

Abstract

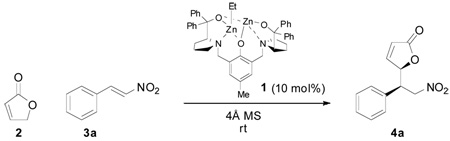
Under dinuclear catalysis, the direct conjugate addition of 2(5H)-furanone to nitroalkenes involves the γ-position of the nucleophile. The synthetically versatile Michael adducts are prepared in good yields, with high levels of diastereo- and enantioselectivity. A model is presented to rationalize the observed stereoselectivity.
The Michael addition is certainly one of the most powerful bond-forming transformations and the diversity in donors and acceptors that can be combined is remarkable. Recent efforts have focused on the development of efficient methods to perform direct, asymmetric Michael addition reactions and notable success has been achieved by using both organocatalysis1 and transition-metal catalysis.2 The self-assembled dinuclear complexes generated from our Bis-ProPhenol ligand and adequate metal sources3 are potentially suitable catalysts.4 The complementary reactivity of the two metal centers allows for dual activation whereas conformational rigidity enables chiral recognition, as illustrated in a variety of direct asymmetric addition reactions and desymmetrization processes.5 Selecting the Michael addition to nitroalkenes as a probe reaction, we decided to incorporate a new concept into our general strategy, namely vinylogous nucleophilicity.6,7 As the donor, 2(5H)-furanone 2 was envisioned to be a challenging yet ideal candidate to demonstrate the synthetic efficiency of our system. Indeed, this commonly utilized nucleophile usually requires pre-activation as a siloxydiene à la Mukaiyama8 and direct approaches are consequently highly valuable from the standpoint of atom economy. While such a strategy was reported in a stereoselective vinylogous 1,2-addition,9 the direct use of 2 in asymmetric conjugate addition has not been precedented.
As previously reported,3 the dinuclear zinc complex 1 was prepared by treating the commercially available (S,S)-Bis-ProPhenol ligand with 2 equiv. of Et2Zn. Early optimization with Michael acceptor 3a proved that 1 was competent at promoting the alkylation of butenolide 2 at the γ-position at room temperature. Among the solvents that were screened, THF gave the highest diastereoselectivity (Table 1, entries 1–4). Interestingly, the reaction proceeded faster in toluene with similar yield and enantioselectivity. Dilution had a beneficial effect on the diastereoselectivity: Michael adduct 4a was obtained with 10:1 dr by lowering the concentration of nitroalkene to 0.25 m (entry 5). Upon dilution to 0.125 m, diastereoselectivity could be further improved to 17:1 dr, albeit at the expense of an extended reaction time (entry 6).
Table 1.
Selected Optimization Resultsa
 | ||||||
|---|---|---|---|---|---|---|
| entry | solvent | [nitroalkene] | time (h) | yield (%)b | drc | ee (%)d |
| 1 | THF | 0.5 m | 19 | 75 | 8:1 | 91 |
| 2 | toluene | 0.5 m | 8 | 71 | 5:1 | 92 |
| 3 | Et2O | 0.5 m | 26 | 40 | 1:1 | 65 |
| 4 | dioxane | 0.5 m | 22 | 69 | 6:1 | 93 |
| 5 | THF | 0.25 m | 19 | 76 | 10:1 | 91 |
| 6 | THF | 0.125 m | 35 | 71 | 17:1 | 92 |
All reactions were carried out using 1 equiv. of 3a (0.50 mmol), 2 equiv. of 2, 0.10 equiv. of (S,S)-complex 1 and 100 mg 4Å MS.
Refers to the isolated mixture of diastereoisomers.
Determined by 1H NMR of the crude reaction mixture.
Enantiomeric excess of the major diastereoisomer, determined by chiral HPLC.
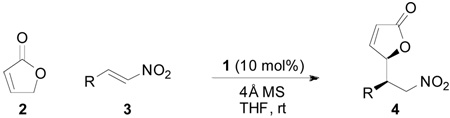
Adopting the conditions described in Table 1, entry 5 as the optimal compromise between reactivity and stereoselectivity, the generality of the method was demonstrated by evaluating a variety of nitroalkenes (Table 2). β-Nitrostyrenes (entries 1–5) tolerated substitution at any position of the aromatic ring and both electron-donating and electron-withdrawing functionalities were compatible. The electron-rich substrate 3d gave the best results: the corresponding adduct 4d was obtained in 78% yield with 20:1 dr and 96% ee (entry 3). In the case of the 1-naphthyl derivative 3g, excellent diastereo- and enantioselectivity were achieved as well (entry 6). Nitroalkenes bearing heteroaromatic β-substituents were also suitable substrates as exemplified by the preparation of both regioisomers 4h and 4i in 95% ee (entries 7 and 8). Similarly good yields and stereoselectivities were observed with the thiophene counterparts (entries 9 and 10). The indole-substituted nitroalkene 3l proved to be a more challenging acceptor under the optimized conditions. However, switching the solvent to toluene improved the yield and the enantioselectivity (entry 11). As it could be expected from the optimization studies, toluene was also more appropriate for the less reactive aliphatic substrate 3m and the corresponding Michael adduct was isolated in 47% yield with 4:1 dr and 83% ee (entry 12). Similarly, starting from the 1-nitro-1,3-diene 3n the 1,4-addition product 4n was obtained with moderate yield and diastereoselectivity but with high enantioselectivity (entry 13). In several instances, the stereoisomeric purity of the Michael adducts could be enhanced by recrystallization. For example, product 4a could be isolated with 20:1 dr and 98% ee with excellent mass recovery (65% yield from 3a). Moreover, crystals from chloride 4e were suitable for an X-ray analysis which established the syn stereochemical outcome of the reaction as well as the absolute configuration. The stereochemistry of the other Michael adducts 4 was assigned by analogy.
Table 2.
Variation of the nitroalkenea
 | |||||
|---|---|---|---|---|---|
| entry | R | product | yield (%)b | drc | ee (%)d |
| 1 | 2-Me-Ph (3b) | 4b | 74 | 8:1 | 92 |
| 2 | 4-Me-Ph (3c) | 4c | 70 | 17:1 | 95 |
| 3 | 4-MeO-Ph (3d) | 4d | 78 | 20:1 | 96 |
| 4 | 4-Cl-Ph (3e) | 4e | 70 | 9:1 | 90 |
| 5 | 3-Br-Ph (3f) | 4f | 72 | 7:1 | 87 |
| 6 | 1-naphthyl (3g) | 4g | 75 | > 20:1 | 94 |
| 7 | 2-furanyl (3h) | 4h | 77 | 18:1 | 95 |
| 8 | 3-furanyl (3i) | 4i | 65 | 14:1 | 95 |
| 9 | 2-thiophenyl (3j) | 4j | 69 | > 20:1 | 94 |
| 10 | 3-thiophenyl (3k) | 4k | 71 | 17:1 | 95 |
| 11e,f | N-Boc-3-indolyl (3l) | 4l | 73 | 6:1 | 85 |
| 12e | PhCH2CH2 (3m) | 4m | 47 | 4:1g | 83 |
| 13e | PhCHCH (3n) | 4n | 52 | 3:1 | 91 |
All reactions were carried out using 1 equiv. of nitroalkene 3 (0.50 mmol, 0.25 m), 2 equiv. of 2, 0.10 equiv. of (S,S)-complex 1 and 100 mg 4Å MS.
Refers to the isolated mixture of diastereoisomers.
Determined by 1H NMR of the crude reaction mixture.
Enantiomeric excess of the major diastereoisomer, determined by chiral HPLC.
Reaction performed in toluene instead of THF.
Results using THF: 60% yield, 7:1 dr, 62% ee.
Determined by chiral HPLC.
A tentative catalytic cycle that accounts for the observed stereoselectivity is depicted in Scheme 1. Binding of the nucleophile as a bidentate bridging aromatic enolate3b,10 would ensure diastereoselection in the attack on the electrophile activated by complexation to the Lewis acidic zinc atom in the indicated orientation. Formation of the new C–C bond within this highly organized environment would lead to a zinc nitronate intermediate. Finally, proton transfer with an incoming molecule of nucleophile would release product 4 and complete the catalytic cycle. The fact that open coordination sites remain on the zinc that may allow additional entities present to ligate and thereby modify the nature of the chiral space may account for the dilution effect.3d
Scheme 1.

Proposed Catalytic Cycle
The Michael adducts 4 are versatile building blocks as one can envision further elaboration of both the butenolide moiety and the nitro functionality. Compound 4a served as a model to straightforwardly illustrate this synthetic potential (Scheme 2). This, ruthenium-catalyzed cis-dihydroxylation11 of the conjugated olefin led to diol 5 in 76% yield with complete diastereoselectivity.12 In this way, excellent control was achieved over four adjacent stereocenters, newly created in only two steps from 3a. The hydroxyl groups were masked as silyl ethers to avoid handling of otherwise highly polar products resulting from the reduction of the nitro substituent. The latter transformation proceeded smoothly under standard conditions to afford the densely functionalized primary amine 6. Spontaneously, upon standing neat at rt the isolated amine slowly evolved into lactam 7. Interestingly, similar polyhydroxyazepanones are being actively investigated in search of potent glycosidases inhibitors.13
Scheme 2.

Elaboration of a vinylogous Michael adducta
a Conditions: (a) RuCl3•6H2O (7 mol%), NaIO4 (1.5 equiv.), CH3CN:H2O 5:1, 0 °C, 76% yield; (b) TBSOTf (3 equiv.), 2,6-lutidine (3 equiv.), CH2Cl2, 0 °C to rt, 84% yield; (c) 10% Pd/C, 1 atm H2, MeOH, rt, 69% yield; (d) neat, rt, 7d.
In summary, synthetically versatile γ-substituted butenolides were prepared stereoselectively by direct asymmetric Michael addition to nitroalkenes.14 This extension of the scope of our dinuclear zinc catalyst showcases its ability to promote vinylogous nucleophilicity. We believe this reactivity feature paves the way for a wide diversity of potential donors and further exploration is currently underway.
Supplementary Material
Detailed experimental procedures and characterization data for compounds 4–7; CIF file for compound 4e and 5. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgment
We thank NSF and the NIH (GM13598) for their generous support. J.H. acknowledges the Ministère Français des Affaires Etrangères et Européennes for a Lavoisier postdoctoral fellowship.
References
- 1.Recent review: Tsogoeva SB. Eur. J. Org. Chem. 2007:1701.
- 2.Reviews: Krause N, Hoffmann-Röder A. Synthesis. 2001:171. Christoffers J, Koripelly G, Rosiak A, Rössle M. Synthesis. 2007:1279. Selected examples involving nitroalkenes as acceptors: Barnes DM, Ji J, Fickes MG, Fitzgerald MA, King SA, Morton HE, Plagge FA, Preskill M, Wagaw SH, Wittenberger SJ, Zang J. J. Am. Chem. Soc. 2002;124:13097. doi: 10.1021/ja026788y. Watanabe M, Ikagawa A, Wang H, Murata K, Ikariya T. J. Am. Chem. Soc. 2004;126:11148. doi: 10.1021/ja046296g. Lu S-F, Du D-M, Xu J, Zhang S-W. J. Am. Chem. Soc. 2006;128:7418. doi: 10.1021/ja0604008. Evans DA, Mito S, Seidel D. J. Am. Chem. Soc. 2007;129:11583. doi: 10.1021/ja0735913.
- 3. Trost BM, Ito H. J. Am. Chem. Soc. 2000;122:12003. Trost BM, Ito H, Silcoff ER. J. Am. Chem. Soc. 2001;123:3367. doi: 10.1021/ja003871h. Xiao Y, Wang Z, Ding K. Chem. Eur. J. 2005;11:3668. doi: 10.1002/chem.200401159. (d) For the effects of additional ligation see Trost BM, Fettes A, Shireman BT. J. Am. Chem. Soc. 2004;126:2660. doi: 10.1021/ja038666r.
- 4.The use of self-assembled multimetallic chiral catalysts was pioneered by Shibasaki: Sasai H, Arai T, Satow Y, Houk KN, Shibasaki M. J. Am. Chem. Soc. 1995;117:6194. Shibasaki M. Pure Appl. Chem. 1996;68:523.
- 5.Latest examples: Trost BM, Müller C. J. Am. Chem. Soc. 2008;130:2438. doi: 10.1021/ja711080y. Trost BM, Malhotra S, Mino T, Rajapaksa NS. Chem. Eur. J. 2008;14:7648. doi: 10.1002/chem.200800623. Trost BM, O'Boyle B. J. Am. Chem. Soc. 2008;130:16190. doi: 10.1021/ja807127s.
- 6.Fuson R. Chem. Rev. 1935;16:1. [Google Scholar]
- 7.(a) Xue D, Chen Y-C, Wang Q-W, Cun L-F, Zhu J, Deng J-G. Org. Lett. 2005;7:5293. doi: 10.1021/ol052283b. [DOI] [PubMed] [Google Scholar]; (b) Jiang L, Zheng H-T, Liu T-Y, Yue L, Chen Y-C. Tetrahedron. 2007;63:5123. [Google Scholar]
- 8.For conjugate additions of 2-siloxyfuran, only α,β-unsaturated acyloxazolidin-2-ones have been used as acceptors. Review: Casiraghi G, Rassu G. Synthesis. 1995:607. Selected examples: Szlosek M, Figadère B. Angew. Chem. Int. Ed. 2000;39:1799. doi: 10.1002/(sici)1521-3773(20000515)39:10<1799::aid-anie1799>3.0.co;2-z. Desimoni G, Faita G, Filippone S, Mella M, Zampori MG, Zema M. Tetrahedron. 2001;57:10203. Onitsuka S, Matsuoka Y, Irie R, Katsuki T. Chem. Lett. 2003;32:974. Carswell EL, Snapper ML, Hoveyda AH. Angew. Chem. Int. Ed. 2006;45:7230. doi: 10.1002/anie.200603496. Salvador González A, Gómez Arrayás R, Rodríguez Rivero M, Carretero JC. Org. Lett. 2008;10:4335. doi: 10.1021/ol8019082.
- 9.During the course of this work, Shibasaki et al. reported their studies on a related 1,2-addition, highlighting the challenges associated with the direct use of γ-butenolides as nucleophiles: Yamaguchi A, Matsunaga S, Shibasaki M. Org. Lett. 2008;10:2319. doi: 10.1021/ol800756r.
- 10.Examples of polynuclear zinc complexes featuring bridging bidentate ligands: Uhlenbrock S, Wegner R, Krebs B. J. Chem. Soc., Dalton Trans. 1996:3731. Sakiyama H, Mochizuki R, Sugawara A, Sakamoto M, Nishida Y, Yamasaki M. J. Chem Soc., Dalton Trans. 1999:997.
- 11.Shing TKM, Tam EKW, Tai VW-F, Chung IHF, Jiang Q. Chem. Eur. J. 1996;2:50. [Google Scholar]
- 12.The stereochemistry of diol 5 was established by X-ray analysis.
- 13.(a) Chaveriat L, Stasik I, Demailly G, Beaupère D. Tetrahedron. 2004;60:2079. doi: 10.1016/j.carres.2004.05.004. [DOI] [PubMed] [Google Scholar]; (b) Gireaud L, Chaveriat L, Stasik I, Wadouachi A, Beaupère D. Tetrahedron. 2006;62:7455. [Google Scholar]
- 14.At the present time, this study has been limited to 2(5H)-furanone 2 as the nucleophile.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Detailed experimental procedures and characterization data for compounds 4–7; CIF file for compound 4e and 5. This material is available free of charge via the Internet at http://pubs.acs.org.