

Abstract
Human carboxypeptidase N (CPN) was discovered in the early 1960s as a plasma enzyme that inactivates bradykinin and was identified 8 years later as the major “anaphylatoxin inactivator” of blood. CPN plays in important role in protecting the body from excessive buildup of potentially deleterious peptides that normally act as local autocrine or paracrine hormones. This review summarizes the structure, enzymatic properties and function of this important human enzyme, including insights gained by the recent elucidation of the crystal structure of the CPN catalytic subunit and structural modeling of the non-catalytic regulatory 83 kDa subunit. We also discuss its physiological role in cleaving substrates such as kinins, anaphylatoxins, creatine kinase, plasminogen receptors, hemoglobin and stromal cell-derived factor-1α (SDF-1α).
1. History
Carboxypeptidases were initially isolated from pancreatic extracts and so were associated with protein and peptide degradation in the digestive tract [1–4]. However, work beginning in the early 1960’s by Erdös and Sloane [5] revealed the presence of a novel carboxypeptidase in the blood that plays a regulatory role by inactivating bradykinin via removal of its C-terminal Arg residue. (Subsequent research some 2 decades later led to the discovery of a second kinin B1 receptor activated by the carboxypeptidase metabolite that is its endogenous agonist - see below). Initially it was assumed that the enzyme represents a circulating form of pancreatic carboxypeptidase B, but this was disproved by the characterization of its enzymatic properties [5–8], which was later borne out by biochemical studies, sequence analysis [9–15] and ultimately X-ray crystallography [16]. To differentiate it from the pancreatic enzyme, it was named carboxypeptidase N (CPN; EC 3.4.17.3) and was the first member of a family of carboxypeptidases now known as the regulatory or CPN/E subfamily of metallocarboxypeptidases [17–19]. It has since been re-discovered many times as other substrates were found and so has acquired aliases such as creatine kinase conversion factor, plasma carboxypeptidase B, arginine carboxypeptidase, lysine carboxypeptidase and protaminase [8, 17, 20, 21]. The most notable was the finding by Bokisch and Müller-Eberhard in 1969 – 70 [22, 23] that the human plasma “anaphylatoxin inactivator” is identical with CPN. This finding explains our interest in the work of Dr. Tony Hugli, who has made many seminal contributions to the understanding of the structure and function of anaphylatoxins [24–27]. In fact, Dr. Hugli’s research produced the first in vivo evidence for the important role of CPN in protecting laboratory animals from the lethal consequences of anaphylatoxin generation [28]. These findings, coupled with the facts that no person has ever been found to completely lack the enzyme and patients with low enzyme levels are rare [29, 30] have led to the conclusion that CPN may be essential to the sustenance of life.
2. Localization
CPN is synthesized by the liver and secreted into the blood where its concentration is high, approximately 30 μg/ml ( 10−7 M) [8, 31]. As determined by Northern analysis (Tan and Skidgel, unpublished)[32], the liver is its only site of synthesis, however, only low levels of CPN can be extracted from the organ itself [9]. Probably it is not stored there, but secreted shortly after synthesis. Although some reports claimed the presence of CPN in tissues or non-hepatic derived cells [33–35], contribution of CPN from the blood in tissues or serum used to grow cells was not ruled out. Possibly, membrane-bound carboxypeptidase M (CPM) could have been detected as these two enzymes cleave the same substrates. For example, we found that human pulmonary arterial endothelial cell membranes have high levels of CPM, but no detectable CPN as determined by immunoprecipitation with specific antiserum [36]. RNA and protein analyses in mouse embryos showed that expression of the active subunit of CPN occurrs as early as 8.5 days and can be found in erythroid progenitor cells in the embryonic liver at day 10.5 and in hepatocytes at day 16.5 [37].
3. Physical Properties and Subunit Structure
Purified native human CPN has an apparent molecular weight of about 270 to 330 kDa although 280 kDa is the most frequently cited value [9, 13, 20, 38]. When separated under denaturing conditions, it dissociates into three major protein bands of 83 kDa, 55 kDa and 48 kDa [13, 20, 38]. The 83 kDa protein is a non-catalytic regulatory subunit whereas the 55 kDa and 48 kDa proteins represent the native and proteolytically cleaved forms of the active subunit [13, 20, 38]. The 280 kDa form of CPN is a dimer of heterodimers, with each heterodimer containing one catalytic subunit and one 83 kDa subunit. The 83 kDa subunit is highly glycosylated (about 28% by weight) whereas the active subunit was thought to lack carbohydrate [13, 38]. However, the recent X-ray crystal structure of the active subunit revealed the surprising presence of O-linked carbohydrate [16] (see below). The subunits of CPN are held together by non-covalent forces as they can be dissociated with 3 M guanidine and separated by gel filtration [13, 20].
Because of its small size and relative instability at 37°C [13, 39], the active subunit would not survive by itself in the circulation for very long. In vitro, the 83 kDa subunit stabilizes the active subunit at 37°C and at low pH [13]. This indicates that, although the 83 kDa subunit lacks enzymatic activity, it is important because it carryies and stabilizes the active subunit in the blood. In addition, the 83 kDa subunit may enhance the interaction of the catalytic subunit with larger substrates such as anaphylatoxin C3a and inhibitors (protamine) [17, 40]. However, most of the enzymatic properties of the intact 280 kDa tetramer are preserved in the isolated catalytic subunits [13, 41]
Both subunits are sensitive to proteolysis by serine proteases in their native conformation [13, 38]. When intact CPN tetramer is exposed to plasmin or trypsin the 83 kDa subunit is cleaved at Arg457-Ser458, releasing a 13 kDa peptide, resulting in dissociation of the tetramer into two active 142 kDa heterodimers [42] (Fig. 1A). When exposed to trypsin or plasmin, the catalytic subunit either alone or bound to the 83 kDa subunit is cleaved at the C-terminus to reduce its size from 55 kDa to 48 kDa [13, 42](Fig. 1A). Proteolysis at these sites appears to occur constitutively in the blood or during processing and secretion from the liver as CPN quickly isolated from fresh blood with added high concentrations of protease inhibitors, still contains the 55 kDa and 48 kDa subunits at a ~ 1:1 ratio [13, 20]. This also applies to CPN secreted from HepG2 cells and detected directly by [S35]-methionine labeling without purification [43]. The C-terminal truncation does not affect specific activity or tetramer formation indicating that the proteolytically generated 48 kDa form of the active subunit is a normal component of the active, circulating CPN tetramer. Subsequent cleavage of this subunit at Arg218–Arg219 increases catalytic activity and generates an active two-chain form held together by non-covalent bonds [13, 42] (Fig. 1A). This two-chain form may play a role in pathological conditions where proteases are activated. It is not found in normal plasma or in a purified protein preparation as long as protease inhibitors are used during the purification.
Figure 1. Plasmin cleavage of CPN and primary structure of the 83 kDa subunit.

(A) A representation of the CPN tetramer is shown with the three major domains of the 83 kDa subunit designated as follows: clear box, N-terminal domain; black box, leucine-rich repeats; hatched box, C-terminal domain. Without carbohydrate, the calculated Mr of the regulatory subunit is 58,287. Dashed lines represent inter- and intra-chain non-covalent interactions. The sites cleaved by plasmin are denoted by stars and the amino acids at the cleavage sites are marked. The conversion of the catalytic subunit from its 55 kDa to 48 kDa form is not shown for sake of clarity. (B) Amino acid sequence and domain structure of the 83 kDa subunit of CPN is shown, defining the location of the N-terminal domain, leucine-rich repeats (LRR) and C-terminal domain containing the 13kDa “docking” peptide. Highly conserved residues within the 24 amino acid leucine-rich repeats are boxed. The plasmin cleavage site at R457–S458 is marked by the dashed arrow. Cys residues are circled and Asn residues that are potential glycosylation sites are marked with a box. The consensus LRR Cys-rich N- and C-flanking sequences are shaded. Reproduced with permission from Quagraine et al., 2005, Biochem. J. 388: 81–91. © the Biochemical Society.
4. Comparison to related carboxypeptidases
Molecular cloning [15] revealed that the 50 kDa subunit has sequence similarities to other metallocarboxypeptidases [17, 21, 31, 32] although the homology is much higher with the members of the regulatory subfamily (~38 – 49%) than with those in the pancreatic CPA/B subfamily (~14 – 19%). However, most of the active site residues are conserved among the metallocarboxypeptidases and all require zinc as cofactor for activity [17, 21, 31].
The C-terminal region of the catalytic subunit of CPN contains numerous basic residues, and this differs from all other family members. In addition, the segment containing the Arg218–Arg219 protease sensitive site noted above, is also unique to CPN because of 4 – 12 extra residues located on an exposed surface loop (see below) compared with other CPN/E family members.
5. Structure of the 83 kDa subunit
CPN is unique among mammalian metallocarboxypeptidases in having a non-catalytic subunit as an essential component of its native form [13, 21, 38]. The cloning and sequencing of the 83 kDa subunit showed that it encodes a 59 kDa protein with no sequence similarity to the 50 kDa active subunit or any other carboxypeptidase and identified it as a member of the leucine-rich repeat (LRR) family of proteins [44]. This repeating pattern was first noticed in the sequence of the leucine-rich α2-glycoprotein and had been found in only a few other mammalian proteins at the time (e.g. platelet GPIb, GP V and GP IX, proteoglycans, RNAse inhibitor, luteinizing hormone receptor, oligodendrocyte/myelin glycoprotein and U2 snRNP-A′) [44–46]. The LRR motif has now been found in over 2000 proteins from a variety of species [47, 48] and these proteins comprise a subset of the larger “solenoid protein” superfamily [48]. Although very few members of this family have been crystallized, the LRR forms a structure important for mediating binding interactions [47, 48].
The primary sequence of the 83 kDa subunit can be divided into 3 major domains with the following features (Fig. 1B): 1. a 52 residue N-terminal domain that contains a cysteine-rich LRR N-flanking region (amino acids 1–27); 2. a 312 residue central domain that consists of 13 tandem leucine-rich repeats of 24 residues each (amino acids 53–364); 3. a C-terminal domain of 145 residues that contains a 26 residue cysteine-rich LRR C-flanking region (amino acids 400 –425).
Although the 3 dimensional structure of the 83 kDa subunit has not been determined, the other known LRR structures adopt an overall horseshoe shape with a curved β-sheet lining the inner, concave surface and other repeated secondary structures, such as α-helix or β-turn, flanking the outer circumference [47, 48]. Homology modeling of the 83 kDa subunit was accomplished using the ESyPred3D program [49], yielding a structure within the LRR domain similar to those of other LRR proteins (Fig. 2). Among these, the 83 kDa subunit has the highest sequence identity (26.3%) with Lingo-1, a transmembrane protein in the central nervous system that inhibits axon growth whose structure was very recently solved [50]. Lingo-1 contains an Ig-like domain at the C-terminus of the LRR and the interface between this domain and the C-terminal end of the LRR domain contains binding sites mediating tetramer formation [50]. Interestingly, the 83 kDa model, based on the Lingo-1 structure, also has a C-terminal Ig-like domain (Fig. 2) containing the protease sensitive site noted above that, when cleaved, leads to dissociation of the CPN tetramer into heterodimers. Thus, interactions between the 83 kDa subunits in this region may mediate formation of the CPN tetramer (Fig. 2).
Figure 2. Molecular Model of the 83 kDa subunit of CPN.
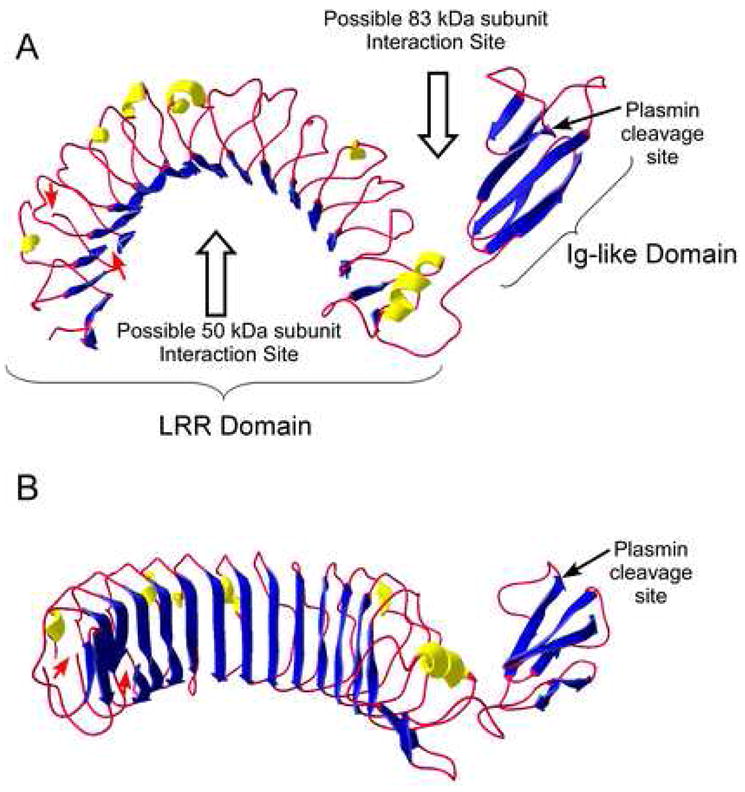
A three-dimensional homology model of the 83 kDa subunit was generated using ESyPred3D [49], based on the structure of Lingo-1 [50]. Helices are colored yellow, β-strands blue and the residual chain as a red rope. Because of additional residues in the 83 kDa subunit (E44– F67 and V83–T106) not found in Lingo-1, the model in the N-terminal region is inaccurate as indicated by gaps in the structure and denoted by the red arrows. The C-terminal 18 residues in the 83 kDa subunit also have no homology with Lingo-1and are not represented in the model. The plasmin cleavage site in the Ig-like domain is indicated by the arrow. A. “Top” view showing the overall curvature of the molecule and the orientation of the C-terminal Ig-like domain. The potential binding sites for the 50 kDa subunit (to form the heterodimer) and another 83 kDa subunit (to form the tetramer) are denoted by the open arrows. B. “Front” view showing the characteristic concave β-sheet surface of the LRR domain.
6. Structure of the catalytic subunit
The recent determination of the crystal structure of the CPN catalytic subunit has revealed some surprising features and important residues likely to be involved in hydrolysis and binding to the regulatory 83 kDa subunit [16]. The catalytic subunit has an overall pear-like shape with a 319 residue N-terminal catalytic domain abutting a cylindrically shaped 79 residue C-terminal β sandwich transthyretin (TT) domain (Fig. 3). The structure of the CPN catalytic subunit is topologically similar to those of the duck CPD-2 fragment [51] and human CPM [52]. The polypeptide chain of the C-terminally truncated CPN catalytic subunit was traced in the final electron density to Ser398 except for nine residues around the Arg218-Arg219 protease-sensitive cleavage site (Fig. 3). This site is in the β6-α6 loop, which is three or six residues longer than in CPM or CPD-2, also without the Arg-Arg sequence. Although previously considered to be non-glycosylated, we were surprised to find three Thr residues of the distal TT edge of the subunit that were O-linked to N-acetyl-glucosamines [16] (Fig. 3). The data do, however, explain a historical paradox. The molecular weights reported for intact and truncated catalytic subunit are 55–56 kDa and 48–49 kDa by SDS-PAGE [13, 38, 42], whereas the calculated molecular weights based on the sequence are only 50 kDa or 45 kDa. Additional mass added by the O-linked carbohydrate can resolve this discrepancy.
Figure 3.

Stereo image of the structure of the catalytic subunit of human CPN. The ribbon representation shows the catalytic domain on top and the cylindrical TT domain at the bottom. Helices (α1 to α9) and β-strands (β1 to β15) are represented as golden helices and blue arrows, and the residual chain as a red rope. Also shown are the zinc ion cofactor inserted according to the CPM structure [52] (purple sphere), the two disulfide bridges (yellow stick model), the three glycosylated residues Thr380, Thr382 and Thr389 (orange stick models), and the O-linked proximal sugars (green stick models) in the TT domain. The figure was prepared with Pymol (http://pymol.sourceforge.net/). This figure is reprinted from [16] with permission from Elsevier.
Modeling of the C-terminal Pro-Phe-Arg of bradykinin into the active site showed how the S1′ pocket might accommodate P1′-Lys better than Arg, in agreement with CPN’s preference for cleaving C-terminal Lys [16]. The relative preference for medium sized penultimate (P1) substrate residues over Gly (see below) might result from enhanced flexibility of bound substrates containing P1 Gly [16].
A region on the catalytic subunit that might interact with the 83 kDa subunit was identified based on several criteria [16]: the ready accessibility to proteases and proteolysis of the C-terminus of the TT domain in tetrameric CPN; the presence of O-linked carbohydrates on the distal edge of the TT domain; the accessibility to proteases of the Arg218–Arg219 cleavage site in the β6-α6 loop; accessibility of the active site groove to large protein substrates (e.g., creatine kinase) in the tetramer; the hydrophobic nature of the catalytic subunit after separation from the 83 kDa subunit. These considerations restrict the likely interaction surface of the catalytic subunit to the interface between the carboxypeptidase and TT domains (Fig. 4). Thus, in tetrameric CPN, a unique hydrophobic surface patch wrapping around the catalytic domain–TT interface and possibly spreading over parts of the CP and the TT domain (Fig. 4), might interact with the concave inner β-sheet of the LRR domain of the 83 kDa subunit to form the heterodimer (Fig. 2).
Figure 4.
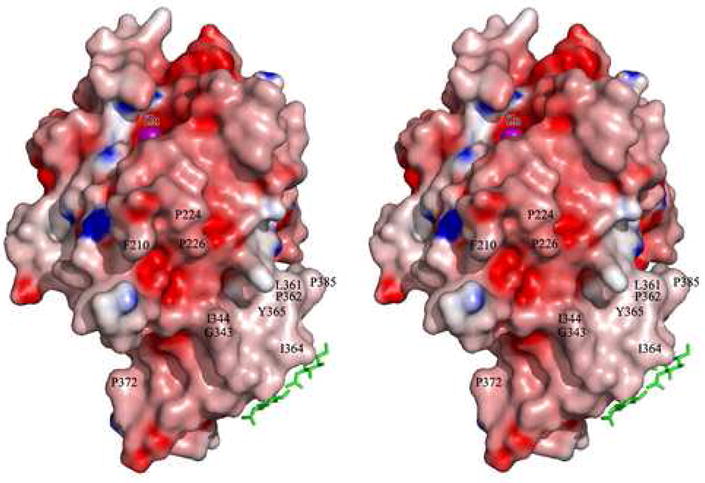
Stereo view of the CPN catalytic subunit, represented by its Connolly surface. The surface is colored according to the electrostatic surface potential, ranging from negative (red, −10 ek−1T−1) to positive (blue, 10 ek−1T−1). The catalytic zinc ion (purple sphere) has been inserted to indicate the active-site groove. The three O-linked sugars are shown as green sticks. Some exposed hydrophobic residues forming a belt-like hydrophobic surface patch wrapping around the catalytic domain/TT interface (possibly representing the attachment site for the 83 kDa subunit) are labeled. The figure was prepared with GRASP [95]. This figure is reprinted from [16] with permission from Elsevier.
7. Enzymatic Properties
CPN contains zinc as a required cofactor, thus it is inhibited by chelating agents such as EDTA and o-phenanthroline [6, 8, 53]. Replacement of Zn++ in the active center by Co++ activates CPN by about 2 to 6-fold at neutral pH depending on the substrate[41, 53–55]. The increased activity is primarily due to an increase in the kcat [41]. In contrast, Cd++ inhibits the enzyme.[6, 53]. CPN has a neutral pH optimum and its activity drops off dramatically at lower pH, retaining about 7% at pH 5.5 [53, 55]. This is likely due to a loss of zinc as added Co++ is still able to activate the enzyme at pH 5.5 to 1.5-fold of that measured at pH 7.5 without Co++ [55]. Relatively high affinity bi-product analog inhibitors of CPN have been developed; guanidinoethylmercaptosuccinic acid with a Ki = 1 μM and DL-2-mercaptomethyl-3-guanidinoethylthiopropanoic acid with a Ki of 2 nM [56, 57]. However, these are not specific for CPN as they also inhibit other B-type carboxypeptidases with similarly high affinity.
CPN cleaves only C-terminal Arg or Lys from peptides or proteins [21, 31, 39]. In general, the enzyme cleaves off C-terminal Lys faster than Arg due to a higher turnover number. For example, for the synthetic substrate FA-Ala-Lys, the Km = 340 μM and kcat = 5820 min−1 whereas for FA-Ala-Arg the Km = 260 μM and kcat = 1860 min−1 [54]. This is also true for biologically active peptides such as Arg6-Met5-enkephalin (Km = 49 μM, kcat = 1024 min−1) and Lys6-Met5-enkephalin (Km = 216 μM, kcat = 6204 min−1) [41]. The penultimate residue in substrates also plays an important role, with Ala or Met being preferred; the presence of Gly greatly slows down hydrolysis [8, 20]. For example, the specificity constant (kcat/Km) for Bz-Gly-Lys is only 0.7 μM−1min−1 but is 60.3 μM−1min−1 for Bz-Ala-Lys. The large effect of the penultimate residue has in vivo significance as the very rapid hydrolysis of anaphylatoxin C3a (C-terminal sequence -Leu-Ala-Arg) by CPN results in a significantly shorter plasma half-life compared with anaphylatoxin C5a with C-terminal -Leu-Gly-Arg [26].
8. Hydrolysis of biologically relevant peptides in vivo
Cleavage of the C-terminal Arg from bradykinin by CPN led to its discovery as the first kininase [5, 8]. Bradykinin has the lowest Km (19 μM) of all peptide substrates tested, although the turnover number (kcat) is lower than for other naturally occurring substrates [20, 41]. CPN carries out this reaction in vivo as the concentration of des-Arg9-bradykinin in human blood is 3-fold higher than that of bradykinin itself [58]. Although CPN contributes in vivo to the degradation of circulating kinins, this pathway is secondary to kinin hydrolysis by angiotensin I converting enzyme (ACE) on pulmonary vascular and other endothelial cells [8]. Consequently, CPN activity takes on added significance in patients receiving ACE inhibitors. Because peptide hormones like kinins are primarily autocrine or paracrine hormones and their sites of action are restricted to a small local area, the levels of circulating peptides are likely to represent “spillover” of peptides generated at local sites. High concentrations of kinins in the blood can have noxious effects, thus blood enzymes like CPN can be protective and prevent the buildup of potentially harmful levels in the circulation [17, 21, 31]. CPN exists at a relatively high level in blood (~30 μg/ml) and is also of obvious relevance in the inactivation of anaphylatoxins [23, 31]. In fact, no one has been found who completely lacks the enzyme and even patients with low enzyme levels are rarely encountered [29, 30]. Genetically determined blood levels of about 20% of normal resulted from decreased hepatic synthesis in one patient [30, 59], likely due to two mutations, one causing a frameshift mutation in the catalytic subunit [60]. This was associated with repeated attacks of angioedema in the patient, possibly due to the increased half-life of kinins and/or anaphylatoxins [30, 59]. Conditions that affect hepatic plasma protein synthesis also alter plasma CPN levels such as a decrease in cirrhosis of the liver or an increase in pregnancy [29]. Elevations in enzyme level have been noted in certain types of cancer and in the blood and synovial fluid of arthritic patients [29, 30, 61, 62]. The possible function of CPN in these disease states is not known.
A potentially more important function of CPN is the alteration of the receptor specificity of kinins [39]. The actions of bradykinin or kallidin (Lys-bradykinin) are mediated via activation of the B2 receptor which is critically dependent on the presence of the C-terminal arginine [63]. However, des-Arg9-bradykinin or des-Arg10-kallidin generated by CPN has a variety of activities mediated through a different receptor designated “B1” [64, 65]. The B1 receptor system is upregulated in response to injury or inflammatory mediators and may be part of the acute phase reaction [63, 65]. Thus, conversion of kinins to des-Arg-kinins to produces the endogenous agonists for the B1 receptor, of potential importance in inflammatory or pathological responses. Activation of B1 receptors initiates increased ERK activation, prostaglandin production [65], high output NO generation and inhibits PKCε activity in cytokine-treated human endothelial cells [66–68].
As noted above, eight years after the discovery of CPN as a kininase, the enzyme was identified as the plasma anaphylatoxin inactivator [22, 23]. Although the potential clinical significance of this finding was clearly understood, direct testing of the concept was not possible until high affinity CPN inhibitors were developed [56, 57]. Dr. Hugli’s group obtained the first convincing in vivo evidence for the importance of this role in studies on guinea pigs in which the complement cascade was activated by the injection of cobra venom factor [28]. The subsequent release of anaphylatoxins had no effect on control guinea pigs because of the inactivation by CPN, but the reaction had dramatic results in guinea pigs when they were pretreated with the potent carboxypeptidase inhibitor DL-2-mercaptomethyl-3-guanidinoethylthiopropanoic acid [28]. In these animals, cobra venom factor induced a lethal reaction within 5 min, due primarily to intense bronchoconstriction and asphyxiation [28]. In addition, histologic sections from these lungs showed small airway constriction, vasoconstriction, cellular aggregates in vessels, pulmonary edema and interstitial infiltrates of mononuclear cells [28]. Whether pulmonary carboxypeptidase M might also play a role in inactivating anaphylatoxins generated in the blood is not clear, but the MGTA used would have also inhibited this enzyme that is highly expressed in lungs [36]. Later studies clearly showed that the carboxypeptidase inhibitor enhanced anaphylatoxin but not bradykinin effects as ACE inhibitors were inactive in guinea pig anaphylaxis while CPN inhibitor pretreatment resulted in high mortality [69].
Evidence for a similar protective function of CPN in humans is, necessarily, somewhat indirect but the protamine-reversal syndrome is one condition where low CPN activity may be involved. In some patients, intravenous protamine given to neutralize the anti-thrombotic effects of heparin after extracorporeal circulation caused a catastrophic reaction consisting of pulmonary vasoconstriction, bronchoconstriction and systemic hypotension [70, 71]. This reaction is attributed to the release of thromboxane and the generation of anaphylatoxins and kinins subsequent to activation of the complement cascade and factor XII which activates plasma kallikrein [71, 72]. We showed that protamine is a potent inhibitor of CPN [40], indicating that the resulting decreased degradation of anaphylatoxins and kinins could contribute to the protamine reversal syndrome. In addition, the CPN level decreased to about half after the initiation of cardiopulmonary bypass, due primarily to dilution of the blood [73]. This syndrome is relatively rare (incidence ~ 1%) indicating that other factors are involved. We also showed that the binding of heparin to protamine reverses the inhibition of CPN [40], thus only when protamine is given in excess would CPN activity be compromised. Indeed, re-administration of heparin reversed protamine reactions in two patients, which was attributed to the reactivation of CPN [74]. In addition, the data of Mathews et al. [30] indicate that CPN levels of 20% of normal or greater might be sufficient for its protective role under normal conditions, thus a 50% reduction after cardiopulmonary bypass may not necessarily cause significant symptoms. However, in patients with abnormally low CPN levels or in those who produce excessive amounts of anaphylatoxins or kinins, a 50% reduction could have serious consequences, especially if protamine is given in excess.
Another complex cascade in blood of crucial importance is the plasminogen system because it functions in thrombolysis/fibrinolysis, extracellular matrix degradation, tissue remodeling and repair. The binding of plasminogen to C-terminal Lys residues on cell surface proteins and fibrin clots increases its activation up to 1000-fold [75–78]. It was shown that plasma CPU and CPN cleave C-terminal Lys residues on these plasminogen “receptors” and thereby downregulate plasminogen activation [77, 78]. CPU (also called TAFI) exists in blood as a proenzyme and once activated by thrombin/thrombomodulin during coagulation has a short half-life [79, 80]. Although CPU reduces plasminogen binding to both cells and in fibrin clots, the effect of CPN is largely restricted to removal of C-terminal Lys residues of plasma membrane proteins [77, 78]. By decreasing cellular plasminogen binding [78], CPN can reduce plasmin-dependent extracellular matrix degradation and cellular migration [77, 81]. For example, freshly isolated monocytes, exposed to CPN in the plasma, bind about 30-fold less plasminogen than monocytes cultured for 18 h or longer [82]. As described above, plasmin can cleave both subunits of CPN, causing increased activity; proteolytically cleaved CPN is also more effective in reducing cellular plasminogen binding than native CPN [78]. This may serve as a feedback mechanism to limit cellular plasminogen activation. Because CPN cleaves preferentially C-terminal Lys over Arg, it is better suited for this function than membrane-bound CPM which releases C-terminal Arg faster [17, 21, 83]. Apart from enhancing CPN activity by cleavage of the catalytic subunit, plasmin conversion of CPN tetramers to heterodimers [42] might enhance CPN’s access to cell surface proteins. Alternatively, conversion to heterodimers may double the effective concentration of CPN in this process as in the tetramer, only one active site at a time may be functional to hydrolyze a fixed substrate such as a cell surface protein of limited diffusion capability.
CPN also cleaves off C-terminal Lys residues from other large protein substrates; it is the “creatine kinase conversion factor” in blood that liberates the C-terminal Lys from both the M and B subunits of creatine kinase, released from the heart after myocardial infarction [84, 85]. Determination of the ratio of the Lys- containing isoform to the des-Lys isoform increased the specificity and sensitivity of blood creatine kinase measurements for the diagnosis of heart attacks [86, 87]. CPN can also remove Arg141 from the α chain of human hemoglobin [88]. This modification accelerated the dissociation of the tetramer into dimers, increased its oxygen affinity over 3-fold and reduced the Hill coefficient (reflecting lower cooperativity) by 2-fold [88]. Des-α-Arg141 hemoglobin also caused greater vasoconstriction than hemoglobin which was mediated by interference with vasodilation by NO and eicosanoids [88].
Mass spectrometry-based surveys of the low molecular weight plasma proteome is a possible source of novel biomarkers for various diseases. Recently, over 250 peptides up to a mass of 5500 d were identified as derivatives of plasma proteins released by plasmin, thrombin or complement proteases [89]. In almost all cases there were peptide isoforms lacking the predicted C-terminal Arg or Lys residues and it was considered to be evidence of hydrolysis by CPN [89]. Whether this cleavage has physiological significance is unknown as the biological activities of these peptides have not been tested, but it should be taken into consideration when measuring peptide levels as biomarkers.
CPN was recently identified as the plasma enzyme responsible for releasing the C-terminal Lys residue from stromal cell-derived factor-1α (SDF-1α) and that reduces the ability of this chemokine to act as a pre-B-cell growth factor and chemoattractant [90, 91]. SDF-1αis important in hematopoietic stem cell trafficking and has received recent attention because it enhances endothelial progenitor cell homing, engraftment and angiogenesis in ischemic tissues [92, 93]. It follows that the regulation of its activity by CPN should be of significance when using endothelial progenitor cells to treat ischemic heart disease [94].
Acknowledgments
Some of the studies reported here were supported by NIH Grants DK41431 (to R.A.S.) and HL36473 (to E.G.E).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Waldschmidt-Leitz E, Purr A. Über Proteinase und Carboxy-polypeptidase aus Pankreas. (XVII. Mitteilung zur spezifität tierischer Proteasen) Ber chem Ges. 1929;62B:956–962. [Google Scholar]
- 2.Waldschmidt-Leitz E, Ziegler F, Schäffner A, Weil L. Über die Struktur der Protamine. I. Protaminase und die Produkte ihrer Einwirkung auf Clupein und Salmin. Z Physiol Chem. 1931;197:219–236. [Google Scholar]
- 3.Anson ML. Carboxypeptidase. I. The preparation of crystalline carboxypeptidase. J Gen Physiol. 1937;20:663–669. doi: 10.1085/jgp.20.5.663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Folk JE, Gladner JA. Carboxypeptidase B. I. Purification of the zymogen and specificity of the enzyme. J Biol Chem. 1958;231:379–391. [PubMed] [Google Scholar]
- 5.Erdös EG, Sloane EM. An enzyme in human blood plasma that inactivates bradykinin and kallidins. Biochem Pharmacol. 1962;11:582 – 592. doi: 10.1016/0006-2952(62)90119-3. [DOI] [PubMed] [Google Scholar]
- 6.Erdös EG, Yang HY, Tague LL, Manning N. Carboxypeptidase in blood and other fluids. 3. The esterase activity of the enzyme. Biochem Pharmacol. 1967;16:1287–1297. doi: 10.1016/0006-2952(67)90159-1. [DOI] [PubMed] [Google Scholar]
- 7.Erdös EG, Yang HY, Miwa I. Kininases. Hoppe Seylers Z Physiol Chem. 1968;349:928. [PubMed] [Google Scholar]
- 8.Erdös EG. Kininases. In: Erdös EG, editor. Handbook of Experimental Pharmacology. Suppl. Vol. 25. Heidelberg: Springer-Verlag; 1979. pp. 427–487. [Google Scholar]
- 9.Oshima G, Kato J, Erdös EG. Plasma carboxypeptidase N, subunits and characteristics. Arch Biochem Biophys. 1975;170:132–138. doi: 10.1016/0003-9861(75)90104-6. [DOI] [PubMed] [Google Scholar]
- 10.Oshima G, Kato J, Erdös EG. Subunits of human plasma carboxypeptidase N (kininase I; anaphylatoxin inactivator) Biochim Biophys Acta. 1974;365:344–348. [Google Scholar]
- 11.Erdös EG. Some old and some new ideas on kinin metabolism. J Cardiovasc Pharmacol. 1990;15 (Suppl 6):S20–24. [PubMed] [Google Scholar]
- 12.Skidgel RA, Bennett CD, Schilling JW, Tan FL, Weerasinghe DK, Erdös EG. Amino acid sequence of the N-terminus and selected tryptic peptides of the active subunit of human plasma carboxypeptidase N: comparison with other carboxypeptidases. Biochem Biophys Res Commun. 1988;154:1323–1329. doi: 10.1016/0006-291x(88)90284-7. [DOI] [PubMed] [Google Scholar]
- 13.Levin Y, Skidgel RA, Erdös EG. Isolation and characterization of the subunits of human plasma carboxypeptidase N (kininase I) Proc Natl Acad Sci U S A. 1982;79:4618–4622. doi: 10.1073/pnas.79.15.4618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Plummer TH, Jr, Erdös EG. Human plasma carboxypeptidase N. Methods Enzymol. 1981;80(Pt C):442–449. doi: 10.1016/s0076-6879(81)80038-9. [DOI] [PubMed] [Google Scholar]
- 15.Gebhard W, Schube M, Eulitz M. cDNA cloning and complete primary structure of the small, active subunit of human carboxypeptidase N (kininase 1) Eur J Biochem. 1989;178:603–607. doi: 10.1111/j.1432-1033.1989.tb14488.x. [DOI] [PubMed] [Google Scholar]
- 16.Keil C, et al. Crystal structure of the human carboxypeptidase N (kininase I) catalytic domain. J Mol Biol. 2007;366:504–516. doi: 10.1016/j.jmb.2006.11.025. [DOI] [PubMed] [Google Scholar]
- 17.Skidgel RA. Structure and function of mammalian zinc carboxypeptidases. In: Hooper NM, editor. Zinc Metalloproteases in Health and Disease. London: Taylor and Francis Ltd; 1996. pp. 241–283. [Google Scholar]
- 18.Fricker LD, Leiter EH. Peptides, enzymes and obesity: new insights from a ‘dead’ enzyme. Trends Biochem Sci. 1999;24:390–393. doi: 10.1016/s0968-0004(99)01448-6. [DOI] [PubMed] [Google Scholar]
- 19.Vendrell J, Querol E, Aviles FX. Metallocarboxypeptidases and their protein inhibitors. Structure, function and biomedical properties. Biochim Biophys Acta. 2000;1477:284–298. doi: 10.1016/s0167-4838(99)00280-0. [DOI] [PubMed] [Google Scholar]
- 20.Skidgel RA. Human carboxypeptidase N: lysine carboxypeptidase. Methods Enzymol. 1995;248:653–663. doi: 10.1016/0076-6879(95)48042-0. [DOI] [PubMed] [Google Scholar]
- 21.Skidgel RA, Erdös EG. Lysine carboxypeptidase. In: Barrett AJ, Rawlings ND, Woessner JF, editors. Handbook of Proteolytic Enzymes. 2. Vol. 1. London: Elsevier; 2004. pp. 837–840. [Google Scholar]
- 22.Bokisch VA, Müller-Eberhard HJ, Cochrane CG. Isolation of a fragment (C3a) of the third component of human complement containing anaphylatoxin and chemotactic activity and description of an anaphylatoxin inactivator of human serum. J Exp Med. 1969;129:1109–1130. doi: 10.1084/jem.129.5.1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bokisch VA, Müller-Eberhard HJ. Anaphylatoxin inactivator of human plasma: its isolation and characterization as a carboxypeptidase. J Clin Invest. 1970;49:2427–2436. doi: 10.1172/JCI106462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hugli TE. Structure and function of C3a anaphylatoxin. Curr Top Microbiol Immunol. 1990;153:181–208. doi: 10.1007/978-3-642-74977-3_10. [DOI] [PubMed] [Google Scholar]
- 25.Hugli TE. Biochemistry and biology of anaphylatoxins. Complement. 1986;3:111–127. doi: 10.1159/000467889. [DOI] [PubMed] [Google Scholar]
- 26.Hugli TE. The structural basis for anaphylatoxin and chemotactic functions of C3a, C4a, and C5a. In: Atassi MZ, editor. Crit Rev Immunol. Vol. 1. Boca Raton: Chemical Rubber Company; 1981. pp. 321–366. [PubMed] [Google Scholar]
- 27.Hugli TE, Müller-Eberhard HJ. Anaphylatoxins: C3a and C5a. Adv Immunol. 1978;26:1–53. doi: 10.1016/s0065-2776(08)60228-x. [DOI] [PubMed] [Google Scholar]
- 28.Huey R, Bloor CM, Kawahara MS, Hugli TE. Potentiation of the anaphylatoxins in vivo using an inhibitor of serum carboxypeptidase N (SCPN). I. Lethality and pathologic effects on pulmonary tissue. Am J Pathol. 1983;112:48–60. [PMC free article] [PubMed] [Google Scholar]
- 29.Erdös EG, Wohler IM, Levine MI, Westerman P. Carboxypeptidase in blood and other fluids. Values in human blood in normal and pathological conditions. Clin Chim Acta. 1965;11:39–43. doi: 10.1016/0009-8981(65)90087-2. [DOI] [PubMed] [Google Scholar]
- 30.Mathews KP, Pan PM, Gardner NJ, Hugli TE. Familial carboxypeptidase N deficiency. Ann Intern Med. 1980;93:443–445. doi: 10.7326/0003-4819-93-3-443. [DOI] [PubMed] [Google Scholar]
- 31.Matthews KW, Mueller-Ortiz SL, Wetsel RA. Carboxypeptidase N: a pleiotropic regulator of inflammation. Mol Immunol. 2004;40:785–793. doi: 10.1016/j.molimm.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 32.Matthews KW, Wetsel RA. Characterization of mouse carboxypeptidase N small active subunit gene structure. J Immunol. 2001;166:6196–6202. doi: 10.4049/jimmunol.166.10.6196. [DOI] [PubMed] [Google Scholar]
- 33.Tsukahara Y, Itakura A, Ohno Y, Ando H, Mizutani S. Umbilical plasma kininase I activity in fetal hypoxia. Horm Metab Res. 2003;35:583–587. doi: 10.1055/s-2003-43503. [DOI] [PubMed] [Google Scholar]
- 34.Ito Y, Mizutani S, Kurauchi O, Kasugai M, Narita O, Tomoda Y. Purification and properties of microsomal carboxypeptidase N (kininase I) in human placenta. Enzyme. 1989;42:8–14. doi: 10.1159/000469001. [DOI] [PubMed] [Google Scholar]
- 35.Ryan US, Ryan JW. Cell biology of pulmonary endothelium. Circulation. 1984;70:III46–62. [PubMed] [Google Scholar]
- 36.Nagae A, Abe M, Becker RP, Deddish PA, Skidgel RA, Erdös EG. High concentration of carboxypeptidase M in lungs: presence of the enzyme in alveolar type I cells. Am J Respir Cell Mol Biol. 1993;9:221–229. doi: 10.1165/ajrcmb/9.2.221. [DOI] [PubMed] [Google Scholar]
- 37.Matthews KW, Drouin SM, Liu C, Martin JF, Skidgel RA, Wetsel RA. Expression of the third complement component (C3) and carboxypeptidase N small subunit (CPN1) during mouse embryonic development. Developmental & Comparative Immunology. 2004;28:647–655. doi: 10.1016/j.dci.2003.10.005. [DOI] [PubMed] [Google Scholar]
- 38.Plummer THJ, Hurwitz MY. Human plasma carboxypeptidase N. Isolation and characterization. J Biol Chem. 1978;253:3907–3912. [PubMed] [Google Scholar]
- 39.Skidgel RA. Basic carboxypeptidases: regulators of peptide hormone activity. Trends Pharmacol Sci. 1988;9:299–304. doi: 10.1016/0165-6147(88)90015-6. [DOI] [PubMed] [Google Scholar]
- 40.Tan F, Jackman H, Skidgel RA, Zsigmond EK, Erdös EG. Protamine inhibits plasma carboxypeptidase N, the inactivator of anaphylatoxins and kinins. Anesthesiology. 1989;70:267–275. doi: 10.1097/00000542-198902000-00015. [DOI] [PubMed] [Google Scholar]
- 41.Skidgel RA, Johnson AR, Erdös EG. Hydrolysis of opioid hexapeptides by carboxypeptidase N. Presence of carboxypeptidase in cell membranes. Biochem Pharmacol. 1984;33:3471–3478. doi: 10.1016/0006-2952(84)90122-9. [DOI] [PubMed] [Google Scholar]
- 42.Quagraine MO, Tan F, Tamei H, Erdös EG, Skidgel RA. Plasmin alters the activity and quaternary structure of human plasma carboxypeptidase N. Biochem J. 2005;388:81–91. doi: 10.1042/BJ20041471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Grimwood BG, Plummer TH, Jr, Tarentino AL. Characterization of the carboxypeptidase N secreted by Hep G2 cells. J Biol Chem. 1988;263:14397–14401. [PubMed] [Google Scholar]
- 44.Tan F, et al. The deduced protein sequence of the human carboxypeptidase N high molecular weight subunit reveals the presence of leucine-rich tandem repeats. J Biol Chem. 1990;265:13–19. [published erratum appears in J Biol Chem 1990 Jul 25;265(21):12749] [PubMed] [Google Scholar]
- 45.Skidgel RA, Tan F. Structural features of two kininase I-type enzymes revealed by molecular cloning. Agents Actions Suppl. 1992;38:359–367. doi: 10.1007/978-3-0348-7321-5_45. [DOI] [PubMed] [Google Scholar]
- 46.Kobe B, Deisenhofer J. Crystal structure of porcine ribonuclease inhibitor, a protein with leucine-rich repeats. Nature. 1993;366:751–756. doi: 10.1038/366751a0. [DOI] [PubMed] [Google Scholar]
- 47.Enkhbayar P, Kamiya M, Osaki M, Matsumoto T, Matsushima N. Structural principles of leucine-rich repeat (LRR) proteins. Proteins. 2003;54:394–403. doi: 10.1002/prot.10605. [DOI] [PubMed] [Google Scholar]
- 48.Kobe B, Kajava AV. The leucine-rich repeat as a protein recognition motif. Curr Opin Struct Biol. 2001;11:725–732. doi: 10.1016/s0959-440x(01)00266-4. [DOI] [PubMed] [Google Scholar]
- 49.Lambert C, Leonard N, De Bolle X, Depiereux E. ESyPred3D: Prediction of proteins 3D structures. Bioinformatics. 2002;18:1250–1256. doi: 10.1093/bioinformatics/18.9.1250. [DOI] [PubMed] [Google Scholar]
- 50.Mosyak L, et al. The structure of the Lingo-1 ectodomain, a module implicated in central nervous system repair inhibition. J Biol Chem. 2006;281:36378–36390. doi: 10.1074/jbc.M607314200. [DOI] [PubMed] [Google Scholar]
- 51.Gomis-Ruth FX, et al. Crystal structure of avian carboxypeptidase D domain II: a prototype for the regulatory metallocarboxypeptidase subfamily. EMBO J. 1999;18:5817–5826. doi: 10.1093/emboj/18.21.5817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Reverter D, Maskos K, Tan F, Skidgel RA, Bode W. Crystal structure of human carboxypeptidase M, a membrane-bound enzyme that regulates peptide hormone activity. J Mol Biol. 2004;338:257–269. doi: 10.1016/j.jmb.2004.02.058. [DOI] [PubMed] [Google Scholar]
- 53.Erdös EG, Sloane EM, Wohler IM. Carboxypeptidase in blood and other fluids - I. Properties, distribution, and partial purification of the enzyme. Biochem Pharmacol. 1964;13:893–905. doi: 10.1016/0006-2952(64)90033-4. [DOI] [PubMed] [Google Scholar]
- 54.Plummer TH, Jr, Kimmel MT. An improved spectrophotometric assay for human plasma carboxypeptidase N1. Anal Biochem. 1980;108:348–353. doi: 10.1016/0003-2697(80)90598-9. [DOI] [PubMed] [Google Scholar]
- 55.Deddish PA, Skidgel RA, Erdös EG. Enhanced Co2+ activation and inhibitor binding of carboxypeptidase M at low pH. Similarity to carboxypeptidase H (enkephalin convertase) Biochem J. 1989;261:289–291. doi: 10.1042/bj2610289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.McKay TJ, Plummer TH., Jr By-product analogues for bovine carboxypeptidase B. Biochemistry. 1978;17:401–405. doi: 10.1021/bi00596a003. [DOI] [PubMed] [Google Scholar]
- 57.Plummer TH, Jr, Ryan TJ. A potent mercapto bi-product analogue inhibitor for human carboxypeptidase N. Biochem Biophys Res Commun. 1981;98:448–454. doi: 10.1016/0006-291x(81)90860-3. [DOI] [PubMed] [Google Scholar]
- 58.Odya CE, Wilgis FP, Walker JF, Oparil S. Immunoreactive bradykinin and [des-Arg9]-bradykinin in low-renin essential hypertension--before and after treatment with enalapril (MK 421) J Lab Clin Med. 1983;102:714–721. [PubMed] [Google Scholar]
- 59.Mathews KP, Curd JG, Hugli TE. Decreased synthesis of serum carboxypeptidase N (SCPN) in familial SCPN deficiency. J Clin Immunol. 1986;6:87–91. doi: 10.1007/BF00915368. [DOI] [PubMed] [Google Scholar]
- 60.Cao H, Hegele RA. DNA polymorphism and mutations in CPN1, including the genomic basis of carboxypeptidase N deficiency. J Hum Genet. 2003;48:20–22. doi: 10.1007/s100380300003. [DOI] [PubMed] [Google Scholar]
- 61.Chercuitte F, Beaulieu AD, Poubelle P, Marceau F. Carboxypeptidase N (kininase I) activity in blood and synovial fluid from patients with arthritis. Life Sci. 1987;41:1225–1232. doi: 10.1016/0024-3205(87)90200-1. [DOI] [PubMed] [Google Scholar]
- 62.Schweisfurth H, Schmidt M, Brugger E, Maiwald L, Thiel H. Alterations of serum carboxypeptidases N and angiotensin-I-converting enzyme in malignant diseases. Clin Biochem. 1985;18:242–246. doi: 10.1016/s0009-9120(85)80049-7. [DOI] [PubMed] [Google Scholar]
- 63.Bhoola KD, Figueroa CD, Worthy K. Bioregulation of kinins: kallikreins, kininogens, and kininases. Pharmacol Rev. 1992;44:1–80. [PubMed] [Google Scholar]
- 64.Regoli D, Barabe J. Pharmacology of bradykinin and related kinins. Pharmacol Rev. 1980;32:1–46. [PubMed] [Google Scholar]
- 65.Leeb-Lundberg LM, Marceau F, Muller-Esterl W, Pettibone DJ, Zuraw BL. International union of pharmacology. XLV. Classification of the kinin receptor family: from molecular mechanisms to pathophysiological consequences. Pharmacol Rev. 2005;57:27–77. doi: 10.1124/pr.57.1.2. [DOI] [PubMed] [Google Scholar]
- 66.Ignjatovic T, Stanisavljevic S, Brovkovych V, Skidgel RA, Erdös EG. Kinin B1 receptors stimulate nitric oxide production in endothelial cells: Signaling pathways activated by angiotensin I-converting enzyme inhibitors and peptide ligands. Mol Pharmacol. 2004;66:1310–1316. doi: 10.1124/mol.104.001990. [DOI] [PubMed] [Google Scholar]
- 67.Sangsree S, Brovkovych V, Minshall RD, Skidgel RA. Kininase I-type carboxypeptidases enhance nitric oxide production in endothelial cells by generating bradykinin B1 receptor agonists. Am J Physiol Heart Circ Physiol. 2003;284:H1959–1968. doi: 10.1152/ajpheart.00036.2003. [DOI] [PubMed] [Google Scholar]
- 68.Stanisavljevic S, et al. Angiotensin I-Converting Enzyme Inhibitors Block Protein Kinase C{epsilon} by Activating Bradykinin B1 Receptors in Human Endothelial Cells. J Pharmacol Exp Ther. 2006;316:1153–1158. doi: 10.1124/jpet.105.093849. [DOI] [PubMed] [Google Scholar]
- 69.Ryan JW, Berryer P, Hart MA, Ryan US. Aggregate anaphylaxis and carboxypeptidase N. Adv Exp Med Biol. 1986;198(Pt A):435–443. doi: 10.1007/978-1-4684-5143-6_59. [DOI] [PubMed] [Google Scholar]
- 70.Lowenstein E, et al. Catastrophic pulmonary vasoconstriction associated with protamine reversal of heparin. Anesthesiology. 1983;59:470–473. doi: 10.1097/00000542-198311000-00022. [DOI] [PubMed] [Google Scholar]
- 71.Morel DR, et al. C5a and thromboxane generation associated with pulmonary vaso- and broncho-constriction during protamine reversal of heparin. Anesthesiology. 1987;66:597–604. doi: 10.1097/00000542-198705000-00002. [DOI] [PubMed] [Google Scholar]
- 72.Colman RW. Humoral mediators of catastrophic reactions associated with protamine neutralization. Anesthesiology. 1987;66:595–596. [PubMed] [Google Scholar]
- 73.Rabito SF, Anders R, Soden W, Skidgel RA. Carboxypeptidase N concentration during cardiopulmonary bypass in humans. Can J Anaesth. 1992;39:54–59. doi: 10.1007/BF03008673. [DOI] [PubMed] [Google Scholar]
- 74.Lock R, Hessel EA., 2nd Probable reversal of protamine reactions by heparin administration. J Cardiothorac Anesth. 1990;4:604–608. doi: 10.1016/0888-6296(90)90410-h. [DOI] [PubMed] [Google Scholar]
- 75.Ranby M. Studies on the kinetics of plasminogen activation by tissue plasminogen activator. Biochim Biophys Acta. 1982;704:461–469. doi: 10.1016/0167-4838(82)90068-1. [DOI] [PubMed] [Google Scholar]
- 76.Hoylaerts M, Rijken DC, Lijnen HR, Collen D. Kinetics of the activation of plasminogen by human tissue plasminogen activator. Role of fibrin. J Biol Chem. 1982;257:2912–2919. [PubMed] [Google Scholar]
- 77.Plow EF, Allampallam K, Redlitz A. The plasma carboxypeptidases and the regulation of the plasminogen system. Trends Cardiovasc Med. 1997;7:71–75. doi: 10.1016/S1050-1738(97)00012-1. [DOI] [PubMed] [Google Scholar]
- 78.Redlitz A, Tan AK, Eaton DL, Plow EF. Plasma carboxypeptidases as regulators of the plasminogen system. J Clin Invest. 1995;96:2534–2538. doi: 10.1172/JCI118315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bouma BN, Marx PF, Mosnier LO, Meijers JC. Thrombin-activatable fibrinolysis inhibitor (TAFI, plasma procarboxypeptidase B, procarboxypeptidase R, procarboxypeptidase U) Thromb Res. 2001;101:329–354. doi: 10.1016/s0049-3848(00)00411-4. [DOI] [PubMed] [Google Scholar]
- 80.Leurs J, Hendriks D. Carboxypeptidase U (TAFIa): a metallocarboxypeptidase with a distinct role in haemostasis and a possible risk factor for thrombotic disease. Thromb Haemost. 2005;94:471–487. doi: 10.1160/TH04-07-0454. [DOI] [PubMed] [Google Scholar]
- 81.Namiranian S, Naito Y, Kakkar VV, Scully MF. Bound plasminogen is rate-limiting for cell-surface-mediated activation of plasminogen by urokinase. Biochem J. 1995;309:977–982. doi: 10.1042/bj3090977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Felez J, Miles LA, Plescia J, Plow EF. Regulation of plasminogen receptor expression on human monocytes and monocytoid cell lines. J Cell Biol. 1990;111:1673–1683. doi: 10.1083/jcb.111.4.1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Skidgel RA, Erdös EG. Cellular carboxypeptidases. Immunol Rev. 1998;161:129–141. doi: 10.1111/j.1600-065x.1998.tb01577.x. [DOI] [PubMed] [Google Scholar]
- 84.Michelutti L, Falter H, Certossi S, Marcotte B, Mazzuchin A. Isolation and purification of creatine kinase conversion factor from human serum and its identification as carboxypeptidase N. Clin Biochem. 1987;20:21–29. doi: 10.1016/s0009-9120(87)80093-0. [DOI] [PubMed] [Google Scholar]
- 85.Prager NA, Suzuki T, Jaffe AS, Sobel BE, Abendschein DR. Nature and time course of generation of isoforms of creatine kinase, MB fraction in vivo. J Am Coll Cardiol. 1992;20:414–419. doi: 10.1016/0735-1097(92)90111-y. [DOI] [PubMed] [Google Scholar]
- 86.Puleo PR, et al. Use of a rapid assay of subforms of creatine kinase-MB to diagnose or rule out acute myocardial infarction. N Engl J Med. 1994;331:561–566. doi: 10.1056/NEJM199409013310901. [DOI] [PubMed] [Google Scholar]
- 87.Erdös EG, Skidgel RA. More on subforms of creatine kinase MB [letter; comment] N Engl J Med. 1995;333:390. doi: 10.1056/NEJM199508103330617. [DOI] [PubMed] [Google Scholar]
- 88.Michel B, Igic R, Leray V, Deddish PA, Erdös EG. Removal of Arg141 from the alpha chain of human hemoglobin by carboxypeptidases N and M. Circ. Res. 1996;78:635–642. doi: 10.1161/01.res.78.4.635. [DOI] [PubMed] [Google Scholar]
- 89.Koomen JM, et al. Direct tandem mass spectrometry reveals limitations in protein profiling experiments for plasma biomarker discovery. J Proteome Res. 2005;4:972–981. doi: 10.1021/pr050046x. [DOI] [PubMed] [Google Scholar]
- 90.Davis DA, et al. Identification of carboxypeptidase N as an enzyme responsible for C-terminal cleavage of stromal cell-derived factor-1alpha in the circulation. Blood. 2005;105:4561–4568. doi: 10.1182/blood-2004-12-4618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.De La Luz Sierra M, et al. Differential processing of stromal-derived factor-1alpha and stromal-derived factor-1beta explains functional diversity. Blood. 2004;103:2452–2459. doi: 10.1182/blood-2003-08-2857. [DOI] [PubMed] [Google Scholar]
- 92.Schober A, Karshovska E, Zernecke A, Weber C. SDF-1[alpha]-mediated tissue repair by stem cells: A promising tool in cardiovascular medicine? Trends Cardiovasc. Med. 2006;16:103–108. doi: 10.1016/j.tcm.2006.01.006. [DOI] [PubMed] [Google Scholar]
- 93.Yamaguchi J, et al. Stromal cell-derived factor-1 effects on ex vivo expanded endothelial progenitor cell recruitment for ischemic neovascularization. Circulation. 2003;107:1322–1328. doi: 10.1161/01.cir.0000055313.77510.22. [DOI] [PubMed] [Google Scholar]
- 94.Walter DH, et al. Impaired CXCR4 signaling contributes to the reduced neovascularization capacity of endothelial progenitor cells from patients with coronary artery disease. Circ Res. 2005;97:1142–1151. doi: 10.1161/01.RES.0000193596.94936.2c. [DOI] [PubMed] [Google Scholar]
- 95.Nicholls A, Sharp KA, Honig B. Protein folding and association: insights from the interfacial and thermodynamic properties of hydrocarbons. Proteins. 1991;11:281–296. doi: 10.1002/prot.340110407. [DOI] [PubMed] [Google Scholar]