguous deletions where only one presented with GI symptoms prior to age two [48].
JPS-HHT
Symptoms consistent with HHT have been reported in individuals with JPS in multiple studies [14]. These symptoms include telangiectasia, epistaxis, and arteriovenous malformations (AVM’s) (often located in the GI tract resulting in bleeding). Gallione et al. first reported individuals specifically with SMAD4 mutations with symptoms of both JPS and HHT and recommended they be reclassified as having a combined syndrome, JPS-HHT [59]. The presence of juvenile polyposis and anemia (due to AVMs) are the primary clinical features of this JPS subtype [60]. To date, the SMAD4 mutations in these individuals have all been located toward the 5’ end of the gene (exons 8-11) [60]. HHT alone is most commonly caused by mutations in the ENG or ALK1 genes (60%-93%) [60]. In a recent study, 3 out of 30 unselected HHT patients (24 met clinical criteria, 6 likely had HHT), were found to have SMAD4 mutations [60].
Gastric polyposis and SMAD4 mutations
In 2002, Friedl et al. identified a correlation between significant gastric polyposis (involving often large polyps) and individuals with JPS due to a SMAD4 mutation [61]. Out of the 7 unrelated JPS patients in their study group who had SMAD4 mutations, four of them had massive gastric polyposis (Figure 3) necessitating partial or total gastrectomy. This phenotypic difference remained consistent in examining the affected family members of the study group – individuals with SMAD4 mutations had much higher rates of gastric polyposis than BMPR1A mutation carriers and the gastric polyposis was much more severe. This finding has the distinction of being the first genotype/phenotype correlation identified in JPS. Of note, massive gastric polyposis is often complicated by GI bleeding and has a significant risk for gastric cancer.
Figure 3.
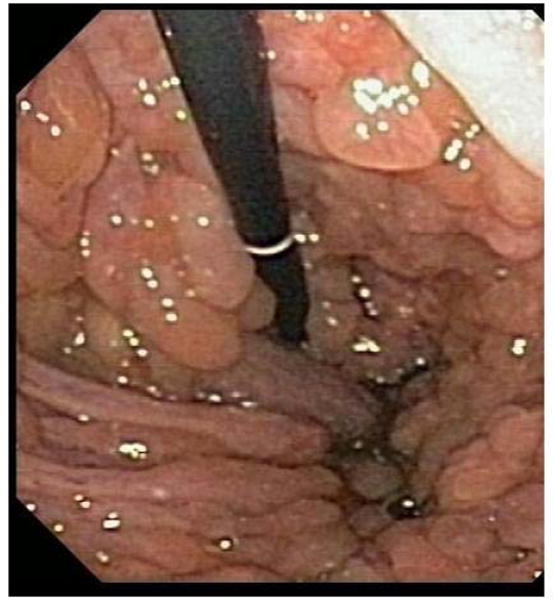
Gastric polyposis in JPS due to SMAD4 mutation
Cancer risks
The primary cancer risks associated with JPS are confined to the GI tract: stomach, colon, and small intestine (see Table 1). The gastric cancer risk is predominately seen in individuals with JPS who develop multiple gastric polyps. As noted, the risk of gastric polyps in SMAD4 mutation carriers is much higher than what is seen in BMPR1A mutation carriers.
Management
Given the genotype/phenotype correlations delineated above, the management recommendations for individuals diagnosed with JPS can differ significantly (see Table 1). General endoscopy recommendations for individuals with JPS include colonoscopy and upper endoscopy starting at age 15, unless symptoms warrant earlier intervention [2, 49]. This screening should be repeated annually if polyps are found, or every two to three years if no polyps are identified [2, 49]. If the polyp burden in the colon and/or stomach cannot be managed with endoscopic removal, surgery is indicated (colectomy, gastrectomy, or small intestine resection) [49]. Multiple enterotomies with polypectomy and intraoperative enteroscopy with polypectomy are often performed for small bowel polyps to minimize loss of small bowel.
Individuals with JPS with an identified SMAD4 mutation should also have appropriate screening for AVMs consistent with published guidelines for HHT management [60, 62]. If AVMs are detected, additional periodic imaging will be necessary to monitor the AVMs (particularly in the lungs to check for progression) [62]. Surgery may be needed to occlude the AVMs in order to prevent potentially life-threatening rupture.
Infants with JPS should be managed according to their symptoms [48, 57]. No standard guidelines specific to the management of JPI have been issued, given the rarity of this condition.
Overall Genetic Evaluation and Testing for Hamartomous Polyposis Syndromes
For individuals with suspected hamartomatous polyposis, correct diagnoses hinge on multiple pieces of information. Review of medical records should include endoscopic/surgical reports detailing the number, location, and age of onset of the individual’s polyps, plus the histologic type of these polyps and any cancers (GI or non-GI related). Reliable pathology reports on the removed/biopsied polyps are crucial. As discussed, hamartomatous polyps are not always reported as such in pathology reports [50]. While PJS polyps are distinctive, a pathologist who is unfamiliar with the features may not report them as PJS polyps. Having an individual’s polyp slides reviewed by a GI pathologist can assist with proper polyp type identification and reduce diagnostic delays.
The content of the individual’s physical examination and questions regarding personal health history can both be tailored to focus on extra-GI manifestations of hamartomatous polyposis syndromes (i.e. macrocephaly, oral pigmentation, problems with bleeding, etc.). Elicitation of family history should be directed toward family members’ GI polyp history, cancer diagnoses, and relevant extra-GI manifestations. By evaluating as much of this information as possible, a hamartomatous polyposis differential can typically be narrowed and genetic testing for one syndrome can be prioritized. If genetic testing cannot identify the causative mutation, the individual can be managed on the basis of his/her manifestations alone or by a working clinical diagnosis of a specific syndrome.
Summary
This review highlights the characteristics and management of the hamartomatous polyposis syndromes. These conditions represent a diverse group of inherited polyposis syndromes associated with high rates of benign and malignant complications, both gastro-intestinal and extra-intestinal. Appropriate diagnosis and treatment requires a team approach including medical and surgical specialists, genetic counselors, and pathologists. While the classic presentations of these syndromes allow them to be easily distinguished, many individuals present with features that overlap one or more of the conditions. Careful assessment of the phenotype, family history, and pathology is needed for clinical diagnosis and to determine the appropriate gene(s) to be assessed for genetic diagnosis. The initial identification of individuals with one of these syndromes most often occurs from endoscopic findings. But once a diagnosis is made multiple specialists are needed for management in view of the benign complications and cancer risks in and outside of the digestive system.
There are still many unanswered questions regarding the optimal management approaches. Many screening and surveillance recommendations are based on consensus rather than being evidence based. The development of chemopreventive agents, which would prevent or reduce the development of hamartomas and reduce cancer risk and the need for surgery would be of great benefit in this population. Several centers have established registries of families with these syndromes. Due to the rarity of these conditions, collaboration between multiple research registries is needed to better formulate management of the hamartomatous polyposis syndromes.
Acknowledgments
This research is supported by NCI grants P01-CA073992(RWB), R01-CA040641(RWB) and Huntsman Cancer Foundation.
Footnotes
Conflict of Interest Statement Randall Burt is a consultant for Myriad Genetics Laboratories, Salt Lake City, Utah. Myriad provided no support the work reported, however, and no Myriad Genetics Laboratories products are discussed.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Amanda Gammon, Huntsman Cancer Institute, 2000 Circle of Hope, Salt Lake City, UT 84112-5550, Phone: 801-585-5938, Fax: 801-585-2980, Amanda.gammon@hci.utah.edu.
Kory Jasperson, Huntsman Cancer Institute, 2000 Circle of Hope, Salt Lake City, UT 84112-5550, Phone: 801-581-7316, Fax: 801-585-2980, Kory.jasperson@hci.utah.edu.
Wendy Kohlmann, Huntsman Cancer Institute, 2000 Circle of Hope, Salt Lake City, UT 84112-5550, Phone: 801-587-5556, Fax: 801-585-2980, Wendy.kohlmann@hci.utah.edu.
Randall W. Burt, Huntsman Cancer Institute, 2000 Circle of Hope, Salt Lake City, UT 84112-5550, Phone: 801-585-3281, Fax: 801-581-3389, Randall.burt@hci.utah.edu.
References
- 1.Kinzler KW, Vogelstein B. Landscaping the cancer terrain. Science. 1998;280(5366):1036–1037. doi: 10.1126/science.280.5366.1036. comment. [DOI] [PubMed] [Google Scholar]
- *2.Schreibman IR, Baker M, Amos C, McGarrity TJ. The hamartomatous polyposis syndromes: A clinical and molecular review. American Journal of Gastroenterology. 2005;100:476–490. doi: 10.1111/j.1572-0241.2005.40237.x. [DOI] [PubMed] [Google Scholar]
- *3.Lindor N, McMaster M, Lindor C, et al. Concise handbook of familial cancer susceptibility syndromes-second edition. J Natl Cancer Inst Monogr. 2008;38:1–93. doi: 10.1093/jncimonographs/lgn001. [DOI] [PubMed] [Google Scholar]
- 4.McGarrity TJ, Kulin HE, Zaino RJ. Peutz-Jeghers syndrome. Am J Gastroenterol. 2000;95(3):596–604. doi: 10.1111/j.1572-0241.2000.01831.x. [DOI] [PubMed] [Google Scholar]
- 5.Westerman AM, Entius MM, de Baar E, et al. Peutz-Jeghers syndrome: 78-year follow-up of the original family. Lancet. 1999;353(9160):1211–1215. doi: 10.1016/s0140-6736(98)08018-0. [DOI] [PubMed] [Google Scholar]
- 6.Amos C, Keitheri-Cheteri M, Sabripour M, et al. Genotype-phenotype correlations in Peutz-Jeghers syndrome. J Med Genet. 2004;41:327–333. doi: 10.1136/jmg.2003.010900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tomlinson IP, Houlston RS. Peutz-Jeghers syndrome. J Med Genet. 1997;34(12):1007–1011. doi: 10.1136/jmg.34.12.1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *8.Giardiello FM, Trimbath J. Peutz-Jeghers syndrome and management recommendations. Clinical Gastroenterol Hepatol. 2006;4(4):408–415. doi: 10.1016/j.cgh.2005.11.005. [DOI] [PubMed] [Google Scholar]
- 9.Giardiello FM, Brensinger JD, Tersmette AC, et al. Very high risk of cancer in familial peutz-jeghers syndrome. Gastroenterology. 2000;119(6):1447–1453. doi: 10.1053/gast.2000.20228. In Process Citation. [DOI] [PubMed] [Google Scholar]
- 10.Hearle N, Schumacher V, Menko F, et al. Frequency and spectrum of cancers in the Peutz-Jeghers syndrome. Clin Cancer Res. 2006;12(10):3209–3215. doi: 10.1158/1078-0432.CCR-06-0083. [DOI] [PubMed] [Google Scholar]
- 11.Lim W, Olschwang S, Keller J, Westerman A, et al. Relative frequency and morphology of cancers in STK11 mutation carriers. Gastroenterology. 2004;126(7):1788–1794. doi: 10.1053/j.gastro.2004.03.014. [DOI] [PubMed] [Google Scholar]
- 12.Young R, Welch W, Dickersin, Scully R. Ovarian sex cord tumor with annular tubules: review of 74 cases including 27 with Peutz-Jeghers syndrome and four with adenoma malignum of the cervix. Cancer. 1982;50(7):1384–1402. doi: 10.1002/1097-0142(19821001)50:7<1384::aid-cncr2820500726>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 13.Connolly D, Katabuchi H, Cliby W, Cho K. Somatic mutations in the STK11/LKB1 gene are uncommon in rare gynecological tumor types associated with Peutz-Jegher’s syndrome. Am J Pathol. 2000;1565(1):339–345. doi: 10.1016/S0002-9440(10)64735-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Calva D, Howe J. Hamartomatous polyposis syndromes. Surg Clin North Am. 2008;88:779–817. doi: 10.1016/j.suc.2008.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mehenni H, Resta N, Guanti G, et al. Molecular and clinical characteristics in 46 families affected with Peutz-Jeghers syndrome. Dig Dis Sci. 2007;52:1924–1933. doi: 10.1007/s10620-006-9435-3. [DOI] [PubMed] [Google Scholar]
- *16.Aretz S, Stienen D, Uhlhaas S, et al. High proportion of large genomic STK11 deletions in Peutz-Jeghers syndrome. Hum Mutat. 2005;26(6):513–519. doi: 10.1002/humu.20253. [DOI] [PubMed] [Google Scholar]
- 17.Kitaoka F, Shiogama T, Mizutani A, et al. A solitary Peutz-Jeghers-type hamartomatous polyp in the duodenum. A case report including results of mutational analysis. Digestion. 2004;69(2):79–82. doi: 10.1159/000077392. [DOI] [PubMed] [Google Scholar]
- 18.Boardman L, Couch F, Burgart L, et al. Genetic heterogeneity in Peutz-Jegher syndrome. Hum Mutat. 2000;16:23–30. doi: 10.1002/1098-1004(200007)16:1<23::AID-HUMU5>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 19.Hearle N, Schumacher V, Menko F, et al. STK11 status and intussusception risk in Peutz-Jeghers syndrome. J Med Genet. 2006;43(8):e41. doi: 10.1136/jmg.2005.040535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Burke CA, Santisi J, Church J, Levinthal G. The utility of capsule endoscopy small bowel surveillance in patients with polyposis. Am J Gastroenterol. 2005;100(7):1498–1502. doi: 10.1111/j.1572-0241.2005.41506.x. [DOI] [PubMed] [Google Scholar]
- 21.Mata A, Llach J, Castells A, et al. A prospective trial comparing wireless capsule endoscpy and barium contrast series for small-bowel surveillance in hereditary GI polyposis syndromes. Gastrointest Endosc. 2005;61:721–725. doi: 10.1016/s0016-5107(05)00289-0. [DOI] [PubMed] [Google Scholar]
- 22.Soares J, Lopes L, Vilas Boas G, Phiho C. Wireless capsule endoscopy for evaluation of phenotypic expression of small-bowel polyps in patients with Peutz-Jeghers syndrome and in symptomatic first degree relatives. Endoscopy. 2004;36(12):1060–1066. doi: 10.1055/s-2004-826038. [DOI] [PubMed] [Google Scholar]
- 23.Rangiah D, Cox M, Richardson M, et al. Small bowel tumours: a 10 year experience in four Syndney teaching hospitals. ANZ J Surg. 2004;74(9):788–792. doi: 10.1111/j.1445-1433.2004.03150.x. [DOI] [PubMed] [Google Scholar]
- 24.Hruban RH, Canto MI, Yeo CJ. Prevention of pancreatic cancer and strategies for management of familial pancreatic cancer. Dig Dis. 2001;19:76–84. doi: 10.1159/000050656. [DOI] [PubMed] [Google Scholar]
- 25.Canto MI, Goggins M, Hruban RH, et al. Screening for early pancreatic neoplasia in high-risk individuals: A prospective controlled study. Clin Gastroenterol Hepatol. 2006;4:766–781. doi: 10.1016/j.cgh.2006.02.005. [DOI] [PubMed] [Google Scholar]
- 26.Eng C. PTEN: one gene, many syndromes. Hum Mutat. 2003;22(3):183–198. doi: 10.1002/humu.10257. [DOI] [PubMed] [Google Scholar]
- 27.Gorlin RJ, Cohen MM, Jr, Condon LM, Burke BA. Bannayan-Riley-Ruvalcaba syndrome. Am J Med Genet. 1992;44(3):307–314. doi: 10.1002/ajmg.1320440309. [DOI] [PubMed] [Google Scholar]
- 28.Lloyd KM, 2nd, Dennis M. Cowden’s disease. A possible new symptom complex with multiple system involvement. Ann Intern Med. 1963;58:136–142. doi: 10.7326/0003-4819-58-1-136. [DOI] [PubMed] [Google Scholar]
- 29.Nelen MR, Padberg GW, Peeters EA, et al. Localization of the gene for Cowden disease to chromosome 10q22-23. Nat Genet. 1996;13(1):114–116. doi: 10.1038/ng0596-114. [DOI] [PubMed] [Google Scholar]
- 30.Li FP, Abramson DH, Tarone RE, et al. Hereditary retinoblastoma, lipoma, and second primary cancers. J Natl Cancer Inst. 1997;89(1):83–84. doi: 10.1093/jnci/89.1.83. [DOI] [PubMed] [Google Scholar]
- 31.Nelen MR, Kremer H, Konings IB, et al. Novel PTEN mutations in patients with Cowden disease: absence of clear genotype-phenotype correlations. Eur J Hum Genet. 1999;7(3):267–273. doi: 10.1038/sj.ejhg.5200289. [DOI] [PubMed] [Google Scholar]
- 32.Schrager CA, Schneider D, Gruener AC, et al. Clinical and pathological features of breast disease in Cowden’s syndrome: an underrecognized syndrome with an increased risk of breast cancer. Hum Pathol. 1998;29(1):47–53. doi: 10.1016/s0046-8177(98)90389-6. [DOI] [PubMed] [Google Scholar]
- 33.Pilarski R, Eng C. Will the real Cowden syndrome please stand up (again)? Expanding mutational and clinical spectra of the PTEN hamartoma tumour syndrome. J Med Genet. 2004;41(5):323–326. doi: 10.1136/jmg.2004.018036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Salem OS, Steck WD. Cowden’s disease (multiple hamartoma and neoplasia syndrome). A case report and review of the English literature. J Am Acad Dermatol. 1983;8(5):686–696. doi: 10.1016/s0190-9622(83)70081-2. [DOI] [PubMed] [Google Scholar]
- 35.Starink TM, van der Veen JP, Arwert F, et al. The Cowden syndrome: a clinical and genetic study in 21 patients. Clin Genet. 1986;29(3):222–233. doi: 10.1111/j.1399-0004.1986.tb00816.x. [DOI] [PubMed] [Google Scholar]
- 36.Hildenbrand C, Burgdorf WH, Lautenschlager S. Cowden syndrome - diagnostic skin signs. Dermatology. 2001;202(4):362–366. doi: 10.1159/000051684. [DOI] [PubMed] [Google Scholar]
- 37.Derrey S, Proust F, Debono B, et al. Association between Cowden syndrome and Lhermitte-Duclos disease: report of two cases and review of the literature. Surg Neurol. 2004;61(5):447–454. doi: 10.1016/S0090-3019(03)00576-7. discussion 454. [DOI] [PubMed] [Google Scholar]
- 38.Kato M, Mizuki A, Hayashi T, et al. Cowden’s disease diagnosed through mucocutaneous lesions and gastrointestinal polyposis with recurrent hematochezia, unrevealed by initial diagnosis. Intern Med. 2000;39(7):559–563. doi: 10.2169/internalmedicine.39.559. [DOI] [PubMed] [Google Scholar]
- 39.McGarrity TJ, Wagner Baker MJ, Ruggiero FM, et al. GI polyposis and glycogenic acanthosis of the esophagus associated with PTEN mutation positive Cowden syndrome in the absence of cutaneous manifestations. Am J Gastroenterol. 2003;98(6):1429–1434. doi: 10.1111/j.1572-0241.2003.07496.x. [DOI] [PubMed] [Google Scholar]
- 40.Bannayan GA. Lipomatosis, angiomatosis, and macrencephalia. A previously undescribed congenital syndrome. Arch Pathol. 1971;92(1):1–5. [PubMed] [Google Scholar]
- 41.Ruvalcaba RH, Myhre S, Smith DW. Sotos syndrome with intestinal polyposis and pigmentary changes of the genitalia. Clin Genet. 1980;18(6):413–416. doi: 10.1111/j.1399-0004.1980.tb01785.x. [DOI] [PubMed] [Google Scholar]
- 42.Marsh DJ, Kum JB, Lunetta KL, et al. PTEN mutation spectrum and genotype-phenotype correlations in Bannayan-Riley-Ruvalcaba syndrome suggest a single entity with Cowden syndrome. Hum Mol Genet. 1999;8(8):1461–1472. doi: 10.1093/hmg/8.8.1461. [DOI] [PubMed] [Google Scholar]
- 43.Zori RT, Marsh DJ, Graham GE, et al. Germline PTEN mutation in a family with Cowden syndrome and Bannayan-Riley-Ruvalcaba syndrome. Am J Med Genet. 1998;80(4):399–402. [PubMed] [Google Scholar]
- *44.Lachlan KL, Lucassen AM, Bunyan D, Temple IK. Cowden syndrome and Bannayan Riley Ruvalcaba syndrome represent one condition with variable expression and age-related penetrance: results of a clinical study of PTEN mutation carriers. J Med Genet. 2007;44(9):579–585. doi: 10.1136/jmg.2007.049981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Eng C. Constipation, polyps, or cancer? Let PTEN predict your future. Am J Med Genet A. 2003;122A(4):315–322. doi: 10.1002/ajmg.a.20477. [DOI] [PubMed] [Google Scholar]
- 46.Marsh DJ, Coulon V, Lunetta KL, et al. Mutation spectrum and genotype-phenotype analyses in Cowden disease and Bannayan-Zonana syndrome, two hamartoma syndromes with germline PTEN mutation. Hum Mol Genet. 1998;7(3):507–515. doi: 10.1093/hmg/7.3.507. [DOI] [PubMed] [Google Scholar]
- 47.Butler MG, Dasouki MJ, Zhou XP, et al. Subset of individuals with autism spectrum disorders and extreme macrocephaly associated with germline PTEN tumour suppressor gene mutations. J Med Genet. 2005;42(4):318–321. doi: 10.1136/jmg.2004.024646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Menko FH, Kneepkens CM, de Leeuw N, et al. Variable phenotypes associated with 10q23 microdeletions involving the PTEN and BMPR1A genes. Clin Genet. 2008;74(2):145–154. doi: 10.1111/j.1399-0004.2008.01026.x. [DOI] [PubMed] [Google Scholar]
- *49.Zbuk KM, Eng C. Hamartomatous polyposis syndromes. Nature clinical practice. 2007;4(9):492–502. doi: 10.1038/ncpgasthep0902. [DOI] [PubMed] [Google Scholar]
- 50.Sweet K, Willis J, Zhou XP, et al. Molecular classification of patients with unexplained hamartomatous and hyperplastic polyposis. JAMA. 2005;294(19):2465–2473. doi: 10.1001/jama.294.19.2465. [DOI] [PubMed] [Google Scholar]
- 51.Burt R, Neklason DW. Genetic testing for inherited colon cancer. Gastroenterology. 2005;128(6):1696–1716. doi: 10.1053/j.gastro.2005.03.036. [DOI] [PubMed] [Google Scholar]
- 52.Jass JR, Williams CB, Bussey HJ, Morson BC. Juvenile polyposis--a precancerous condition. Histopathology. 1988;13(6):619–630. doi: 10.1111/j.1365-2559.1988.tb02093.x. [DOI] [PubMed] [Google Scholar]
- 53.Giardiello FM, Hamilton SR, Kern SE, et al. Colorectal neoplasia in juvenile polyposis or juvenile polyps. Arch Dis Child. 1991;66(8):971–975. doi: 10.1136/adc.66.8.971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *54.Aretz S, Stienen D, Uhlhaas S, et al. High proporation of large genomic deletions and a genotype phenotype update in 80 unrelated families with juvenile polyposis. J Med Genet. 2007;44(11):702–709. doi: 10.1136/jmg.2007.052506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *55.van Hattem WA, Brosens LA, de Leng WW, et al. Large genomic deletions of SMAD4, BMPR1A and PTEN in juvenile polyposis. Gut. 2008;57(5):623–627. doi: 10.1136/gut.2007.142927. [DOI] [PubMed] [Google Scholar]
- 56.Calva-Cerqueira D, Chinnathambi S, Pechman B, et al. The rate of germline mutations and large deletions of SMAD4 and BMPR1A in juvenile polyposis. Clin Genet. 2008 doi: 10.1111/j.1399-0004.2008.01091.x. [DOI] [PubMed] [Google Scholar]
- *57.Delnatte C, Sanlaville D, Mougenot JF, et al. Contiguous gene deletion within chromosome arm 10q is associated with juvenile polyposis of infancy, reflecting cooperation between the BMPR1A and PTEN tumor-suppressor genes. Am J Hum Genet. 2006;78(6):1066–1074. doi: 10.1086/504301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Salviati L, Patricelli M, Guariso G, et al. Deletion of PTEN and BMPR1A on chromosome 10q23 is not always associated with juvenile polyposis of infancy. Am J Hum Genet. 2006;79(3):593–596. doi: 10.1086/507151. author reply 596-597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *59.Gallione CJ, Repetto GM, Legius E, et al. A combined syndrome of juvenile polyposis and hereditary haemorrhagic telangiectasia associated with mutations in MADH4 (SMAD4) Lancet. 2004;363(9412):852–859. doi: 10.1016/S0140-6736(04)15732-2. [DOI] [PubMed] [Google Scholar]
- 60.Gallione CJ, Richards JA, Letteboer TG, et al. SMAD4 mutations found in unselected HHT patients. J Med Genet. 2006;43(10):793–797. doi: 10.1136/jmg.2006.041517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Friedl W, Uhlhaas S, Schulmann K, et al. Juvenile polyposis: massive gastric polyposis is more common in MADH4 mutation carriers than in BMPR1A mutation carriers. Hum Genet. 2002;111(1):108–111. doi: 10.1007/s00439-002-0748-9. [DOI] [PubMed] [Google Scholar]
- 62.Bayrak-Toydemir P, Mao R, Lewin S, McDonald J. Hereditary hemorrhagic telangiectasia: an overview of diagnosis and management in the molecular era for clinicians. Genet Med. 2004;6(4):175–191. doi: 10.1097/01.gim.0000132689.25644.7c. [DOI] [PubMed] [Google Scholar]