

Abstract
Antibodies to myelin oligodendrocyte glycoprotein (MOG) have been implicated in Multiple Sclerosis demyelination through activation of complement and/or macrophage-effector processes. We presented a novel mechanism, whereby MOG on oligodendrocytes, when cross-linked with anti-MOG and secondary antibody resulted in its repartitioning into lipid rafts, and changes in protein phosphorylation and morphology. Here, we show that similar events occur when anti-MOG is cross-linked with Fc receptors (FcRs) present on microglia but not with complement. These results indicate that FcRs are endogenous antigen/antibody cross-linkers in vitro, suggesting that FcRs could be physiologically relevant in vivo and possible targets for therapy in Multiple Sclerosis.
Keywords: oligodendrocytes, myelin, MOG, microglia, Multiple Sclerosis, complement, Fc receptors
1. INTRODUCTION
Multiple Sclerosis (MS) is an inflammatory demyelinating disease affecting the central nervous system (CNS). It is characterized by areas of demyelination, often with associated axonal degeneration, causing severe, generally irreversible, functional consequences (Lassmann, 2004). Inflammation is mediated by T cells, leading to local activation of microglia and impairment of the integrity of the blood brain barrier (Noseworthy et al., 2000). In addition, B cell responses are implicated in MS pathogenesis. For example, immunoglobulin deposits and opsonized myelin debris are detected at the active edge of demyelinating lesions (Genain et al., 1999; Lassmann, 2004; von Budingen et al., 2004), and plasma exchange dramatically reduces clinical disease in a subset of patients (Kieseier and Hartung, 2003). In particular, antibodies to myelin oligodendrocyte glycoprotein (MOG), a highly encephalitogenic glycoprotein exposed to the extracellular environment on the outer lamella of the myelin sheath (Linington et al., 1984), are found in the cerebrospinal fluid and in disintegrating myelin around axons in lesions of acute MS patients (Genain et al., 1999). Although there is still some controversy on the specificity of this antibody response in MS patients, the role of anti-MOG in those patients with pattern II demyelination has been clearly demonstrated (Lassmann et al., 2001).
We have proposed a novel mechanism for anti-MOG-induced demyelination, wherein cross-linking MOG on the surface of oligodendrocytes (OLs) in culture with demyelinating antibodies against MOG followed by secondary cross-linking antibody, rapidly (minutes) and sequentially induces (a) repartitioning of MOG into detergent insoluble microdomains characteristic of lipid rafts, (b) alterations in the phosphorylation state of key proteins related to a cellular stress response and cytoskeletal instability, and (c) dramatic changes in cell morphology including a retraction of cell processes (without triggering cell death) (Marta et al., 2003; Marta et al., 2005a ). These results were observed by using either a monoclonal antibody against the extracellular domain of MOG ( Marta et al., 2003; Marta et al., 2005a ) or with pathogenic anti-MOGs purified from mice with a B-cell mediated EAE induced by immunization with human MOG, but not with non-pathogenic anti-MOGs from mice with a B-cell independent EAE induced by immunization with rat MOG (Marta et al., 2005b ). Recognizing the important implications of these data for understanding B-cell mediated disease in MS, we sought to identify endogenous activating cross-linkers of anti-MOG/MOG complexes that would be present in human MS brain, testing the hypothesis that molecules capable of binding to the Fc portion of pathogenic IgGs can mimic the effects triggered by a secondary cross-linking antibody. We focused on microglial Fc receptors (FcRs) and complement, Fc-binding components identified as effectors of antibody-mediated demyelination (Lassmann, 2004; Noseworthy et al., 2000).
2. MATERIALS AND METHODS
Cell culture
Rat mixed primary cultures were prepared and maintained, and enriched populations of either mature OLs or microglia were obtained by a differential adhesion protocol (Bansal et al., 1996; Pfeiffer et al., 1993); purified OLs were grown in modified N2 medium (serum-free) for 6–7 days to obtain MOG-expressing OLs (Bottenstein and Sato, 1979; Gard and Pfeiffer, 1989). Freshly prepared microglia were resuspended in modified N2 medium for OL exposure (see below).
MOG cross-linking ( Marta et al., 2003; Marta et al., 2005a ; Marta, 2005b)
Purified OLs or mixed primary cultures were incubated with anti-MOG mAb 8-18C5 (IgG1) (Schluesener et al., 1987) (156 μg/ml; C. Linington, Aberdeen, Scotland) or anti-MOG Z12 (IgG2a) (Piddlesden et al., 1993) (10 μg/ml; S. Amor-P. Smith, Rijswijk, NL) for 15 min at 37°C. MOG/anti-MOG complexes were then treated with either goat anti-mouse IgG, complement components or microglia (see below). Some mixed primary cultures were only exposed to anti-MOG 8-18C5 for 15, 30 or 60 min without any further cross-linking.
Cross-linking with microglia
Following anti-MOG treatment, OLs were incubated with a suspension containing microglia (number of microglia/OLs = 0.5, 1, 2, 4 final ratio) for different intervals of time (15–60 min), or with microglia (ratio = 2) that had been previously incubated with anti-CD16/CD32 (1–10 μg/ml, mouse BD Fc Block™, BD Biosciences, Palo Alto, CA) (McMahon et al., 2002) for 30 min at 37°C. In some experiments, anti-MOG 8-18C5 was pre-crosslinked with microglia for 30 min at 37°C and then the mixture was added to OL cultures for an additional 30 min at 37°C.
Cross-linking with complement components
Following anti-MOG, anti-MAG (mouse IgG1; R Quarles, NIH, Md, USA) (Marta et al., 2004) or anti-galactocerebroside (mouse IgM, O1, 1:25) treatment (15 min, 37°C), OLs were incubated with 10–50 μg/ml of C1q (Sigma, St. Louis, MI) or 1:60 complement sera from guinea pig (Sigma) for 15, 30 or 60 min at 37°C. To deplete cholesterol, OLs were incubated with methyl-β-cyclodextrin (5 mM, Sigma, St Louis, MI) for 15 min at 37°C prior to antibody and complement treatments.
Immunofluorescence microscopy
To examine changes in morphology, mature OLs were stained with O4 (live cells) or with anti-myelin basic protein (fixed cells), as previously described (Bansal et al., 1989; Marta et al., 2005b ). The diameter of randomly chosen cells (100 cells per experiment) was determined by using a calibrated microscopic grid. FcR expression in cultured rat microglia was evaluated by staining with anti-CD64 (FcR I), -CD32 (FcR II) or -CD16 (FcR III) (BD Biosciences).
Evaluation of cell death
OL death following MOG cross-linking treatments was evaluated by propidium iodine staining (Hewett et al., 2000). The total cell number was determined by Hoechst dye staining. The percent of total cells that became propidium iodine positive was estimated by analyses of ≥30 fields from three independent experiments (100 cells were analyzed in each experiment).
Preparation of cell lysate and detergent extraction
OLs were washed three times with cold phosphate buffered saline (PBS) (microglia were detached from OLs following these washes). OLs were scraped into 150 mM NaCl, 5 mM EDTA, 25 mM Tris-Cl buffer (pH 7.5) containing 1 mM PMSF, 10 μg/ml leupeptin/aprotinin, 50 mM NaF, 10 mM NaP2O7, 1 mM Na o-Vanadate and 1% Triton X-100, centrifuged to separate them into detergent insoluble pellet and detergent soluble supernatant fractions and processed for SDS-PAGE (Marta et al., 2003).
SDS-PAGE and immunoblot analysis
Equal volumes of soluble and insoluble fractions of detergent extracts were solubilized in 50 mM Tris-HCl pH 6.8, 2.5% glycerol, 5% SDS, 4 M urea, 0.01% bromophenol blue, 10 mM DTT for SDS-PAGE (Marta et al., 2003), followed by immunoblot with anti-MOG 8–18C5, anti-phosphotyrosine (for detection of phospho-β-tubulin, 4G10, Transduction Laboratories, San Diego, CA), anti-phospho-elongation factor 2 (EF-2) (Cell Signaling Technology, Beverly, MA) or anti-active MAPK (Promega, Madison, WI).
Statistical Analysis
Means and SEMs were calculated, and evaluation of statistical significance between conditions was made using Student’s two-tail t-test.
3. RESULTS
Microglia Fc Receptors Mediate MOG Cross-Linking Induced Events
We examined the functional cross-linking ability of microglia, cells involved in both MS and the animal model EAE pathology (Raivich and Banati, 2004). Microglia (brain resident) and macrophages (recruited from blood) are rapidly concentrated at sites of inflammation, where they present antigens to T cells (Lassmann, 2004). Fc receptors on the cell surface bind to the Fc region of antibodies deposited on myelin, often leading to antibody-dependent cell-mediated cytotoxicity (ADCC), damage to myelin and phagocytosis and clearance of myelin debris. Deficiency of Fc receptor γ chain ameliorates EAE, probably due to inefficient ADCC (Abdul-Majid et al., 2002).
While cross-linking secondary antibody was required to repartition MOG from a detergent soluble into an insoluble fraction in purified OL cultures treated with anti-MOG, we noted that this requirement was eliminated in mixed primary cultures containing not only OLs, but also astrocytes and microglia (Hewett et al., 1999) (Fig. 1). Hypothesizing that microglia were the likely functional cross-linkers in these cultures, we added purified microglia as a function of dose (ratios of microglia/OLs = 0.5, 1, 2, 4) and time (15–60 min) to highly enriched cultures of OLs treated with anti-MOG (Fig. 2, and data not shown). Under these conditions, even in the absence of cross-linking secondary antibody, MOG was repartitioned into detergent insoluble microdomains (Fig. 2a) followed by the expected cascade of events characteristic of cross-linking with secondary antibody (Marta et al., 2005), including dephosphorylation of β-tubulin and increased phosphorylation of EF-2 (Fig. 2b), and morphological alterations (Fig. 2c, d). MOG was repartitioned when OLs were incubated either sequentially with first anti-MOG and then microglia (30min, ratio = 2, Fig. 2a), or with anti-MOG previously bound to microglia (data not shown). There was no indication of augmented OL death (data not shown). Treatment with microglia alone was ineffective (Fig. 2). MOG cross-linking was next carried out using microglia in which surface FcRs were blocked by anti-FcR antibodies. The three classic FcRs for IgG, FcR-I, -II and –III (Szalai and Barnum, 2004), were detected in the microglial cultures. In agreement with previous observations (Kurlander et al., 1984), after incubating microglia with blocking antibody against FcR II and III (anti-CD32/CD16), only residual surface expression of FcRI was detected (Fig. 3). When MOG cross-linking was carried out using microglia previously treated with 1 μg/ml (&MOG+Bl1) (Fig. 2a, 2b, 2cE, 2dE), 5 μg/ml (&MOG+Bl5) (Fig. 2a, 2b, 2dF) or 10 μg/ml (Fig. 2dG) of anti-CD32/CD16, the expected cascade of events (MOG repartitioning into detergent insoluble domains, phosphorylation changes in β-tubulin and EF-2, and morphological alterations) was not observed. We conclude that microglial FcRs can mediate MOG cross-linking induced events. Note that all the experiments described above were carried out in the absence of serum; therefore, effects observed do not require serum-derived components.
Figure 1.
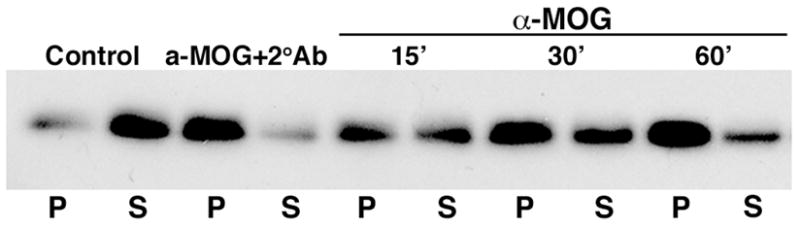
MOG repartitioning occurs independently of a secondary cross-linking antibody in mixed primary cultures. MOG immunoblot shows repartitioning of MOG into a TX-100 insoluble fraction (P) after treatment with anti-MOG 8-18C5 (15 min, 37°C) followed by anti-mouse IgG (15 min, 37°C) (α-MOG +2°Ab), or after treatment with anti-MOG alone for 15, 30 or 60 min. MOG is TX-100 soluble (S) under control conditions. The results shown are characteristic of three independent experiments.
Figure 2.
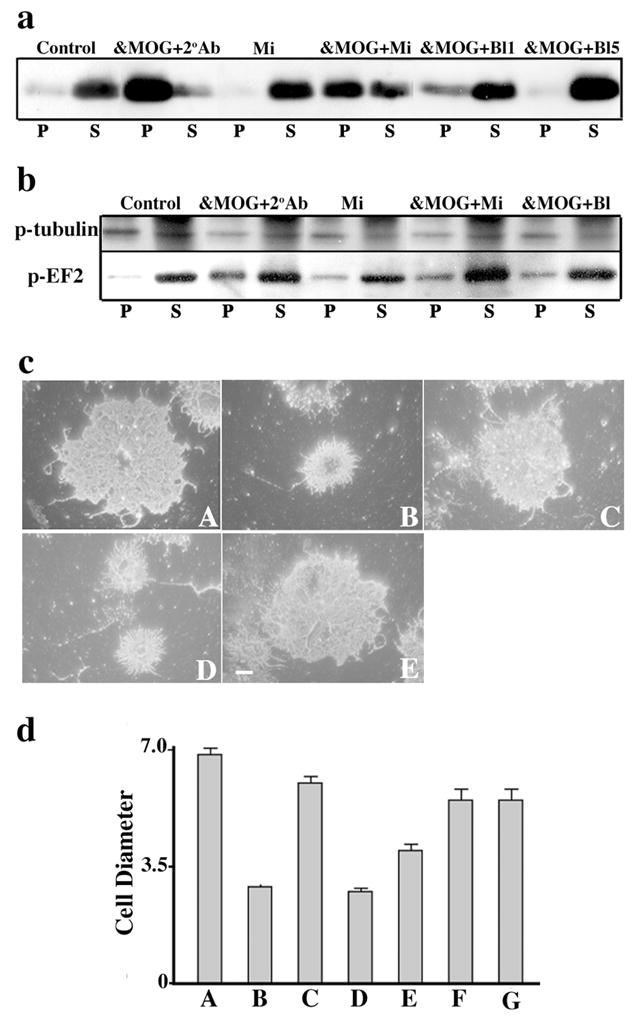
Fc Receptors mediate MOG cross-linking effects. Immunoblots show (a) MOG repartitioning into a TX-100 insoluble fraction (P) and (b) dephosphorylation of β-tubulin and increased phosphorylation of EF-2 in the pellet fraction, and (c) O4 staining shows process retraction (quantification of cell diameters shown in d) in OLs after treatment with anti-MOG 8-18C5 followed by either anti-mouse IgG (α-MOG+2°Ab, cB, dB) or microglia (α-MOG+Mi, cD, dD), but not after 30 min incubation with microglia alone (Mi, cC, dC) or anti-MOG plus microglia previously blocked with 1 (α-MOG+Bl1, dE), 5 (α-MOG+Bl5, cE, dF) or 10 (dG) μg/ml of anti-CD32/CD16. Under control conditions, MOG is mostly TX-100 soluble (S) and cells show normal morphology (cA, dA). Bar, 5 μm. dD vs. dE P < 0.05, dD vs. dF. p < 0.005 (Student’s t-test). The results shown are characteristic of three independent experiments.
Figure 3.

FcR I, II and III are expressed in cultured microglia. Immunostaining with anti-CD64 (A,D), anti-CD32 (B,E) or anti-CD16 (C,F) shows microglial surface expression of FcR I (A), II (B) and III (C), respectively. Only low levels of FcR I are detected after FcR block by anti-CD32/CD16 (5 μg/ml, D–F). Asterisks show position of cell nuclei. Bar, 10 μm.
Complement Does Not Mediate MOG Cross-Linking Effects
Complement activation products are deposited in areas of demyelination in both MS and EAE, causing cell lysis and damage to myelin membrane (Compston et al., 1989; von Budingen et al., 2004). Further, depletion of complement components ameliorates EAE in rats (Linington et al., 1989). On the other hand, complement can enhance OL survival (Soane et al., 2001). Gene manipulation of specific complement components in rodents has produced contradictory results. For example, although deficiency of complement component C6 reduces disease (Tran et al., 2002), down-regulation of component C4 does not affect the progression of EAE (Boos et al., 2005). Further, and of particular interest to the present study, MOG-related damage to myelin-like OL membranes in vitro occurs even in the absence of complement (Dyer and Matthieu, 1994).
The first component of the complement cascade activation, C1, is a complex of three proteins, C1q, C1r and C1s. Binding of multivalent C1q to either a single bound IgM or multiple bound IgGs leads to sequential activation of C1r and C1s, launching the full cascade. Therefore, we reasoned that cross-linking MOG by treatment with anti-MOG followed by C1q could in principle mimic secondary cross-linking antibody (above). However, upon treating OLs with anti-MOG monoclonal antibody 8-18C5, an IgG1 that fixes complement (Scolding et al., 1989), followed by C1q (10 μg/ml, 15–30 min) neither MOG redistribution into detergent insoluble microdomains (Fig. 4a), tyrosine dephosphorylation of β-tubulin (Fig. 4b), nor retraction of OL processes (Fig. 4eE) was observed. As a control for cellular physiological integrity, the activation of MAPK that results from exposure of OLs to anti-MOG without secondary cross-linking (Marta et al., 2005a ) was still observed after anti-MOG plus C1q treatment (Fig. 4b). Similar results were obtained using higher doses of C1q (data not shown),
Figure 4.

Neither C1q nor whole complement mimic the effects of a secondary cross-linking antibody. Immunoblots show (a,c) MOG repartitioning into a TX-100 insoluble fraction (P) and (b,d) dephosphorylation of β-tubulin, and (e) O4 staining shows process retraction in OLs after treatment with anti-MOG 8-18C5 followed by anti-mouse IgG (α-MOG+2°Ab, eD). This was not the case after incubation with C1q alone (15 min, a,b,eB) or complement alone (30 min, c,d,eC), or after anti-MOG followed by C1q for 15 (a,b,eE) or 30 (a) min. (b,d) MAPK activation is observed upon treatment with either anti-MOG or complement. Although MOG in OLs is mostly TX-100 soluble (S) and there is no change in the phosphorylation of β-tubulin after treatment with anti-MOG followed by complement for 15 (c), 30 (c,d) or 60 (c) min, OLs show significant morphological alterations (eF). This was non-specific and occurred after treatment with either anti-galactocerebroside (eG) or anti-MAG (eH) followed by complement. Under control conditions, cells show normal morphology (eA). Bar, 5 μm. (f) MOG is also repartitioned into the TX-100 insoluble fraction (P) after treatment with anti-MOG Z12 followed by anti-mouse IgG (as for 8-18C5 + 2°Ab), but not after anti-MOG Z12 followed by complement. The results shown are characteristic of three independent experiments.
Reasoning that these results could be due to an inefficiency of C1q alone to cross-link MOG/anti-MOG, we examined whole guinea pig complement at sublytic doses (1:60, 15–60 min), whereby other components (e.g., C1r, C1s) may cooperate with C1q to cross-link MOG. Again, this treatment did not increase the association of MOG with detergent insoluble microdomains or lead to dephosphorylation of tubulin (Fig 4c, d), nor were the phosphorylation states of other signaling molecules responding to MOG cross-linking with secondary antibody (Marta et al., 2005a ) modified (data not shown). At these doses and times of exposure, complement alone did not significantly increase OL death (data not shown). However, two confounding factors were that, as previously reported, whole complement alone caused an increase in the phosphorylation of MAPK (Soane et al., 2001) (Fig. 4d), and OLs treated with anti-MOG plus complement underwent significant morphological alterations (Fig. 4eF) (Scolding et al., 1989). However, it is to be noted that OLs treated with other antibodies plus complement also underwent similar morphological alterations [e.g., anti-galactocerebroside (Fig. 4eG), anti-MAG (Fig. 4eH)], suggesting that complement cross-linking is a different phenomenon than second antibody cross-linking (Marta et al., 2004). Specifically, in the case of second antibody cross-linking, morphological alterations had occurred specifically upon treatment with anti-MOG but not with anti-myelin associated glycoprotein (MAG) (Marta et al., 2004) showing specificity of these interactions. Further, while morphological alterations induced by anti-MOG plus secondary antibody were prevented by prior disruption of lipid rafts [pre-treatment with methyl-β-cyclodextrin (MβCD, Fig. 5F) (Marta et al., 2005a )], those triggered by complement were not (Fig. 5H). We carried out similar experiments using anti-MOG Z12 (IgG2a), an antibody that fixes complement more efficiently than 8-18C5 (Piddlesden et al., 1993). Again, although MOG repartitioning was observed when anti-MOG Z12 was cross-linked with secondary antibody, this was not the case when cross-linking was attempted using complement (Fig. 4f). We conclude that complement is unable to mimic the MOG cross-linking effects observed using a secondary antibody or microglia.
Figure 5.
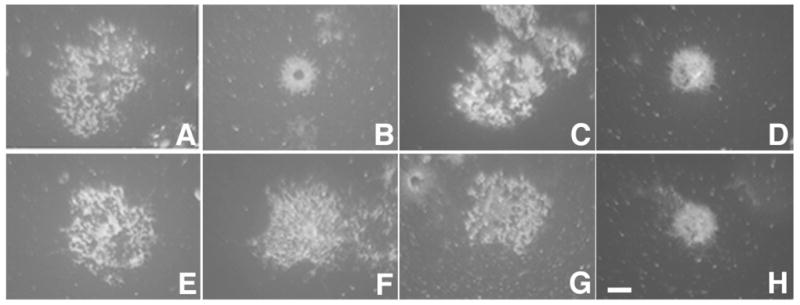
Complement induces morphological alterations by a different mechanism than a secondary cross-linking antibody. Myelin basic protein staining shows process retraction of OLs treated with anti-MOG plus secondary antibody (B) or OLs treated with anti-MOG plus complement [with (H) or without (D) pre-treatment with methyl-β-cyclodextrin (MβCD)]. Cells show normal morphology under control conditions (A) or after treatment with complement alone (C), MβCD alone (E), or MβCD plus complement (G). In agreement with previous observations, pre-treatment with MβCD prevented the morphological alterations in OLs treated with anti-MOG plus secondary antibody (F). Bar, 5 μm.
4. DISCUSSON
These data demonstrate that the anti-MOG mediated perturbations of OL physiology we have shown previously using secondary cross-linking antibody (Marta et al., 2005a ; Marta et al., 2005b ; Marta et al., 2003) can be mediated by Fc receptors on the surface of microglia. This suggests a novel function for microglial/macrophage Fc receptors during the process of demyelination.
In addition to inflammatory events in MS plaques per se, more subtle leukocytic infiltrates, demyelination, gliosis and axonal damage also can be detected in ‘normal appearing white matter’ (NAWM), suggesting that the difference between plaques and NAWM is a quantitative one (Bjartmar et al., 2003; Griffin et al., 2002). The mechanism we describe here could contribute to early pathological events in these areas, including loss of myelin integrity. Under these conditions, complement-mediated lysis and ADCC would not predominate, but rather resident microglia and small macrophage infiltrates (Raivich and Banati, 2004) would be sufficient to trigger anti-MOG lipid raft-mediated signaling and the associated downstream events observed in our previous model by cross-linking with a secondary antibody (Marta et al., 2003; Marta et al., 2005 a; Marta et al., 2005b ). This cascade of events does not result in increased OL death, as observed in MS plaques (Lassmann, 2004), but rather leads to the initiation of a cellular stress response and instability of the cytoskeleton (Marta et al., 2005a ), events that can significantly contribute to MS pathology.
Thus, a model can be proposed in which upon disruption of the permeability of the blood brain barrier in early MS, high levels of anti-MOG antibodies access the brain. While in active sites of inflammation, abundant infiltrates of macrophages, activated microglia and complement form complexes with anti-MOG antibodies to trigger demyelination and OL death, in sites with relatively intact blood brain barrier and low inflammation, circulating anti-MOG antibodies could get cross-linked by resident FcRs leading to the series of events described in this study.
In summary, we show that microglial Fc receptors are a likely source of a functional, endogenous cross-linker, one clearly present, and in fact elevated, in MS. In addition to providing a viable, physiologically relevant model for B-cell mediated disease in MS, these data suggest an important cellular/molecular focus for possible novel therapeutic interventions.
Acknowledgments
We thank Christopher Linington (University of Aberdeen, UK), and Paul Smith and Sandra Amor (Rijswijk, NL), for providing anti-MOG antibodies 8-18C5 and Z12 anti-MOG, respectively. This work was supported by grants NS10861 and NS41078 from the National Institutes of Health (SP, RB), and RG 3704-A-11 (SP, RB) and FG1423A (CBM) from the National Multiple Sclerosis Society. We would like to dedicate this manuscript to the memory of Dr Steven Pfeiffer who was an excellent mentor and continuous source of scientific inspiration.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abdul-Majid KB, Stefferl A, Bourquin C, Lassmann H, Linington C, Olsson T, Kleinau S, Harris RA. Fc receptors are critical for autoimmune inflammatory damage to the central nervous system in experimental autoimmune encephalomyelitis. Scand J Immunol. 2002;55:70–81. doi: 10.1046/j.1365-3083.2002.01024.x. [DOI] [PubMed] [Google Scholar]
- Bansal R, Kumar M, Murray K, Morrison RS, Pfeiffer SE. Regulation of FGF receptors in the oligodendrocyte lineage. Mol Cell Neurosci. 1996;7:263–275. doi: 10.1006/mcne.1996.0020. [DOI] [PubMed] [Google Scholar]
- Bansal R, Warrington AE, Gard AL, Ranscht B, Pfeiffer SE. Multiple and novel specificities of monoclonal antibodies O1, O4, and R- mAb used in the analysis of oligodendrocyte development. J Neurosci Res. 1989;24:548–557. doi: 10.1002/jnr.490240413. [DOI] [PubMed] [Google Scholar]
- Bjartmar C, Wujek JR, Trapp BD. Axonal loss in the pathology of MS: consequences for understanding the progressive phase of the disease. J Neurol Sci. 2003;206:165–171. doi: 10.1016/s0022-510x(02)00069-2. [DOI] [PubMed] [Google Scholar]
- Boos LA, Szalai AJ, Barnum SR. Murine complement C4 is not required for experimental autoimmune encephalomyelitis. Glia. 2005;49:158–160. doi: 10.1002/glia.20093. [DOI] [PubMed] [Google Scholar]
- Bottenstein JE, Sato GH. Growth of a rat neuroblastoma cell line in serum-free supplemented medium. Proceedings of the National Academy of Sciences of the United States of America. 1979;76:514–517. doi: 10.1073/pnas.76.1.514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Compston DA, Morgan BP, Campbell AK, Wilkins P, Cole G, Thomas ND, Jasani B. Immunocytochemical localization of the terminal complement complex in multiple sclerosis. Neuropathol Appl Neurobiol. 1989;15:307–316. doi: 10.1111/j.1365-2990.1989.tb01231.x. [DOI] [PubMed] [Google Scholar]
- Dyer CA, Matthieu JM. Antibodies to myelin/oligodendrocyte-specific protein and myelin/oligodendrocyte glycoprotein signal distinct changes in the organization of cultured oligodendroglial membrane sheets. J Neurochem. 1994;62:777–787. doi: 10.1046/j.1471-4159.1994.62020777.x. [DOI] [PubMed] [Google Scholar]
- Gard AL, Pfeiffer SE. Oligodendrocyte progenitors isolated directly from developing telencephalon at a specific phenotypic stage: myelinogenic potential in a defined environment. Development. 1989;106:119–132. doi: 10.1242/dev.106.1.119. [DOI] [PubMed] [Google Scholar]
- Genain CP, Cannella B, Hauser SL, Raine CS. Identification of autoantibodies associated with myelin damage in multiple sclerosis. Nat Med. 1999;5:170–175. doi: 10.1038/5532. [DOI] [PubMed] [Google Scholar]
- Griffin CM, Chard DT, Parker GJ, Barker GJ, Thompson AJ, Miller DH. The relationship between lesion and normal appearing brain tissue abnormalities in early relapsing remitting multiple sclerosis. J Neurol. 2002;249:193–199. doi: 10.1007/pl00007864. [DOI] [PubMed] [Google Scholar]
- Hewett JA, Hewett SJ, Winkler S, Pfeiffer SE. Inducible nitric oxide synthase expression in cultures enriched for mature oligodendrocytes is due to microglia. J Neurosci Res. 1999;56:189–198. doi: 10.1002/(sici)1097-4547(19990415)56:2<189::aid-jnr8>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- Hewett SJ, Uliasz TF, Vidwans AS, Hewett JA. Cyclooxygenase-2 contributes to N-methyl-D-aspartate-mediated neuronal cell death in primary cortical cell culture. J Pharmacol Exp Ther. 2000;293:417–425. [PubMed] [Google Scholar]
- Kieseier BC, Hartung HP. Current disease-modifying therapies in multiple sclerosis. Semin Neurol. 2003;23:133–146. doi: 10.1055/s-2003-41138. [DOI] [PubMed] [Google Scholar]
- Kurlander RJ, Ellison DM, Hall J. The blockade of Fc receptor-mediated clearance of immune complexes in vivo by a monoclonal antibody (2.4G2) directed against Fc receptors on murine leukocytes. J Immunol. 1984;133:855–862. [PubMed] [Google Scholar]
- Lassmann H. Cellular Damage and Repair in Multiple Sclerosis. In: Lazzarini RA, editor. Myelin Biology and Disorders. Vol. 2. Elsevier Academic Press; San Diego: 2004. pp. 733–762. [Google Scholar]
- Lassmann H, Bruck W, Lucchinetti C. Heterogeneity of multiple sclerosis pathogenesis: implications for diagnosis and therapy. Trends Mol Med. 2001;7:115–121. doi: 10.1016/s1471-4914(00)01909-2. [DOI] [PubMed] [Google Scholar]
- Linington C, Morgan BP, Scolding NJ, Wilkins P, Piddlesden S, Compston DA. The role of complement in the pathogenesis of experimental allergic encephalomyelitis. Brain. 1989;112 ( Pt 4):895–911. doi: 10.1093/brain/112.4.895. [DOI] [PubMed] [Google Scholar]
- Linington C, Webb M, Woodhams PL. A novel myelin associated glycoprotein defined by a mouse monoclonal antibody. J Neuroimmunol. 1984;6:387–396. doi: 10.1016/0165-5728(84)90064-x. [DOI] [PubMed] [Google Scholar]
- Marta CB, Taylor CM, Coetzee T, Kim T, Winkler S, Bansal R, Pfeiffer SE. Antibody cross-linking of myelin oligodendrocyte glycoprotein leads to its rapid repartitioning into detergent-insoluble fractions, and altered protein phosphorylation and cell morphology. J Neurosci. 2003;23:5461–5471. doi: 10.1523/JNEUROSCI.23-13-05461.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marta CB, Taylor CM, Cheng S, Quarles R, Bansal R, Pfeiffer SE. Myelin associated glycoprotein cross-linking triggers its partitioning into lipid rafts, specific signaling events and cytoskeletal rearrangements in oligodendrocytes. Neuron Glia Biology. 2004;1:35–46. doi: 10.1017/s1740925x04000067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marta CB, Montano MB, Taylor CM, Taylor AL, Bansal R, Pfeiffer SE. Signaling cascades activated upon antibody cross-linking of myelin oligodendrocyte glycoprotein: potential implications for multiple sclerosis. J Biol Chem. 2005 a;280:8985–8993. doi: 10.1074/jbc.M413174200. [DOI] [PubMed] [Google Scholar]
- Marta CB, Oliver AR, Sweet RA, Pfeiffer SE, Ruddle NH. Pathogenic myelin oligodendrocyte glycoprotein antibodies recognize glycosylated epitopes and perturb oligodendrocyte physiology. Proc Natl Acad Sci. 2005 b;102:13992–13997. doi: 10.1073/pnas.0504979102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon EJ, Suzuki K, Matsushima GK. Peripheral macrophage recruitment in cuprizone-induced CNS demyelination despite an intact blood-brain barrier. J Neuroimmunol. 2002;130:32–45. doi: 10.1016/s0165-5728(02)00205-9. [DOI] [PubMed] [Google Scholar]
- Noseworthy JH, Lucchinetti C, Rodriguez M, Weinshenker BG. Multiple sclerosis. N Engl J Med. 2000;343:938–952. doi: 10.1056/NEJM200009283431307. [DOI] [PubMed] [Google Scholar]
- Pfeiffer SE, Warrington AE, Bansal R. The oligodendrocyte and its many cellular processes. Trends Cell Biol. 1993;3:191–197. doi: 10.1016/0962-8924(93)90213-k. [DOI] [PubMed] [Google Scholar]
- Piddlesden SJ, Lassmann H, Zimprich F, Morgan BP, Linington C. The demyelinating potential of antibodies to myelin oligodendrocyte glycoprotein is related to their ability to fix complement. Am J Pathol. 1993;143:555–564. [PMC free article] [PubMed] [Google Scholar]
- Raivich G, Banati R. Brain microglia and blood-derived macrophages: molecular profiles and functional roles in multiple sclerosis and animal models of autoimmune demyelinating disease. Brain Res Brain Res Rev. 2004;46:261–281. doi: 10.1016/j.brainresrev.2004.06.006. [DOI] [PubMed] [Google Scholar]
- Schluesener HJ, Sobel RA, Linington C, Weiner HL. A monoclonal antibody against a myelin oligodendrocyte glycoprotein induces relapses and demyelination in central nervous system autoimmune disease. J Immunol. 1987;139:4016–4021. [PubMed] [Google Scholar]
- Scolding NJ, Morgan BP, Houston WA, Linington C, Campbell AK, Compston DA. Vesicular removal by oligodendrocytes of membrane attack complexes formed by activated complement. Nature. 1989;339:620–622. doi: 10.1038/339620a0. [DOI] [PubMed] [Google Scholar]
- Soane L, Cho HJ, Niculescu F, Rus H, Shin ML. C5b-9 terminal complement complex protects oligodendrocytes from death by regulating Bad through phosphatidylinositol 3-kinase/Akt pathway. J Immunol. 2001;167:2305–2311. doi: 10.4049/jimmunol.167.4.2305. [DOI] [PubMed] [Google Scholar]
- Szalai AJ, Barnum SR. Fc receptors and the common gamma-chain in experimental autoimmune encephalomyelitis. J Neurosci Res. 2004;75:597–602. doi: 10.1002/jnr.20023. [DOI] [PubMed] [Google Scholar]
- Tran GT, Hodgkinson SJ, Carter N, Killingsworth M, Spicer ST, Hall BM. Attenuation of experimental allergic encephalomyelitis in complement component 6-deficient rats is associated with reduced complement C9 deposition, P-selectin expression, and cellular infiltrate in spinal cords. J Immunol. 2002;168:4293–4300. doi: 10.4049/jimmunol.168.9.4293. [DOI] [PubMed] [Google Scholar]
- von Budingen HC, Hauser SL, Ouallet JC, Tanuma N, Menge T, Genain CP. Frontline: Epitope recognition on the myelin/oligodendrocyte glycoprotein differentially influences disease phenotype and antibody effector functions in autoimmune demyelination. Eur J Immunol. 2004;34:2072–2083. doi: 10.1002/eji.200425050. [DOI] [PubMed] [Google Scholar]