

Abstract
Nucleus accumbens-1 (Nac1 or NAC-1) belongs to the BTB/POZ transcription factor family and is a novel protein that potentially participates in self-renewal and pluripotency in embryonic stem cells. In human cancer, NAC-1 is upregulated in several types of neoplasms, but particularly in recurrent chemoresistant ovarian carcinomas, suggesting a biological role for NAC-1 in the development of drug resistance in ovarian cancer. We have assessed this possibility and demonstrated a correlation between NAC-1 expression and ex vivo paclitaxel resistance in ovarian serous carcinoma tissues and cell lines. We found that expression of Gadd45gamma-interacting protein 1 (Gadd45gip1), a downstream target negatively regulated by NAC-1, was reduced in paclitaxel-resistant cells. Ectopic expression of NAC-1 or knockdown of Gadd45gip1 conferred paclitaxel resistance, while NAC-1 knockdown or ectopic expression of Gadd45gip1 increased paclitaxel sensitivity. Furthermore, silencing NAC-1 expression or disrupting NAC-1 homodimerization by a dominant negative NAC-1 protein that contained only the BTB/POZ domain induced expression of Gadd45gamma which interacted with Gadd45gip1. Reducing Gadd45gamma expression by shRNAs partially enhanced paclitaxel resistance. Thus, this study provides new evidence that NAC-1 upregulation and homodimerization contribute to tumor recurrence by equipping ovarian cancer cells with the paclitaxel-resistant phenotype through negative regulation of the Gadd45 pathway.
Keywords: Ovarian, NAC-1, chemoresistance, paclitaxel
Introduction
Ovarian cancer represents the most lethal gynecological malignant disease in the United States with an estimated 22,430 new cases and 15,280 deaths in 2007 (Jemal et al., 2007). Unlike other histological types of ovarian epithelial tumors, high-grade serous carcinoma is the most aggressive and most rapidly growing neoplasm, and has a 5-year survival rate below 30%. Patients with high-grade serous carcinomas always present their diseases at advanced stages. These patients routinely receive cytoreduction surgery followed by a combined carboplatin/paclitaxel chemotherapy regimen. Despite initial responsiveness to the chemotherapeutic drugs, most patients eventually develop chemoresistant tumors and succumb to their diseases. Therefore, treatment failure is often attributed to primary or acquired resistance to chemotherapeutic agents, which remains a clinically formidable problem in managing cancer patients. Understanding chemoresistance at the molecular level should illuminate fundamental properties of drug resistance and provide new therapeutic targets to treat advanced neoplastic diseases.
In an effort to elucidate the molecular etiology of drug resistance, we have previously identified and characterized the nucleus accumbens-1 (Nac1 or NAC-1) as one of the candidate chemoresistance genes in ovarian cancer (Nakayama et al., 2006). NAC-1 is a nuclear protein belonging to the BTB/POZ (Pox virus and Zinc finger/Bric-a-brac Tramtrack Broad complex) domain family. NAC-1 was originally identified and cloned as a novel transcript from the nucleus accumbens, a unique forebrain structure involved in reward motivation and addictive behaviors (Cha et al., 1997; Kalivas et al., 1999; Koob, 1996; Mackler et al., 2000), and it was later shown to be involved in the acute behavioral and neurochemical responses to psychomotor stimulants (Mackler et al., 2008). Recently, NAC-1 has been found to be one of the factors associated with pluripotency in embryonic stem cells. NAC-1 transcriptionally regulates gene expression of other pluripotency factors including Nanog, Oct4, Sox2, Klf4, Sall1 and Sall4, and participates in constituting the protein interaction and transcriptional regulatory networks that are critical to establish and/or maintain embryonic stem cell pluripotency (Kim et al., 2008; Muller et al., 2008; Wang et al., 2006). In human cancer, NAC-1 is upregulated in several neoplasms including ovarian high-grade serous carcinoma, pancreatic carcinoma, colorectal carcinoma, breast carcinoma, renal cell carcinoma, hepatocellular carcinoma and cervical cancer (Nakayama et al., 2006; Yeasmin et al., 2008). In ovarian high-grade serous carcinomas, the levels of NAC-1 expression were significantly higher in recurrent post-chemotherapy samples than in primary previously untreated tumors (Davidson et al., 2007; Nakayama et al., 2006). Furthermore, intense NAC-1 immunoreactivity in primary ovarian serous carcinomas was associated with early recurrence, suggesting that NAC-1 is potentially involved in the pathogenesis of drug resistance.
At the molecular level, NAC-1 proteins form homodimers through the BTB/POZ domain, and this process is essential for tumor cell growth and survival in tumors with NAC-1 overexpression (Nakayama et al., 2006). NAC-1 has been shown to interact with nuclear proteins including CoRest and histone deacetylases, HDAC3 and HDAC4, in which the binding with deacetylases is mediated by a unique BEN domain on NAC-1 protein (Abhiman et al., 2008; Korutla et al., 2007; Korutla et al., 2005). Ectopic expression of full-length NAC-1 proteins is sufficient to enhance tumorigenecity of ovarian surface epithelial cells and of NIH3T3 cells in athymic nu/nu mice. We have previously identified growth arrest and DNA-damage inducible 45-γ interacting protein (Gadd45gip1) as one of the genes negatively regulated by NAC-1, and have demonstrated that NAC-1 contributes to tumor growth and survival at least in part by inhibiting Gadd45gip1 expression (Nakayama et al., 2007). The above findings strongly suggest that NAC-1 is a tumor recurrence-associated gene in high-grade ovarian serous carcinomas. In this study, by correlating NAC-1 expression and drug resistance status, we determined that NAC-1 causally contributes to chemoresistance for paclitaxel and carboplatin and showed that the Gadd45 pathway is involved in NAC-1-mediated chemoresistance in cancer cells.
Results
Correlation of NAC-1 expression and paclitaxel-resistance in ovarian carcinoma
Three ovarian cancer cell lines, SKOV3, OVCAR3, and A2780, were used to generate paclitaxel resistant cells. Dose-dependent growth inhibition effects by paclitaxel are shown in Figure 1A. The IC50s of paclitaxel at 96 hours were less than 0.1 nM in all 3 parental naive cell lines, but were more than 5,000 nM in all 3 paclitaxel-resistant cell lines. Quantitative real-time RT-PCR was used to determine NAC-1 transcript levels in resistant and parental naïve cells from each cell line. As shown in Figure 1B, NAC-1 was highly expressed in paclitaxel resistant cells as compared to parental cells in all three different cell lines (Student’s t test p value < 0.001). To extrapolate the above findings to clinical specimens, we performed immunohistochemistry using an anti-NAC-1 antibody on a total of 113 high-grade ovarian serous carcinomas for which the ex vivo drug response status was available. We showed a correlation between the intensity of NAC-1 immunoreactivity and an extreme paclitaxel resistance status (p= 0.05) (Table 1). In contrast, there was no significant correlation (p= 0.43) between carboplatin resistance and NAC-1 immunointensity. Therefore, we focused on the mechanism underlying how NAC-1 participates in paclitaxel resistance in ovarian cancer.
Fig. 1. Upregulation of NAC-1 transcript in paclitaxel-resistant ovarian cancer cell lines including SKOV3, OVCAR3 and A2780.
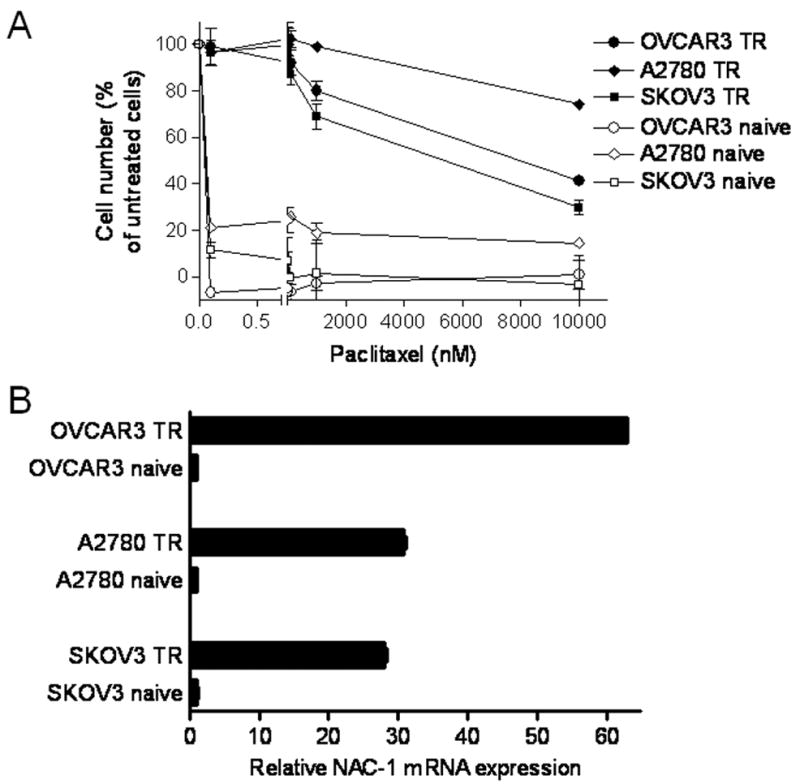
A. Dose-dependent decrease in cell number in the presence of paclitaxel. Paclitaxel-resistant cells (TR) are less sensitive to paclitaxel than are the parental (naïve) controls. B. Paclitaxel-resistant cell lines were analyzed for NAC-1 mRNA expression using quantitative real-time RT-PCR. NAC-1 expression levels were normalized to the GAPDH expression level. NAC-1 is up-regulated in all paclitaxel-resistant cell lines.
Table 1.
Correlation of NAC-1 immunoreactivity and drug resistance status in ovarian cancer tissues.
| Taxol-EDR | Taxol-LDR | |
|---|---|---|
| positive staining | 51 | 41 |
| negative staining | 7 | 14 |
| Carbo-EDR | Carbo-LDR | |
|---|---|---|
| positive staining | 44 | 48 |
| negative staining | 9 | 12 |
Fisher’s Exact Test P -value = 0.05
Fisher’s Exact Test P-value = 0.43
NAC-1 overexpression confers paclitaxel resistance
To assess whether or not NAC-1 expression causally contributes to paclitaxel resistance, we used both forward (overexpression) and reverse (gene knockdown) approaches. First, NIH3T3 cells were transfected with a NAC-1 expression vector and stable NAC-1 expressing clones were selected. As shown in Figure 2A, three independent NAC-1 expressing clones were more resistant to paclitaxel-induced growth suppression than a clone transfected with vector alone. The IC50s of paclitaxel at 96 hours were 785, 755, and 692 nM in NAC-1 expressing clones in contrast to 59 nM in the control clone (Fig. 2A, upper panel). Growth curve analysis demonstrated an increase in proliferation rate of NAC-1-expressing clones as compared to the control cells in the presence of paclitaxel (Fig. 2A, lower panel). Next, we applied shRNAs targeting NAC-1 in SKOV3 cells that expressed high levels of NAC-1 protein. The efficiency of shRNAs in reducing NAC-1 protein expression is shown in Fig. 2B. Similar to our previous finding (Nakayama et al., 2006), NAC-1 knockdown resulted in growth suppression in parental SKOV3 cells but not in paclitaxel resistant SKOV3 cells (supplementary Fig. 1A). The sensitivity to paclitaxel was then determined in control and NAC-1 shRNA treated paclitaxel-resistant SKOV3 cells. We found that NAC-1 knockdown increased paclitaxel sensitivity, especially at higher concentrations of paclitaxel. The IC50s of paclitaxel at 96 hours were 2839 and 3275 nM in shNAC1-1 and shNAC1-3-treated groups, resepectively, and 12,087 nM in the control shRNA-treated cells (Fig. 2C).
Fig. 2. NAC-1 expression is sufficient and essential to induce paclitaxel resistance.

A. Upper panel: three NAC-1 expressing NIH3T3 clones (C1, C2, and C3) show an increased resistance to paclitaxel as compared to the vector only control. Lower panel: the effect of paclitaxel on cell growth in both control and resistant NIH3T3 clones. B. The efficiency of two NAC-1 shRNAs (-1 and -3) in reducing NAC-1 expression was determined by quantitative real-time RT-PCR in SKOV3 cells. C. NAC-1 shRNA significantly sensitized paclitaxel-resistant SKOV3 cells to paclitaxel treatment. The effect is more pronounced at higher drug concentrations.
Gadd45gip1 is associated with paclitaxel resistance in ovarian cancer cells
NAC-1 is thought to be a transcription regulator and has been shown to negatively control Gadd45gip1 expression (Nakayama et al., 2007). Thus, it is likely that Gadd45gip1 may contribute, at least in part, to the paclitaxel resistance phenotype. To test this hypothesis, we first compared the mRNA levels of Gadd45gip1 between paclitaxel-resistant and parental naive ovarian cancer cell lines, and found that the Gadd45gip1 transcript level was significantly reduced in paclitaxel-resistant cells as compared to their naive counterparts in all three tested cell lines (Fig. 3A). Next, we knocked down Gadd45gip1 expression by shRNA to determine if reduced Gadd45gip1 level will enhance paclitaxel resistance. Figure 3B demonstrates that the mRNA levels of Gadd45gip1 were significantly reduced 48 hrs after treating paclitaxel-resistant SKOV3 cells with two Gadd45gip1 shRNA. Growth curve analysis demonstrated that Gadd45gip1 shRNA transfection did not significantly alter the proliferation rate in either paclitaxel-resistant or control SKOV3 cells (supplementary Fig. 1B). However, Gadd45gip1 shRNAs decreased paclitaxel sensitivity in paclitaxel-resistant SKOV3 cells. The IC50s of paclitaxel (measured after 96 hours) were 3,108 nM in the Gadd45gip1-knockdown paclitaxel-resistant cells, and 1,805 nM in the control paclitaxel-resistant cells (Fig. 3C). Likewise, Gadd45gip1 shRNAs also decreased paclitaxel sensitivity in naive SKOV3 cells (data not shown). Furthermore, we overexpressed Gadd45gip1 in paclitaxel-resistant SKOV3 cells using the retrovirus delivery system. Western blot analysis showed Gadd45gip1 protein expression in Gadd45gip1 virus transduced cells but not in PWZL (vector control) virus transduced cells (Fig. 4A). Forty-eight hours after viral infection, cells were incubated with serial concentrations of paclitaxel and the IC50 was measured 4 days later. We found that ectopic Gadd45gip1 expression significantly increased the paclitaxel sensitivity in paclitaxel-resistant SKOV3 cells. The IC50s of paclitaxel were 2,320 nM in Gadd45gip1-overexpressed paclitaxel-resistant cells, and 35,512 nM in the control paclitaxel-resistant cells (Fig. 4B).
Fig. 3. The role of Gadd45gip1 in NAC-1-mediated paclitaxel resistance.

A. The level of Gadd45gip1 mRNA as determined by quantitative real-time RT-PCR was decreased in paclitaxel-resistant OVCAR3, A2780 and SKOV3 cells as compared to the parental naïve counterparts. B. The efficiency of Gadd45gip1 shRNA in reducing its mRNA level is demonstrated by quantitative real-time RT-PCR in paclitaxel-resistant SKOV3 cells. The level of Gadd45gip1 expression was normalized to that of GAPDH.
C. Reducing Gadd45gip1 by shRNA in cells increases paclitaxel resistance as compared to control shRNA treatment.
Fig. 4. Gadd45gip1 overexpression reduced paclitaxel-resistant property in paclitaxel chemoresistant cells.
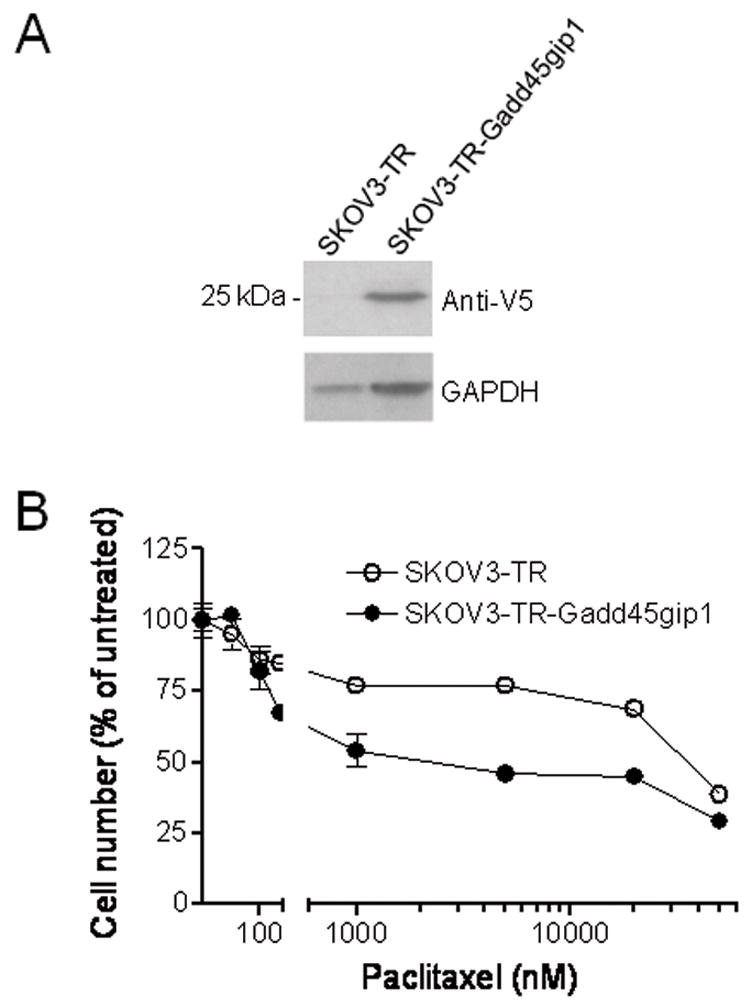
Paclitaxel-resistant SKOV3 (TR) cells were infected with Gadd45gip1-V5 expressing retrovirus or control virus containing only the pWZL vector. A. Western blot demonstrates Gadd45gip1-V5 expression in Gadd45gip1-V5 retrovirus-infected SKOV3-TR cells. B. Forty-eight hours after infection, cells were incubated with a series of concentrations of paclitaxel and the IC50 was measured 4 days later. Ectopic Gadd45gip1 expression increased the paclitaxel sensitivity in SKOV3-TR cells.
The role of Gadd45γ in the development of paclitaxel resistance
Because Gadd45gip1 interacts with all three Gadd45 isoforms including Gadd45α, Gadd45β and Gadd45γ, we determined if Gadd45 protein(s) was also regulated by NAC-1. For these experiments we used SKOV3 cells which were engineered with an N130 Tet-off inducible system. N130 is a truncated NAC-1 protein (1–130 a.a.) containing only the BTB/POZ domain. It has been shown that N130 proteins competitively interact with full-length NAC-1 molecules, preventing them from forming NAC-1 homodimers and induce cell death (Nakayama et al., 2006). We also generated N130mut by random mutagenesis and the sequence of N130mut comparing to the N130wt is shown in supplementary Fig. 2A. We established both N130wt and N130mut inducible SKOV3 cell lines using the Tet-off system. We found that unlike N130wt protein, N130mut protein was not co-localized with endogenous NAC-1 (Fig. 5) and did not exert a growth inhibitory effect (supplementary Fig. 2B). Next, we assessed gene expression in three Gadd45 isoforms after N130wt or N130mut induction based on the assumption that NAC-1 signaling could also affect the expression levels of Gadd45gip1 binding partners for an efficient regulation of the function in Gadd45/Gadd45gip1 complex. As shown in Figure 6A, Gadd45γ expression increased 24 hrs after N130 induction. We also found that Gadd45α mRNA levels increased in the first 24 hrs then decreased afterwards, whereas Gadd45β expression did not show any significant change after N130 induction (data not shown). In contrast, induction of N130mut did not have any effect on the expression of all the Gadd45 isoforms. The effect of N130mut on Gadd45γ is shown in Fig. 6A. Next, we transfected SKOV3 and HeLa cells with NAC-1 shRNA and observed that the copy number of Gadd45γ transcript increased after NAC-1 knockdown (Fig. 6B). In SKOV3 cells, a small reduction in NAC-1 RNA caused a mild (but significant) increase in GADD45-γ expression, while in HeLa cells, a more efficient in NAC-1 knockdown led to a larger increase in GADD45-γ expression. Thus, the findings based on both NAC-1 knockdown and N130mut approaches indicated that NAC-1 expression and homodimerization were essential to suppress Gadd45γ expression. We also determined if Gadd45γ was causally involved in developing paclitaxel resistance based on IC50 curve analysis after gene knockdown. We found that both paclitaxel-resistant and naïve cells in the Gadd45γ knockdown group became more resistant to paclitaxel-induced growth suppression than the control treated group. The IC50s of paclitaxel at 96 hours were ~12,000 nM in Gadd45γ– knockdown paclitaxel-resistant cells, and ~6,000 nM in the control paclitaxel-resistant cells (supplementary Fig. 3).
Fig. 5. N130wt but not N130mut co-localizes with NAC-1.
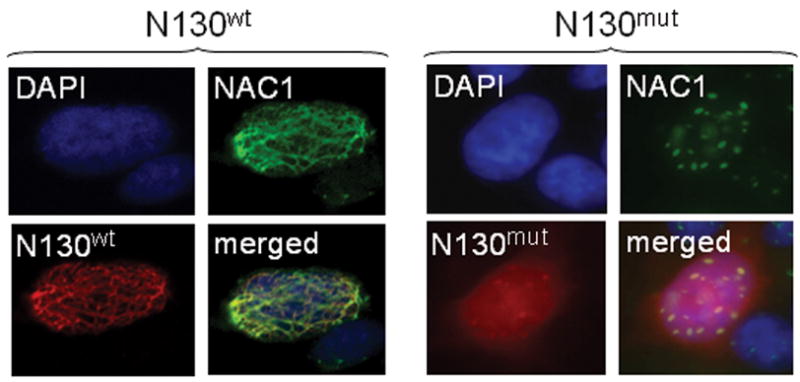
Immunofluorescence staining demonstrates that N130wt (red fluorescence) co-localizes with NAC-1 (green fluorescence) whereas N130mut does not.
Fig. 6. The effects of N130 induction on the expression of Gadd45 family members.

A. Quantitative real-time RT-PCR demonstrates that Gadd45γ is upregulated 24 hours after N130wt induction in SKOV3 cells, while induction of N130mut does not alter Gadd45γ expression. B. NAC-1 shRNA treatment (right panel) resulted in a significant upregulation of Gadd45γ (left panel) in both SKOV3 and HeLa cell lines that overexpress NAC-1.
Discussion
Most ovarian cancer patients are initially responsive to carboplatin-paclitaxel combination chemotherapy, but the majority of them eventually develop recurrent chemoresistant tumors, which account for the high mortality in ovarian cancer patients. In a previous report, we demonstrated that NAC-1 upregulation in ovarian serous carcinomas was significantly associated with early tumor recurrence after cytoreduction therapy and paclitaxel/carboplatin combined chemotherapy (Nakayama et al., 2006). In this study, we determined the biological roles of NAC-1 in developing drug resistance in ovarian cancer. Our results provide evidence that NAC-1 upregulation contributes to tumor recurrence by equipping ovarian cancer cells with the paclitaxel-resistant phenotype through negative regulation of the Gadd45 pathway.
The results of this study have implications regarding the pathogenesis of ovarian cancer. Our finding that NAC-1 participates in the development of paclitaxel resistance in ovarian cancer is consistent with a recent report (Ishibashi et al., 2008), and further supports the view that NAC-1 is a paclitaxel-resistance associated gene. It has been shown that one of the stemness properties of cancer initiating (stem-like) cells is the development of resistance to chemotherapeutic agents. For example, ovarian cancer initiating cells are highly resistant to cisplatin or paclitaxel treatment (Zhang et al., 2008). Acquisition of the stem cell-like phenotype is often associated with up-regulation of several embryonic stem cell markers including Notch-1, Nanog, nestin and Oct-4 as compared to parental tumor cells. Recently, new evidence has demonstrated a role of NAC-1 as an important transcriptional regulator in maintaining embryonic stem (ES) cell pluripotency. An extended regulatory network necessary for preserving the pluripotent state of embryonic stem cells has been recently reported (Kim et al., 2008; Wang et al., 2006), and NAC-1 was demonstrated to be a primary Nanog-interacting protein that is part of the protein regulatory complex responsible for maintaining pluripotency (Wang et al., 2006). NAC-1 protein complex has been shown to transcriptionally regulate approximately 800 genes in ES cells (Kim et al., 2008). Interestingly, NAC-1 itself was determined to regulate transcription of the transcription factors Nanog, Oct4, and Sox2 which appeared to be essential for development and maintenance of the pluripotent state of ES cells (Boyer et al., 2005). Given that NAC-1 likely participates in transcriptional regulation of these genes in the maintenance of cell pluripotency, it is plausible that NAC-1 contributes to drug resistance by enhancing stem-cell-like features of ovarian cancer cells.
Identification of the involvement of the Gadd45 pathway in NAC-1 mediated drug resistance is of great interest because the Gadd45 pathway plays a major role in cell cycle arrest and apoptosis under cellular stress such as in the presence of chemotherapeutic agents (Zerbini & Libermann, 2005; Zerbini & Libermann, 2005). In the current report and our previous study, we have demonstrated that NAC-1 proteins negatively regulate the expression of Gadd45gip1 (Crif1) (Nakayama et al., 2007) and Gadd45γ. Gadd45gip1 (Crif1) has been shown to interact with all Gadd45 isoforms, and importantly, the interaction between Gadd45gip1 and Gadd45 members enhances the functions of the Gadd45 complex (Chung et al., 2003). Expression of Gadd45α and Gadd45γ activates MEKK4/MTK1 which then activates MKK4. MKK4 activation, in turn, leads to p38/JNK activation by phosphorylation, resulting in apoptosis/growth inhibition (De Smaele et al., 2001; Verheij et al., 1996; Zerbini & Libermann, 2005). It has also been shown that Gadd45α and Gadd45γ are down-regulated as a consequence of NF-κB pathway activation through upregulation of c-myc. Suppression of both Gadd45 members is essential for cancer cells to survive (De Smaele et al., 2001; Tang et al., 2001; Zerbini et al., 2004). Thus, survival signals from at least two different oncogenic pathways, i.e., NAC-1 and NF-κB, converge on one molecular “hub” which is Gadd45/Gadd45gip1. For example, inhibition of NF-κB has been shown to sensitize ovarian cancer cells to cisplatin and paclitaxel-induced apoptosis (Liu et al., 2006; Mabuchi et al., 2004). Thus, turning off the Gadd45 pathway appears to be critical for cancer cell survival and to facilitate tumor growth under cellular stress. In addition to the established NF-κB-Gadd45 molecular circuit, downregulation of Gadd45gip1 by NAC-1 overexpression represents another molecular switch to suppress Gadd45 death signals. It is likely that in the evolution of cancer cell species, tumor cells acquire multiple strategies to inactivate Gadd45 cell suppressor signaling. In fact, overexpression of NF-κB (Liu et al., 2006; Mabuchi et al., 2004), gene amplification and transcriptional upregulation of c-myc (Abeysinghe et al., 1999; Baker et al., 1990; Dimova et al., 2006) and Gadd45 promoter methylation (Qiu et al., 2004; Ying et al., 2005; Zerbini & Libermann, 2005) have been reported in human cancers including ovarian cancer cells. Furthermore, our recent study also demonstrated that homozygous deletion and downregulation of MKK-4 were detected in ovarian carcinomas (Nakayama et al., 2006). The above data together with the results reported in this study suggest that cancer cells likely employ at least a dual molecular system to switch off Gadd45-mediated growth suppression and apoptosis, i.e., the NF-kB/c-myc/Gadd45 pathway and the NAC-1/Gadd45gip1 pathway.
The biological roles of NAC-1 in promoting paclitaxel resistance imply that NAC-1 might be used as a surrogate stand-alone marker or in combination with other markers to predict paclitaxel resistance. Such information may aid in individualized selection of effective therapies in ovarian cancer patients. For example, patients with NAC-1 overexpressing tumors might be treated with chemotherapeutic agents other than paclitaxel in future clinical trials. Furthermore, NAC-1 may also serve as a molecular target to enhance paclitaxel sensitivity in ovarian cancer patients. The results from this study suggest that several strategies can be applied for NAC-1 targeted therapy including disruption of NAC-1 homodimerization, silencing NAC-1, or forced expression of Gadd45gip1. Although the above represent our favored view, it should be noted that multiple genes and pathways are likely involved in the development of paclitaxel resistance in cancer cells (Bourguignon et al., 2008; Duan et al., 2003; Duan et al., 2006; Duan et al., 2006; Mabuchi et al., 2004; Materna et al., 2007; Yusuf et al., 2003).
In summary, the findings from this current study provide a possible molecular mechanism by which NAC-1 overexpression contributes to paclitaxel resistance. Our results demonstrate the essential role of NAC-1 homodimerization in conferring paclitaxel resistance and also highlight the critical role of the Gadd45 pathway in mediating this phenotype. It would be of great interest to determine how NAC-1 proteins collaborate with other molecules and pathways in rendering stem cell and drug resistance phenotypes in cancer cells. Furthermore, since NAC-1 may serve as an upstream transcriptional regulator for several signaling pathways, it would be worthwhile to explore the potential of developing NAC-1 targeted therapy as a means to enhance paclitaxel sensitivity in cancer patients. It might be imagined that NAC-1 targeted therapy could be used either as a stand-alone sensitizing agent for chemotherapeutics, or used in combination with other inhibitors of known drug-resistance pathways.
Materials and Methods
Cell lines and culture conditions
Cancer cell lines including 293T, OVCAR3, SKOV3, A2780, and HeLa cells were purchased from ATCC (Rockville, MD). All cell lines used in this study were cultured in RPMI1640 medium supplemented with 5% fetal bovine serum. To generate chemoresistant ovarian cancer cells, SKOV3, A2780, and OVCAR3 cells were selected by continuous treatment with different concentrations of paclitaxel (ranging from 0.2 μM – 2.0 μM) or carboplatin (0.2 μg/ml – 4.0 μg/ml). Resistant colonies of each cell line were pooled and subsequently used for experiments. Chemoresistant cell lines were maintained in selective medium containing the highest concentrations of paclitaxel (0.25 μM for SKOV3, 0.5 μM for A2780 and OVCAR3) or carboplatin (2.0 μg/ml for SKOV3, 1.0 μg/ml for A2780, and 0.5 μg/ml for OVCAR3) that permitted growth.
Retrovirus, shRNA transfection and generation of N130mut
A retrovirus carrying the Gadd45gip1-V5 expression vector was produced by cloning the entire coding sequence into the pWZL-Hygro retroviral vector. High titer retroviral stocks were generated using the Phoenix ecotropic packaging cell line. shRNA plasmids that targeting NAC-1 (pRS-shNAC1) and Gadd45gip1 (pRS-shGadd45gip1) as well as the shRNA plasmid that targeting GFP (pRS-shGFP) were obtained from Origene (Rockville, MD). The shRNA plasmids targeting Gadd45γ and the control plasmid (pLKO.1-puro vector only) were obtained from Sigma (St. Louis, MO). The shRNA sequences that target different genes are listed in supplementary Table 1.
We have previously generated N130 inducible cells that express the first 130 a.a. segment of NAC-1 (N130) which corresponds to the BTB/POZ domain. A mutant of N130, N130mut, was generated by random mutagenesis using the JBS dNTP-mutagenesis kit (Jena Bioscience, Jena, Germany). In order to completely abolish the interaction between N130 and NAC-1, we introduced missense mutations that changed 30 amino acids in the N130 fragment. The mutant N130 sequence was then cloned into pcDNA4-Xp and subcloned into the pTRE inducible vector. The cloning primers for N130 were: N130 5′-tgaccagaattcgccatggcccagacactgcag-3′ (forward) and 5′-tggtcactcgagagtcgcagctcggggagctca-3′ (reverse). To determine if N130mut interacted with NAC-1, we co-transfected 293T cells with NAC-1 tagged with V5 and N130mut tagged with Xpress. Double immunofluorescence was performed as previously described using V5 and Xpress antibodies (Nakayama et al., 2006).
Drug sensitivity and cell viability assays
For drug sensitivity assays, cells were transfected with targeting and control shRNAs and seeded in 384-well plates at a density of 750 cells/well. After overnight culture, the cells were treated with a series of concentrations of paclitaxel. Four days after transfection (i.e., three days after drug treatment), viable cell numbers were measured based on the ability of living cells to convert a redox dye (resazurin) into a fluorescent end product (resorufin) using CellTiter-Blue™ reagent (Promega, Madison, WI). Data read by a fluorescence microplate reader (Fluostar from BMG, Durham, NC) were determined from three replicates, and were expressed as a percent of control group without paclitaxel treatment. IC50 was defined as the concentration that resulted in a 50% decrease in the number of cells as reflected by fluorescence intensity compared to that of the control cultures in the absence of the drug.
Quantitative real-time reverse transcription-PCR
An RNA extraction kit (Qiagen) was used to extract total RNA, and the Superscript II First-strand cDNA synthesis kit (Invitrogen) was used to generate cDNA. Real-time PCR was performed on a Bio-Rad iCyclers (MyIQ, IQ4), and data analysis was performed using the Bio-Rad IQ5 v2 software. The PCR primers used in this study are listed in supplementary Table 1. The mean Ct of the gene of interest was calculated from replicate measurements and normalized with the mean Ct of a control gene, APP (beta-amyloid precursor gene), for which expression is relatively constant.
Immunohistochemistry and Western blot
A mouse monoclonal anti-NAC-1 antibody was obtained from Novus (Littleton, CO) and was used in immunohistochemistry at a dilution of 1:200. The specificity of this antibody has been validated in previous reports (Mao et al., 2006; Shih Ie et al., 2005). An EnVision+System peroxidase kit (DAKO, Carpinteria, CA) was used for staining following the manufacturer’s protocol. Immunohistochemical staining was carried out on tissue microarrays containing 113 anonymous high-grade ovarian serous carcinoma tissues for which ex vivo chemoresponse status was available from Oncotech, Inc. (http://www.oncotech.com/pdfs/edr_4_pager.pdf). The definition of extreme drug resistance status was that tumor cell growth was greater than one standard deviation above the median whereas the low drug resistance was that cell growth was less than the median growth (Holloway et al., 2002; Loizzi et al., 2003). Immunointensity for NAC-1 was scored using a 4-tier system from 0 (undetectable), 1+ (weakly positive), 2+ (moderately positive) to 3+ (intensely positive). This criteria was used in our previous study (Nakayama et al., 2006).
Western blot analysis was conducted using a standard protocol. The antibodies used in this study included a mouse NAC-1 monoclonal antibody (Nakayama et al., 2006) at a 1:200 dilution, an anti-V5 antibody (Invitrogen, Cat# R960-25) at a 1:1000 dilution and a rabbit anti-GAPDH polyclonal antibody (Sigma, Cat # G9545) at a 1:4000 dilution. Similar amounts of total protein from each lysate were loaded and separated on 4–12% Tris-Glycine-SDS polyacrylamide gels (Novex, San Diego, CA) and electroblotted to Millipore Immobilon-P polyvinylidene difluoride membranes. Western blots were developed by chemiluminescence (Pierce, Rockford, IL).
Supplementary Material
Acknowledgments
This work is in memory of Ms. Sean Patrick, the founder of HERA women’s cancer foundation who has fought courageously with recurrent ovarian cancer. The authors appreciate the valuable comments from members in our laboratory. We also appreciate Oncotech, Inc. for providing the anonymous ovarian cancer tissue samples with drug sensitivity data. This work is supported by NIH RO1CA103937 (IMS), NIH RO1CA129080 (IMS) and HERA women’s cancer foundation OSB1 (NJ).
Footnotes
Supplementary Information Supplementary information is available at Oncogene’s website.
References
- Abeysinghe HR, Cedrone E, Tyan T, Xu J, Wang N. Cancer Genet Cytogenet. 1999;114:136–43. doi: 10.1016/s0165-4608(99)00064-3. [DOI] [PubMed] [Google Scholar]
- Abhiman S, Iyer LM, Aravind L. Bioinformatics. 2008;24:458–61. doi: 10.1093/bioinformatics/btn007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker VV, Borst MP, Dixon D, Hatch KD, Shingleton HM, Miller D. Gynecol Oncol. 1990;38:340–2. doi: 10.1016/0090-8258(90)90069-w. [DOI] [PubMed] [Google Scholar]
- Bourguignon LY, Peyrollier K, Xia W, Gilad E. J Biol Chem. 2008;283:17635–51. doi: 10.1074/jbc.M800109200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer LA, Lee TI, Cole MF, Johnstone SE, Levine SS, Zucker JP, Guenther MG, Kumar RM, Murray HL, Jenner RG, Gifford DK, Melton DA, Jaenisch R, Young RA. Cell. 2005;122:947–56. doi: 10.1016/j.cell.2005.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha XY, Pierce RC, Kalivas PW, Mackler SA. J Neurosci. 1997;17:6864–71. doi: 10.1523/JNEUROSCI.17-18-06864.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung HK, Yi YW, Jung NC, Kim D, Suh JM, Kim H, Park KC, Song JH, Kim DW, Hwang ES, Yoon SH, Bae YS, Kim JM, Bae I, Shong M. J Biol Chem. 2003;278:28079–88. doi: 10.1074/jbc.M212835200. [DOI] [PubMed] [Google Scholar]
- Davidson B, Berner A, Trope CG, Wang TL, Shih Ie M. Hum Pathol. 2007;38:1030–6. doi: 10.1016/j.humpath.2006.12.009. [DOI] [PubMed] [Google Scholar]
- De Smaele E, Zazzeroni F, Papa S, Nguyen DU, Jin R, Jones J, Cong R, Franzoso G. Nature. 2001;414:308–13. doi: 10.1038/35104560. [DOI] [PubMed] [Google Scholar]
- Dimova I, Raitcheva S, Dimitrov R, Doganov N, Toncheva D. Eur J Cancer. 2006;42:674–9. doi: 10.1016/j.ejca.2005.11.022. [DOI] [PubMed] [Google Scholar]
- Duan Z, Duan Y, Lamendola DE, Yusuf RZ, Naeem R, Penson RT, Seiden MV. Clin Cancer Res. 2003;9:2778–85. [PubMed] [Google Scholar]
- Duan Z, Foster R, Bell DA, Mahoney J, Wolak K, Vaidya A, Hampel C, Lee H, Seiden MV. Clin Cancer Res. 2006;12:5055–63. doi: 10.1158/1078-0432.CCR-06-0861. [DOI] [PubMed] [Google Scholar]
- Duan Z, Foster R, Brakora KA, Yusuf RZ, Seiden MV. Cancer Chemother Pharmacol. 2006;57:25–33. doi: 10.1007/s00280-005-0026-3. [DOI] [PubMed] [Google Scholar]
- Holloway RW, Mehta RS, Finkler NJ, Li KT, McLaren CE, Parker RJ, Fruehauf JP. Gynecol Oncol. 2002;87:8–16. doi: 10.1006/gyno.2002.6797. [DOI] [PubMed] [Google Scholar]
- Ishibashi M, Nakayama K, Yeasmin S, Katagiri A, Iida K, Nakayama N, Fukumoto M, Miyazaki K. Clin Cancer Res. 2008;14:3149–55. doi: 10.1158/1078-0432.CCR-07-4358. [DOI] [PubMed] [Google Scholar]
- Jemal A, Siegel R, Ward E, Murray T, Xu J, Thun MJ. CA Cancer J Clin. 2007;57:43–66. doi: 10.3322/canjclin.57.1.43. [DOI] [PubMed] [Google Scholar]
- Kalivas PW, Duffy P, Mackler SA. Synapse. 1999;33:153–9. doi: 10.1002/(SICI)1098-2396(199908)33:2<153::AID-SYN5>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- Kim J, Chu J, Shen X, Wang J, Orkin SH. Cell. 2008;132:1049–61. doi: 10.1016/j.cell.2008.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF. Neuron. 1996;16:893–6. doi: 10.1016/s0896-6273(00)80109-9. [DOI] [PubMed] [Google Scholar]
- Korutla L, Degnan R, Wang P, Mackler SA. J Neurochem. 2007;101:611–8. doi: 10.1111/j.1471-4159.2006.04387.x. [DOI] [PubMed] [Google Scholar]
- Korutla L, Wang PJ, Mackler SA. J Neurochem. 2005;94:786–93. doi: 10.1111/j.1471-4159.2005.03206.x. [DOI] [PubMed] [Google Scholar]
- Liu GH, Wang SR, Wang B, Kong BH. Int J Gynecol Cancer. 2006;16:1777–82. doi: 10.1111/j.1525-1438.2006.00652.x. [DOI] [PubMed] [Google Scholar]
- Loizzi V, Chan JK, Osann K, Cappuccini F, DiSaia PJ, Berman ML. Am J Obstet Gynecol. 2003;189:1301–7. doi: 10.1067/s0002-9378(03)00629-x. [DOI] [PubMed] [Google Scholar]
- Mabuchi S, Ohmichi M, Nishio Y, Hayasaka T, Kimura A, Ohta T, Saito M, Kawagoe J, Takahashi K, Yada-Hashimoto N, Sakata M, Motoyama T, Kurachi H, Tasaka K, Murata Y. J Biol Chem. 2004;279:23477–85. doi: 10.1074/jbc.M313709200. [DOI] [PubMed] [Google Scholar]
- Mackler S, Pacchioni A, Degnan R, Homan Y, Conti AC, Kalivas P, Blendy JA. Behav Brain Res. 2008;187:48–55. doi: 10.1016/j.bbr.2007.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackler SA, Korutla L, Cha XY, Koebbe MJ, Fournier KM, Bowers MS, Kalivas PW. J Neurosci. 2000;20:6210–7. doi: 10.1523/JNEUROSCI.20-16-06210.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao TL, Hsu CY, Yen MJ, Gilks B, Sheu JJ, Gabrielson E, Vang R, Cope L, Kurman RJ, Wang TL, Shih Ie M. Hum Pathol. 2006;37:1169–75. doi: 10.1016/j.humpath.2006.04.008. [DOI] [PubMed] [Google Scholar]
- Materna V, Surowiak P, Kaplenko I, Spaczynski M, Duan Z, Zabel M, Dietel M, Lage H. Virchows Arch. 2007;450:187–94. doi: 10.1007/s00428-006-0346-7. [DOI] [PubMed] [Google Scholar]
- Muller FJ, Laurent LC, Kostka D, Ulitsky I, Williams R, Lu C, Park IH, Rao MS, Shamir R, Schwartz PH, Schmidt NO, Loring JF. Nature. 2008;455:401–5. doi: 10.1038/nature07213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama K, Nakayama N, Davidson B, Katabuchi H, Kurman RJ, Velculescu VE, Shih Ie M, Wang TL. Cancer Biol Ther. 2006;5:630–4. doi: 10.4161/cbt.5.6.2675. [DOI] [PubMed] [Google Scholar]
- Nakayama K, Nakayama N, Davidson B, Sheu JJ, Jinawath N, Santillan A, Salani R, Bristow RE, Morin PJ, Kurman RJ, Wang TL, Shih IM. Proc Natl Acad Sci U S A. 2006 doi: 10.1073/pnas.0604083103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama K, Nakayama N, Wang TL, Shih IM. Cancer Res. 2007;67:8058–8064. doi: 10.1158/0008-5472.CAN-07-1357. [DOI] [PubMed] [Google Scholar]
- Qiu W, Zhou B, Zou H, Liu X, Chu PG, Lopez R, Shih J, Chung C, Yen Y. Am J Pathol. 2004;165:1689–99. doi: 10.1016/s0002-9440(10)63425-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih Ie M, Sheu JJ, Santillan A, Nakayama K, Yen MJ, Bristow RE, Vang R, Parmigiani G, Kurman RJ, Trope CG, Davidson B, Wang TL. Proc Natl Acad Sci U S A. 2005;102:14004–9. doi: 10.1073/pnas.0504195102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang G, Minemoto Y, Dibling B, Purcell NH, Li Z, Karin M, Lin A. Nature. 2001;414:313–7. doi: 10.1038/35104568. [DOI] [PubMed] [Google Scholar]
- Verheij M, Bose R, Lin XH, Yao B, Jarvis WD, Grant S, Birrer MJ, Szabo E, Zon LI, Kyriakis JM, Haimovitz-Friedman A, Fuks Z, Kolesnick RN. Nature. 1996;380:75–9. doi: 10.1038/380075a0. [DOI] [PubMed] [Google Scholar]
- Wang J, Rao S, Chu J, Shen X, Levasseur DN, Theunissen TW, Orkin SH. Nature. 2006;444:364–8. doi: 10.1038/nature05284. [DOI] [PubMed] [Google Scholar]
- Yeasmin S, Nakayama K, Ishibashi M, Katagiri A, Iida K, Purwana IN, Nakayama N, Miyazaki K. Clin Cancer Res. 2008;14:1686–91. doi: 10.1158/1078-0432.CCR-07-4085. [DOI] [PubMed] [Google Scholar]
- Ying J, Srivastava G, Hsieh WS, Gao Z, Murray P, Liao SK, Ambinder R, Tao Q. Clin Cancer Res. 2005;11:6442–9. doi: 10.1158/1078-0432.CCR-05-0267. [DOI] [PubMed] [Google Scholar]
- Yusuf RZ, Duan Z, Lamendola DE, Penson RT, Seiden MV. Curr Cancer Drug Targets. 2003;3:1–19. doi: 10.2174/1568009033333754. [DOI] [PubMed] [Google Scholar]
- Zerbini LF, Libermann TA. Clin Cancer Res. 2005;11:6409–13. doi: 10.1158/1078-0432.CCR-05-1475. [DOI] [PubMed] [Google Scholar]
- Zerbini LF, Libermann TA. Cell Cycle. 2005;4:18–20. doi: 10.4161/cc.4.1.1363. [DOI] [PubMed] [Google Scholar]
- Zerbini LF, Wang Y, Czibere A, Correa RG, Cho JY, Ijiri K, Wei W, Joseph M, Gu X, Grall F, Goldring MB, Zhou JR, Libermann TA. Proc Natl Acad Sci U S A. 2004;101:13618–23. doi: 10.1073/pnas.0402069101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Balch C, Chan MW, Lai HC, Matei D, Schilder JM, Yan PS, Huang TH, Nephew KP. Cancer Res. 2008;68:4311–20. doi: 10.1158/0008-5472.CAN-08-0364. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.