

Abstract
AIMS
To characterize the impact of potent CYP3A4 inhibition and induction on lapatinib pharmacokinetics.
METHODS
Two studies were conducted in healthy subjects. One study examined the effect of ketoconazole 200 mg b.i.d. for 7 days on a single 100-mg dose of lapatinib in 22 healthy subjects. The other study examined the effect of carbamazepine titrated up to 200 mg b.i.d. over 20 days on a single 250-mg dose of lapatinib in 24 healthy subjects.
RESULTS
Ketoconazole altered lapatinib AUC, Cmax and half-life, with geometric mean [95% confidence interval (CI)] increases of 3.57-fold (3.07, 4.15), 2.14-fold (1.74, 2.64) and 1.66-fold (1.50, 1.84), respectively, but had no effect on absorption rate. Carbamazepine altered lapatinib AUC, Cmax and absorption rate, with geometric mean (95% CI) decreases of 72% (68, 77), 59% (49, 66) and 28% (4, 46), respectively, but had no effect on half-life.
CONCLUSIONS
Systemic exposure to lapatinib was significantly altered by potent inhibition and induction of CYP3A4. Dose adjustments may be required when lapatinib is administered with orally administered drugs that potently alter the activity of this enzyme.
Keywords: carbamazepine, drug interaction, ketoconazole, lapatinib, pharmacokinetics
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT.
Lapatinib is an orally administered tyrosine kinase inhibitor indicated for use in combination with capecitabine to treat advanced or metastatic breast cancers that overexpress ErbB2 in patients previously treated with anthracyclines, taxanes and trastuzumab.
In vitro studies have suggested that lapatinib is metabolized primarily by CYP3A4, that it is a substrate of the efflux transporter ABCB1, and that it is a modest inhibitor of both CYP3A4 and ABCB1.
However, no data regarding the in vivo metabolism of lapatinib have as of yet been reported.
WHAT THIS STUDY ADDS
The in vivo pharmacokinetics of lapatinib in healthy subjects was determined in the presence of a potent inhibitor (ketoconazole) or inducer (carbamazepine) of CYP3A4.
The results indicated that systemic exposure to lapatinib was significantly altered by inhibition or induction of CYP3A4.
Thus, dose adjustments may be required when lapatinib is administered orally concomitantly with drugs that potently alter CYP3A4 activity.
Introduction
Lapatinib (Tykerb®/Tyverb®) is an orally administered tyrosine kinase inhibitor indicated for use in combination with capecitabine to treat advanced or metastatic breast cancers that overexpress ErbB2 in patients previously treated with anthracyclines, taxanes and trastuzumab. Following oral administration, plasma concentrations appear after a lag time of 15 min and peak approximately 4 h after the dose. The half-life of lapatinib appears to increase with dose from 6 to 14 h after single doses of 10–250 mg, and over time up to 24 h after daily dosing, with accumulation to steady state in 6–7 days [1]. Lapatinib bioavailability is increased three- to four-fold when administered with food [2], and it is therefore recommended to be taken in the fasted state. Lapatinib is highly bound (>99%) to plasma albumin and α1 acid glycoprotein.
In vitro studies have indicated that lapatinib was metabolized primarily by CYP3A and that it was a substrate of the efflux transporter ABCB1 (Pgp) [3], as well as an inhibitor of CYP3A4, CYP2C8 and ABCB1 [3]. It was therefore of interest to quantify the impact of potent CYP3A4 inhibitors and inducers on lapatinib pharmacokinetics in vivo. Ketoconazole, a representative potent inhibitor, and carbamazepine, an inducer with potential for use in patients with brain metastases, were each studied in a clinical drug–drug interaction study to determine the maximal in vivo effect of pretreatment and co-administration on lapatinib pharmacokinetics.
Methods
Study participants
Healthy men and women of nonchildbearing potential, aged 18–55 years, were enrolled in each study after obtaining written informed consent. The protocols for each study were approved by a local independent ethics committee, and each study was conducted in accordance with Good Clinical Practice and the 1996 Declaration of Helsinki. Subjects were excluded from participation if they displayed evidence of any clinically relevant adverse medical condition or had consumed any known or suspected inhibitors or inducers of CYP enzymes within 2 weeks before starting the studies. The use of any prescription or nonprescription drugs (excluding acetaminophen/paracetamol), drugs of abuse, herbal or dietary supplements, and grapefruit juice was prohibited throughout the studies.
Study design
The ketoconazole study (EGF10013) was an open-label, randomized, two-way crossover study. A single 100-mg dose of lapatinib was administered either alone or on day 4 of treatment with ketoconazole 200 mg twice daily (b.i.d.) for 7 days. Treatment periods were separated by at least a 7-day wash-out period.
The carbamazepine study (EGF10018) was an open-label, fixed-sequence, two-period study. In the first period a single oral 250-mg dose of lapatinib was administered alone, followed by a 1-week wash-out period. In the second period, carbamazepine was administered b.i.d. in oral doses of 100 mg on days 1–3, 200 mg on days 4–20, and once again on the morning of day 21 together with a single oral 250-mg dose of lapatinib.
Subjects in both studies underwent medical evaluations within 4 weeks before starting and 2 weeks after completing the study. Subjects were confined to a clinical research unit from the evening before dosing lapatinib until the last blood sample was taken.
Pharmacokinetic sampling and drug concentration assays
Blood (2 ml) was collected for measurement of lapatinib concentration starting prior to each dose and continuing for 72 h after each lapatinib dose in the ketoconazole study, and for 48 h after each lapatinib dose in the carbamazepine study. For both studies, blood samples (2 ml) for determination of lapatinib concentrations were collected predose, 0.25, 0.5, 0.75*, 1, 1.5, 2, 2.5, 3, 4, 6, 8, 10, 12, 16*, 24, 36, 48, 60* and 72* h (*ketoconazole study only) post dose. In the ketoconazole study, 4 ml blood was collected for measurement of ketoconazole concentration 1 h after the ketoconazole and lapatinib doses on day 4. In the carbamazepine study, 2 ml blood was collected for measurement of carbamazepine concentration 1 h after the carbamazepine doses on days 7, 14 and 21. Plasma was separated and frozen until analysis. Lapatinib concentrations were measured after solid-phase extraction by a previously published liquid chromatography–mass spectrometry–mass spectrometry method [4], with 1 ng ml−1 sensitivity, precision within 14%, and accuracy within 4%. Ketoconazole was extracted from 0.1 ml plasma with protein precipitation by acetonitrile and diluted with 30 : 70 acetonitrile : 10 mM ammonium acetate. Separation by high-performance liquid chromatography (HPLC) on a C8 5 µm column was followed by positive-ion electrospray ionization tandem mass spectrometric detection (sensitivity 0.05 µg ml−1). Precision and accuracy were within 15%. Carbamazepine was extracted from 0.5 ml plasma into chloroform : hexane, evaporated, and reconstituted in mobile phase (acetate : methanol : acetonitrile 60 : 18 : 22). Separation by HPLC on a C18 column was followed by ultraviolet detection at 210 nm (sensitivity 0.05 µg ml−1). Precision and accuracy were within 6%.
Pharmacokinetic and statistical analyses
Plasma lapatinib concentration vs. time data were analysed by noncompartmental methods using WinNonlin 4.0 software (Pharsight, Mountain View, CA, USA) to determine area under the curve extrapolated to infinity (AUC), peak concentration (Cmax) and time (tmax), absorption lag time (tlag) and elimination half-life (t1/2). Absorption rate constants (ka) were also determined using a one-compartment model with oral absorption.
Log-transformed AUC, Cmax, t1/2 and ka were compared between treatments using analysis of variance with treatment as a fixed effect and subject as a random effect. Period and sequence were included for the ketoconazole study. Treatment comparisons are presented as geometric least-squares mean ratios and associated 95% confidence intervals (CI). Untransformed tmax and tlag were compared using the Wilcoxon matched-pairs test, and comparisons are presented as median differences and 95% CI.
Results
Demographics
In the ketoconazole study, 22 subjects were enrolled, 21 completed the study, and one did not return for the second period. In the carbamazepine study, 24 subjects were enrolled, 23 completed the study, and one was discontinued for violating the protocol restriction on prohibited medications after the first period. Subject demographics and baseline characteristics for both studies are summarized in Table 1.
Table 1.
Demographic characteristics
| Ketoconazole study | Carbamazepine study | |
|---|---|---|
| Age, years, median (range) | 28 (20–55) | 35 (20–47) |
| Sex, n | ||
| Male | 20 | 22 |
| Female | 1 | 2 |
| Race/ethnicity, n | ||
| White | 16 | 19 |
| Black | 1 | 2 |
| Hispanic | 3 | 3 |
| Other | 2 | 0 |
| Weight, kg, median (range) | 82 (53–102) | 80 (58–96) |
| Body mass index, kg m−2, median (range) | 26 (18–31) | 25 (19–29) |
Pharmacokinetics
Ketoconazole increased lapatinib AUC 257% (3.6-fold), Cmax 114% (2.1-fold) and t1/2 66% (1.7-fold), but had no significant effect on tmax, tlag or ka (Table 2, Figure 1). Plasma concentrations of ketoconazole ranged from 1.7 to 9.4 µg ml−1 (3–18 mM). There was no apparent direct relationship between these effects on lapatinib and ketoconazole plasma concentrations.
Table 2.
Effect of ketoconazole (KTZ) on lapatinib (LAP) pharmacokinetics
| Parameter | LAP alone* (n= 22) | LAP + KTZ* (n= 21) | Comparison† (n= 21) |
|---|---|---|---|
| AUC, h ng−1 ml−1 (range) | 1429 (1198, 1704) | 5242 (4388, 6263) | 3.57 (3.07, 4.15) |
| Cmax, ng ml−1 (range) | 115 (101, 130) | 252 (210, 301) | 2.14 (1.74, 2.64) |
| tmax, h | 4.0 (2.5, 8.0) | 4.0 (2.5, 10.0) | 0.75 (0.00, 1.75) |
| tlag, h | 0.25 (0.00, 0.77) | 0.25 (0.00, 1.00) | 0.12 (0.00, 0.13) |
| ka, h−1 | 0.56 (0.44,. 0.72) | 0.56 (0.44, 0.73) | 1.01 (0.77, 1.32) |
| t1/2, h | 9.6 (8.5, 10.7) | 16.0 (14.0, 18.3) | 1.66 (1.50, 1.84) |
Geometric mean (95% CI) for AUC, Cmax, ka, t1/2; median (range) for tlag and tmax.
Geometric least-squares mean ratio (95% CI) for AUC, Cmax, ka, t1/2; median difference for tmax and tlag.
Figure 1.
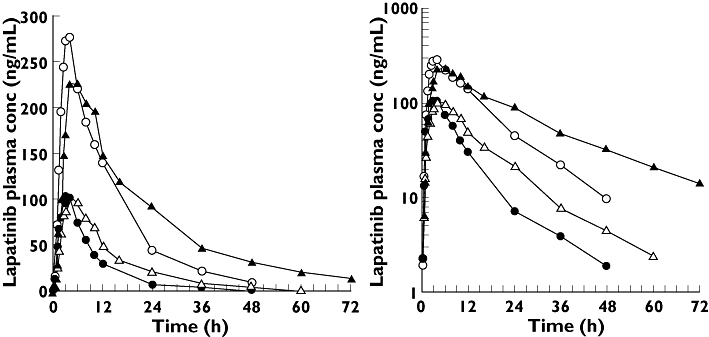
Mean plasma concentrations of lapatinib (LAP) from a 100-mg dose given alone (▵) and after treatment with ketoconazole (KTZ) (▴), and from a 250-mg dose given alone (○) and after treatment with carbamazepine (CBZ) (•). LAP (—○—); LAP + CBZ (—•—); LAP (—▵—); LAP + KTZ (—▴—)
Carbamazepine decreased lapatinib AUC 72% (3.6-fold), Cmax 59% (2.4-fold) and ka 28% (1.3-fold), but had no significant effect on t1/2, tmax or tlag (Table 3, Figure 1). Plasma carbamazepine trough concentrations ranged from 4.0 to 10.8 µg ml−1 on day 7, 3.4 7.4 14 and 3.9 8.7 µg ml−1 21. The geometric mean (95% CI) ratio of concentration 7 was 0.81 (0.78, 0.84) 21 1.04 (1.00, 1.08). There no apparent direct relationship these effects lapatinib concentrations.
Table 3.
Effect of carbamazepine (CBZ) on lapatinib (LAP) pharmacokinetics
| Parameter | LAP alone* (n= 24) | LAP + CBZ* (n= 23) | Comparison† (n= 23) |
|---|---|---|---|
| AUC, h ng−1 ml−1 | 3526 (2888, 4306) | 984 (826, 1171) | 0.28 (0.23, 0.32) |
| Cmax, ng ml−1 | 261 (209, 327) | 110 (90.5, 134) | 0.41 (0.34, 0.51) |
| tmax, h | 4.0 (2.5, 6.0) | 3.0 (1.0, 8.0) | −0.21 (−0.70, 0.76) |
| tlag, h | 0.12 (0.00, 0.50) | 0.25 (0.00, 1.00) | 0.00 (0.00, 0.14) |
| ka, h−1 | 0.70 (0.59, 0.82) | 0.50 (0.39, 0.63) | 0.72 (0.54, 0.96) |
| t1/2, h | 10.2 (9.24, 11.3) | 9.98 (8.12, 12.3) | 0.98 (0.80, 1.19) |
Geometric mean (95% CI) for AUC, Cmax, ka, t1/2; median (range) for tlag and tmax.
Geometric least-squares mean ratio (95% CI) for AUC, Cmax, ka, t1/2; median difference for tmax and tlag.
Clinical safety
Two subjects in the ketoconazole study experienced four drug-related adverse events, the most prominent being loose stools (n= 2). Subjects in the carbamazepine study reported 19 drug-related adverse events, the most prominent being somnolence (n= 11), headache (n= 6), nausea (n= 5) and abdominal pain (n= 3), consistent with known reactions to carbamazepine. In both studies, all drug-related adverse events were mild or moderate in intensity, and resolved without complication.
Discussion
The importance of metabolism in the elimination of lapatinib was demonstrated by these two clinical studies. The results affirmed in vitro data indicating that lapatinib was primarily metabolized by CYP3A4, with lesser contributions from CYP3A5, 2C8 and 2C19 (data on file). 3A4 is the major CYP expressed in the intestine and liver. CYP3A5, 2C8 and 2C19 are also present in enterocytes and hepatocytes, but their activity is much lower and more variable, in part due to polymorphic expression [5, 6].
Ketoconazole inhibits CYP3A4 with Ki in the low nM range, more potently than it inhibits CYP3A5, 2C8 and 2C19 with Ki in the mid to high µM range. In vitro metabolism of lapatinib in human microsomes was completely inhibited at 0.58 µM ketoconazole. It is therefore likely that the mM concentrations of ketoconazole achieved in this study produced complete inhibition of lapatinib metabolism in vivo. If the 3.6-fold increase in lapatinib AUC is interpreted as complete inhibition of metabolic clearance, the corresponding 72% (1/3.6-fold) decrease in total clearance represented metabolism of the absorbed dose. Assuming further that ketoconazole did not alter volume of distribution (Vd), the 66% (1.66-fold) increase in lapatinib t1/2 would indicate a 40% (1/1.66-fold) decrease in systemic CL and a corresponding (0.60/0.28) 2.2-fold increase in bioavailability.
Carbamazepine and its epoxide metabolite are inducers and substrates of CYP3A4 and therefore exhibit autoinduction. There is no clear evidence that carbamazepine induces either CYP2C8 or CYP2C19, and CYP3A5 induction is unlikely because, in contrast to other CYP, its induction is mediated by the glucocorticoid receptor [7]. The magnitude of induction is dependent on the concentrations of carbamazepine and its epoxide metabolite [8]. First-pass metabolism of carbamazepine results in approximately 30% of the dose being converted to the epoxide metabolite [9]. Therefore, the exposure of enterocytes and hepatocytes to inducers does not greatly differ between these routes of administration. However, induction is also time dependent, with an estimated t1/2 of 70 h in the liver but a shorter t1/2 of 24 h in the intestine due to enterocyte turnover, which limits induction in this organ [8]. Based on these half-lives, the 21 days of carbamazepine dosing in this study was sufficient to achieve maximal induction in both the intestine and liver. This was evident as autoinduction in the decline of carbamazepine plasma concentrations from days 7 to 14, but obscured on day 21, presumably by lapatinib inhibition of carbamazepine metabolism by CYP3A4.
The absence of an effect of carbamazepine on the t1/2 of lapatinib was unexpected, because it suggests that systemic CL and Vd were increased to the same extent. Increases in Vd due to induction have been observed previously with carbamazepine [10], where induction increased Vd, possibly through increased synthesis of proteins to which the drug is bound (e.g. α1 acid glycoprotein) [11]. Under these conditions, the occult increase in systemic CL would contribute to the 3.6-fold increase in CL/F. However, it is unlikely that the increase in Vd would be large for a drug as extensively bound as lapatinib. Therefore, the effect of induction appears to be greater on presystemic metabolism.
These studies have revealed the importance of presystemic metabolism to lapatinib bioavailability. In addition, the remarkable similarity in the average magnitude of inhibition and induction, both 3.6-fold changes (Figure 2), as well the decreased interindividual variability of lapatinib AUC, suggest that these changes were approaching a limit (i.e. absolute bioavailability). Complete inhibition by ketoconazole and maximal induction carbamazepine indicate 72% absorbed drug is metabolized. remaining 28% CL/F must therefore be attributed to mechanism other than metabolism.
Figure 2.
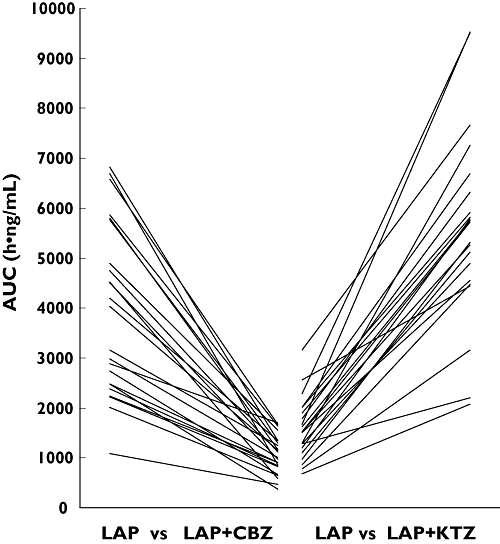
Individual changes in lapatinib AUC due to carbamazepine (CBZ) induction (left) and ketoconazole (KTZ) inhibition (right) of CYP3A4
Lapatinib, like many CYP3A4 substrates, also substrate ABCB1 [12], which is inhibited by ketoconazole [13] and induced by carbamazepine [14]. This transporter mediates intestinal efflux, which decreases the rate of absorption by moving drug in the opposite direction. It could therefore be expected that ketoconazole and carbamazepine would potently increase and decrease absorption rate, respectively; however, ketoconazole did not increase absorption rate, and carbamazepine produced only a small 28% decrease in absorption rate. These observations may be explained by the fact that the rate of lapatinib absorption is two orders of magnitude greater than in vitro passive permeability [3], suggesting that lapatinib absorption is dominated by as yet unidentified uptake transporters. Under these circumstances, ABCB1-mediated intestinal efflux would not be a rate-determining process, such that even complete inhibition of efflux could have little or no effect on the overall rate of absorption. In contrast to inhibition, induction of ABCB1 could increase efflux to a rate no longer dominated by uptake transport and appear as an attenuated decrease. Intestinal efflux transport affects the rate of absorption, but has less impact on the extent of absorption because it can represent a futile cycle in which effluxed drug may be reabsorbed. In contrast, CYP3A4-mediated metabolism represents an irreversible process of elimination. Both of these studies suggest that the role of ABCB1 in the disposition of lapatinib is less important than the role of CYP3A4.
These interactions were characterized in healthy subjects receiving a single 100-mg or 250-mg dose of lapatinib, so their relevance to therapeutic doses in patients with cancer requires extrapolation. Dose-normalized AUC and Cmax in healthy subjects at doses <250 mg are similar to those in patients with cancer at doses up to 1800 mg day−1 (data on file). Therefore, it is likely that co-administration of potent inhibitors and inducers administered orally will produce similar effects in patients.
Conclusions
Potent inhibition or induction of CYP3A4, and to a lesser extent ABCB1, significantly alters lapatinib systemic exposure, illustrating the importance of presystemic metabolism to lapatinib bioavailability. Based on these data, the dose of lapatinib should be adjusted when it is necessary to treat patients simultaneously with potent inhibitors or inducers of CYP3A4, especially when such drugs are administered orally.
Competing interests
DS, KK, NA and CB are employees of GlaxoSmithKline. JH is an employee of Amgen and holds shares in the company. AB is a former employee of GlaxoSmithKline.
These studies were presented at the American Society of Clinical Oncology 2004 meeting, with corresponding abstracts in the Journal of Clinical Oncology 2004; 22 (14S): 3071 and 3081. These studies were funded by GlaxoSmithKline.
REFERENCES
- 1.Bence AK, Anderson EB, Halepota MA, Doukas MA, DeSimone PA, Davis GA, Smith DA, Koch KM, Stead AG, Mangum S, Bowen CJ, Spector NL, Hsieh S, Adams VR. Phase I pharmacokinetic studies evaluating single and multiple doses of oral GW572016, a dual EGFR-erbb2 inhibitor, in healthy subjects. Invest New Drugs. 2005;23:39–49. doi: 10.1023/B:DRUG.0000047104.45929.ea. [DOI] [PubMed] [Google Scholar]
- 2.Reddy N, Cohen R, Whitehead B, Koch KM, Stead A, Beelen AP, Lewis LD. A phase I, open-label, three-period, randomized crossover study to evaluate the effect of food on the pharmacokinetics of lapatinib in cancer patients. Clin Pharmacol Ther. 2007;81:S16–7. [Google Scholar]
- 3.Polli JW, Humphreys JE, Harmon KA, Castellino S, O’Mara MJ, Olson KL, St John-Williams L, Koch KM, Serabjit-Singh CJ. The role of efflux and uptake transporters in N-{3-Chloro-4-[(3-fluorobenzyl)oxy]phenyl}-6-[5-({[2-(methylsulfonyl) ethyl]amino}methyl)-2-furyl]-4-quinazolinamine) ( GW572016, Lapatinib) disposition and drug interactions. Drug Metab Dispos. 2008;36:1–7. doi: 10.1124/dmd.107.018374. [DOI] [PubMed] [Google Scholar]
- 4.Hsieh S, Tobien T, Koch K, Dunn J. Increasing throughput of parallel on-line extraction liquid chromatography/ electrospray ionization tandem mass spectrometry system for GLP quantitative bioanalysis in drug development. Rapid Commun Mass Spectrom. 2004;18:285–92. doi: 10.1002/rcm.1327. [DOI] [PubMed] [Google Scholar]
- 5.Gibbs MA, Thummel KE, Shen DD, Kunze KL. Inhibition of cytochrome P-450 3A (CYP3A) in human intestinal and liver microsomes: comparison of Ki values and impact of CYP3A5 expression. Drug Metab Dispos. 1999;27:180–7. [PubMed] [Google Scholar]
- 6.Lapple F, von Richter O, Fromm MF, Richter T, Thon KP, Wisser H, Griese EU, Eichelbaum M, Kivisto KT. Differential expression and function of CYP2C isoforms in human intestine and liver. Pharmacogenetics. 2003;13:565–75. doi: 10.1097/00008571-200309000-00005. [DOI] [PubMed] [Google Scholar]
- 7.Schuetz EG, Schuetz JD, Strom SC, Thompson MT, Fisher RA, Molowa DT, Li D, Guzelian PS. Regulation of human liver cytochromes P-450 in family 3A in primary and continuous culture of human hepatocytes. Hepatology. 1993;18:1254–62. [PubMed] [Google Scholar]
- 8.Magnusson MO, Dahl M-L, Cederberg J, Karlsson MO, Sandstrom R. Pharmacodynamics of carbamazepine-mediated induction of CYP3A4, CYP1A2, and Pgp as assessed by probe substrates midazolam, caffeine, and digoxin. Clin Pharmacol Ther. 2007;84:52–62. doi: 10.1038/sj.clpt.6100431. [DOI] [PubMed] [Google Scholar]
- 9.Fagiolino P, Vazquez M, Olano I, Delfino A. Systemic and presystemic conversion of carbamazepine to carbamazepine-10,11-epoxide during long term treatment. J Epilepsy Clin Neurophysiol. 2006;12:13–6. [Google Scholar]
- 10.Lucas RA, Gilfillan DJ, Bergstrom RF. A pharmacokinetic interaction between carbamazepine and olanzapine: observations on possible mechanism. Eur J Clin Pharmacol. 1998;54:639–43. doi: 10.1007/s002280050527. [DOI] [PubMed] [Google Scholar]
- 11.Han P, Brichant JF, Pieron F, Pieyns P, Born JD, Lamy M. Elevated plasma alpha 1-acid glycoprotein levels: lack of connection to resistance to vecuronium blockade induced by anticonvulsant therapy. J Neurosurg Anesthesiol. 1997;9:3–7. [PubMed] [Google Scholar]
- 12.Kim RB, Wandel C, Leake B, Cvetkovic M, Fromm MF, Dempsey PJ, Roden MM, Belas F, Chaudhary AK, Roden DM, Wood AJ, Wilkinson GR. Interrelationship between substrates and inhibitors of human CYP3A4 and P-glycoprotein. Pharm Res. 1999;16:408–14. doi: 10.1023/a:1018877803319. [DOI] [PubMed] [Google Scholar]
- 13.Rautio J, Humphreys JE, Webster LO, Balakrishnan A, Keogh JP, Kunta JR, Serabjit-Singh CJ, Polli JW. In vitro p-glycoprotein inhibition assays for assessment of clinical drug interaction potential of new drug candidates: a recommendation for probe substrates. Drug Metab Dispos. 2006;34:786–92. doi: 10.1124/dmd.105.008615. [DOI] [PubMed] [Google Scholar]
- 14.Giessmann T, May K, Modess C, Wegner D, Hecker U, Zschiesche M, Dazert P, Grube M, Schroeder E, Warzok R, Cascorbi I, Kroemer HK, Siegmund W. Carbamazepine regulates intestinal P-glycoprotein and multidrug resistance protein MRP2 and influences disposition of tolamolol in humans. Clin Pharmacol Ther. 2004;76:192–200. doi: 10.1016/j.clpt.2004.04.011. [DOI] [PubMed] [Google Scholar]