

Abstract
Background
The Akt protein kinase is an important mediator of cardiac myocyte growth and survival. To identify factors with novel therapeutic applications in cardiac diseases, we focused on the identification of factors secreted from Akt1-activated cells that have cardioprotective effects through autocrine/paracrine mechanisms.
Methods and Results
Using an inducible Akt1 transgenic mouse model, we have found that follistatin-like 1 (Fstl1) protein and transcript expression are increased 4.0- and 2.0-fold, respectively, by Akt activation in the heart (P<0.05). Fstl1 transcript was also upregulated in response to myocardial stresses including transverse aortic constriction, ischemia/reperfusion injury, and myocardial infarction. Adenovirus-mediated overexpression of Fstl1 protected cultured neonatal rat ventricular myocytes from hypoxia/reoxygenation-induced apoptosis (P<0.01), and this protective effect was dependent on the upregulation of both Akt and ERK activities. Conversely, knockdown of Fstl1 in cardiac myocytes decreased basal Akt signaling and increased the frequency of apoptotic death in vitro (P<0.01). The intravenous administration of an adenoviral encoding Fstl1 to mice resulted in a 66.0% reduction in myocardial infarct size after ischemia/reperfusion injury that was accompanied by a 70.9% reduction in apoptosis in the heart (P<0.01).
Conclusions
These results indicate that Fstl1 is a cardiac-secreted factor that functions as an antiapoptotic protein. Fstl1 could play a role in myocardial maintenance and repair in response to harmful stimuli.
Keywords: apoptosis, myocytes, reperfusion
Clinical Perspective p 3108
The serine/threonine protein kinase Akt is a key regulator of myocardial growth that is essential for cardiac adaptation to diverse stress.1 The cardioprotective and antiapoptotic properties of Akt have been documented in cultured cardiac myocytes2 and in myocardial ischemia/reperfusion and heart failure models.3,4 Furthermore, phosphatidylinositol 3-kinase (PI3K)-Akt signaling plays important roles in cardiac tissue growth.1 Work from a number of laboratories has shown that cardiac-specific overexpression of modified forms of Akt1 or Akt3 will have different effects on heart phenotype ranging from mild hypertrophy and increased cardiac function to extensive hypertrophy associated with interstitial fibrosis and contractile dysfunction.5-8 In addition, overexpression of nuclear localization of a nuclear-targeted form of nonactivated Akt1 increases contractile function and protects myocytes from apoptosis.9,10
A growing body of evidence shows that the heart secretes factors to maintain its performance. Examples include atrial natriuretic peptide, brain natriuretic peptide, cardiotrophin-1, and adrenomedullin.11-15 More recently, Frost and Engelhardt16 employed a yeast secretion trap screen to identify a novel antihypertrophic protein that is secreted by the heart. We have reported that physiological cardiac hypertrophy in Akt transgenic mice is dependent on vascular endothelial growth factor secretion and the coupling of coronary angiogenesis with myocyte growth.8 Furthermore, it has been reported that the injection of Akt-transduced mesenchymal stem cells into infarcted rat hearts leads to an improvement in cardiac function that results from the release of paracrine factors from these cells rather than through their limited ability to fuse with or differentiate into myocytes.17 These data indicate that the autocrine/paracrine effects of factors secreted from Akt-activated cells could play significant roles in cardiac growth and maintenance.
Previously, we constructed a cardiac-specific inducible Akt1 transgenic mouse system that produces a physiological form of hypertrophy after short-term induction and a pathological form of hypertrophy after long-term induction.8 To elucidate the mechanisms by which Akt signaling regulates cardiac phenotype, we performed an Affymetrix microarray analysis on transgenic mouse hearts at different time points after Akt induction to identify differentially regulated transcripts.18 One of these transcripts, follistatin-like 1 (Fstl1), is a member of the follistatin family of factors that bind to transforming growth factor-β (TGF-β) superfamily proteins. Transfection of Fstl1 into cancer cell lines is reported to inhibit proliferation and invasive behavior,19,20 but there are no functional studies on this factor in the heart. Here we report that Akt transgene activation upregulates the expression and secretion of Fstl1 in cardiac myocytes. We also show that Fstl1 is upregulated by injury in the heart and that it functions as a cardioprotective molecule in vitro and in vivo.
Methods
An expanded Methods section can be found in the online-only Data Supplement.
Transgenic Mice
The generation of cardiac-specific inducible myrAkt1 transgenic mice was described previously.8 Briefly, 2 transgenic mouse lines (Tet-myrAkt1 and α-MHC-tTA) were mated to generate double-transgenic mice (DTG), and these mice were maintained with 0.5 mg/mL doxycycline in the drinking water, resulting in the repression of myrAkt1 gene expression. When doxycycline is withdrawn from the drinking water, tTA binds to tetO elements and induces myrAkt1 gene expression in the cardiac myocytes. For these studies, the Akt1 transgene was induced for 2 weeks by withdrawing doxycycline from the drinking water at the age of 12 weeks. Control experiments were performed with MHC-tTA single-transgenic mice that underwent the same doxycycline treatment protocol as DTG mice.
Microarray Analysis
Microarray analysis were performed by Affymetrix GeneChip Mouse Expression Set 430 microarrays and normalized as described in a previous report.18 Gene expression levels were compared before myr-Akt1 induction and 2 weeks after myr-Akt1 induction. Among transcripts that are upregulated by Akt activation, we selected transcripts that have full-length open reading frame cDNAs available in the National Center for Biotechnology Information Web site (http://www.ncbi.nlm.nih.gov/). Amino acid sequences were then examined for signal sequences with the use of Signal IP software (http://www.cbs.dtu.dk/services/SignalP/). Transcripts with signal sequence were analyzed with SOSUI signal beta version software (http://bp.nuap.nagoya-u.ac.jp/sosui/) to predict transcripts with transmembrane domain.
Cloning and In Vitro Transfection Assay of Mouse Fstl1
Full-length Fstl1 cDNA was obtained by polymerase chain reaction (PCR) of RNA isolated from cardiac myocytes and subcloned into pcDNA3.1/V5-His that express mouse Fstl1 as a fusion to the V5 epitope at the C-terminus. To test the secretion of the gene product, the plasmid vector pcDNA3.1/V5-His expressing Fstl1 was transfected into HEK293 cells with the use of Lipofectamin2000 (Invitrogen, Carlsbad, Calif). After cells were incubated with serum-free media for 24 hours, the cell lysate and media were collected. Cells were mock-transfected (no plasmid) as a negative control. The collected media was concentrated ≈10-fold with the use of Microcon (Millipore). Cell lysates and media were separated by SDS-PAGE, and Fstl1 fused with V5 was detected by Western blot analysis with anti-V5 antibody (Invitrogen).
Construction of Adenoviral Vector Expressing Mouse Fstl1
Full-length mouse Fstl1 cDNA was subcloned into an adenovirus shuttle vector. After linearization, shuttle vector was cotransformed into Escherichia coli with the adenoviral backbone plasmid pAdEasy-1. The resultant recombinant adenoviral DNA with Fstl1 cDNA was transfected into HEK 293 cells to produce the recombinant adenoviral vector. For some experiments, an adenoviral vector expressing β-galactosidase (Ad-βgal) was used as a control. Adenoviral vectors were purified by CsCl ultracentrifugation.
RNA Interference in Ventricular Myocytes
The rat Fstl1 small interfering RNA (siRNA) Smart Pool was purchased from Dharmacon Inc, and the second siRNA targeting Fstl1 and unrelated siRNA were from Qiagen. The sequences of siRNAs used in this study were as follows: Fstl1 (Dharmacon): mixture of 4 siRNAs, 5′-UGCAAAUACUUACGGACUUUU-3′, 5′-CAGAUGGAGCUGAGACCGAUU-3′, 5′-CCGUCAACAUCACCGCUUAUU-3′, and 5′-UGGCUAACGGGCAGAGCGAUU-3′; and Fstl1-b (Qiagen): 5′-r(GCAUCUUGAGAUUUAAUCA)dTdT-3′. Neonatal rat ventricular myocytes (NRVMs) were transfected with siRNA by Lipofectamine 2000 according to the manufacturer's protocol. Forty-eight hours after transfection, the protein or mRNA was extracted for Western blot analysis or quantitative real-time polymerase chain reaction (QRT-PCR) or cells were exposed to hypoxia/reoxygenation.
Statistical Analysis
Data are presented as mean±SEM. Group differences were analyzed by 2-tailed Student t test or ANOVA. To compare multiple groups, the Mann-Whitney U test with Bonferroni correction was used. A value of P<0.05 was considered statistically significant.
The authors had full access to the data and take full responsibility for the integrity of the data. All authors have read and agree to the manuscript as written.
Results
Fstl1 Upregulation by Akt Activation in the Heart
Microarray gene expression analysis was compared between control mouse hearts and in hearts 2 weeks after myocardial induction of myristoylated Akt1 by withdrawing doxycycline from the drinking water of DTG mice.18 Transcripts upregulated by 2-week Akt activation with full-length open reading frame cDNAs were selected. Amino acid sequences were then examined for signal sequences with the use of Signal IP software. Transcripts with signal sequence were further analyzed with SOSUI signal beta version software to exclude transcripts predicted to encode proteins with transmembrane domains. Akt-regulated transcripts were identified that contained a predicted signal sequence but lacked a transmembrane domain. Of this subset of proteins, Fstl1 was selected for further analysis because it was upregulated 2.7-fold. To confirm the changes in Fstl1 mRNA level, QRT-PCR was performed with specific primer sets for Fstl1 and normalization of the signal to that of GAPDH (Table). Fstl1 was upregulated 2.0-fold by Akt activation in the heart (P<0.05). In contrast, transcripts encoding other proteins containing the follistatin domain, including follistatin, follistatin-like 3 (Fstl3), and secreted acidic cysteine-rich glycoprotein (SPARC), were not regulated by activation of the Akt transgene in hearts analyzed as assessed by microarray and QRT-PCR analyses (data not shown and Table, respectively). Fstl1 transcript and protein expression in mouse hearts were not affected by the presence or absence of doxycycline in the drinking water of control mice (data not shown).
Table.
Upregulation of Fstl1 Transcript by Cardiac Myocyte-Specific Overexpression of Akt
| Gene Name | Fold Increase (Versus Control) |
|---|---|
| Fstl1 | 2.04±0.10* |
| Follistatin | 0.36±0.13 |
| Fstl3 | 0.68±0.08 |
| SPARC | 0.73±0.07 |
| Akt1 | 7.19±0.83* |
Values are expressed as mean±SE. n=3 for each group.
P<0.05 compared with control.
To better understand the regulation of Fstl1 in cardiac cells, transcript levels of this factor were measured by RT-PCR in hearts that were subjected to various injuries. Fstl1 transcript was upregulated ≈7-fold at the 7-day time point after transverse aortic constriction that induces pressure overload hypertrophy (Figure 1A). Fstl1 transcript was upregulated ≈2-fold in the myocardial area at risk 1 day after ischemia/reperfusion injury (Figure 1B) and ≈13-fold in hearts 3 days after permanent left anterior descending coronary artery (LAD) ligation (Figure 1C). Fstl1 protein levels were analyzed by Western immunoblot analysis after myocardial infarction because the changes in transcript level were robust in this model. LAD ligation led to a substantial increase in Fstl1 protein in the heart (Figure 1D). Fstl1 immunoreactive material appeared as a doublet with electrophoretic mobilities indicative of 37- and 46-kDa proteins. Quantification of the 37-kDa band indicated a 10-fold upregulation of the Fstl1 protein (Figure 1E). Fstl1 protein could also be detected in the serum after Western immunoblot with an electrophoretic mobility that corresponded to that of a 46-kDa protein. After myocardial infarction, the 46-kDa form of Fstl1 increased 3-fold in serum (Figure 1F). Thus both Fstl1 protein and transcript are induced by a variety of stresses in the heart.
Figure 1.

Upregulation of Fstl1 by pathological stimuli in the heart. A to C, QRT-PCR analysis of Fstl1 transcript expression of Fstl1 mRNA after transverse aortic constriction (TAC) (A), ischemia/reperfusion (I/R) injury (B), and myocardial infarction (MI) (C) resulting from permanent LAD ligation. The data are normalized to the intensity of the GAPDH signal and are compared with Fstl1 transcript expression level in sham-operated heart. n=3 to 6. *P<0.05 vs sham. D, Western immunoblot analysis of Fstl1 expression in mouse heart lysate and serum after sham operation (left 2 lanes) and myocardial infarction (right 4 lanes). E, Quantification of Fstl1 protein expression in hearts of mice subjected to sham surgery or myocardial infarction. F, Quantification of Fstl1 protein levels in serum of mice subjected to sham surgery or myocardial infarction.
Western blot analysis was performed to assess the Akt-mediated change in Fstl1 protein expression in the heart. Figure 2A shows Western immunoblots from 2 control hearts and 3 myrAkt1 transgenic hearts after 2 weeks of induction. The expression of Fstl1 protein (both the high- and low-molecular-weight forms) was increased in heart by Akt transgene induction (Figure 2A). The hemagglutinin-tagged Akt transgene product was detected by anti-hemagglutinin antibody, and Akt activation was confirmed by phosphospecific anti-Akt antibody (Figure 2A). After normalization to the α-tubulin signal, Fstl1 protein expression was induced ≈4-fold by Akt activation in the heart (Figure 2B).
Figure 2.
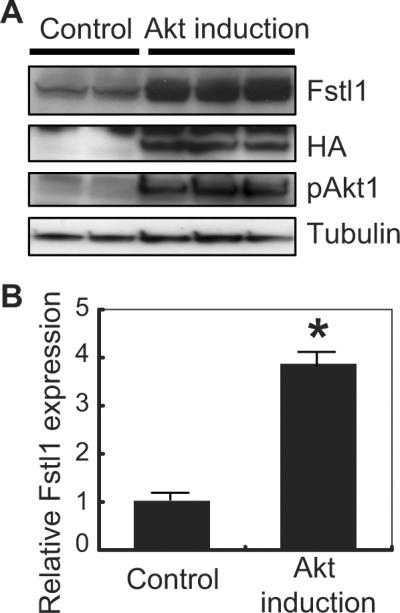
Upregulation of Fstl1 in Akt-activated heart. A, Western blots from transgenic mouse heart lysate. Transgene (myr-Akt tagged with hemagglutinin [HA]) induction was confirmed by probing with antibody against hemagglutinin (middle panel). Two weeks after myr-Akt transgene induction (lanes 3, 4, and 5), Fstl1 expression level was upregulated (top panel) compared with control mice that undergo the same doxycycline administration/withdrawal protocol (lanes 1 and 2). Each lane represents a difference transgenic or control mouse. B, Quantification of the intensity of the bands for Fstl1 normalized with that for α-tubulin. n=3 to 4. *P<0.05.
Fstl1 Is a Secreted Protein
Plasmid and adenovirus vectors expressing Fstl1 were constructed. With the use of cDNA mouse heart, Fstl1 was subcloned into the pcDNA3.1/V5-His expression vector. The vector was transfected into HEK293 cells, and cells were incubated with serum-free media for 24 hours. As shown in Figure 3A, the tagged Fstl1 protein was detected in both the cell pellet lysate and the media, indicating that it is secreted from HEK293 cells. The apparent molecular weight of Fstl1, based on electrophoretic mobility, was greater in the media than in the cell pellet lysate.
Figure 3.

Fstl1 is secreted from cells. A, HEK293 cells were transfected with a plasmid expressing V5 epitope-tagged Fstl1 protein. Western blot analysis using anti-V5 antibody indicates that Fstl1 is present in the cell pellet and the culture media. B, NRVMs were transfected with an adenovirus expressing Fstl1 or β-galactosidase. Western blot analysis using anti-Fstl1 antibody revealed Fstl1 protein expression in the cell pellet and the culture media, and the intensity of these bands increased in cells that were transduced with Ad-Fstl1.
An adenoviral expression vector expressing nontagged Fstl1 was also constructed and tested. NRVMs were transduced with Ad-Fstl1 or Ad-βgal, as a control, and Fstl1 protein was detected in both the cell pellet and the media (Figure 3B). In addition, lower-intensity Fstl1 signals, corresponding to endogenous Fstl1 expression, could be detected in the lysate and media of cells transduced with Ad-βgal (Figure 3B). As with the plasmidencoded Fstl produced by HEK293 cells, the protein detected in the NRVM media exhibited reduced electrophoretic mobility, indicating that the secreted protein is posttranslationally modified. The predicted molecular weight of the protein in the NRVM pellet corresponded to 37 kDa, in good agreement with the predicted molecular weight of 34.4 kDa for the 306-amino acid Fstl1 protein.
Fstl1 Prevents NRVMs From Hypoxia/Reoxygenation-Induced Apoptosis Through Activation of Akt and ERK Signaling
There are no reports of Fstl1-induced changes in intracellular signaling. Thus, NRVMs in serum-free media were transduced with Ad-Fstl1, and Western immunoblot analysis of signaling proteins was performed. Adenovirus-mediated overexpression of Fstl1 in NRVMs led to a marked increase in the activating phosphorylation of Akt at Ser473 (Figure 4). Consistent with an increase in Akt signaling, increases in the phosphorylation of mTOR and FOXO proteins, downstream targets of Akt, were also observed. Transduction with Ad-Fstl1 also led to an increase in ERK phosphorylation in NRVMs.
Figure 4.
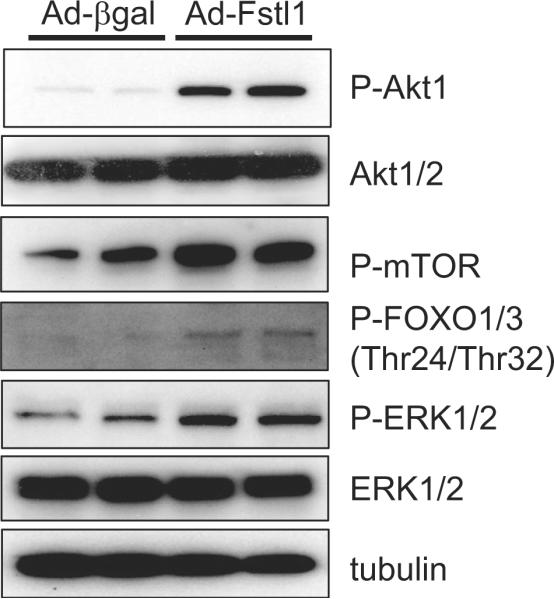
Activation of intracellular signaling pathways by Fstl1. After transfection of Ad-Fstl1, NRVMs were cultured for 36 hours, and cell lysates were prepared for Western blot analysis. Transduction of Ad-Fstl1 led to the activating phosphorylation of Akt1 and 2 and ERK1/2 in NRVMs. Phosphorylation of FOXO1 and 3 and mTOR, downstream targets of Akt signaling, also increased.
Both Akt and ERK signaling promote cardiomyocyte survival.2,21 Thus, we examined the effect of Fstl1 expression on apoptosis after hypoxia/reoxygenation in NRVMs. Cells were transduced with Fstl1-expressing and control adenoviral vectors and then exposed to media without serum. Parallel cultures were maintained in serum-free media and exposed to normoxia or 12 hours of hypoxia followed by 24 hours of reoxygenation (hypoxia/reoxygenation). Terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) staining was performed to evaluate the effect of Fstl1 on cardiac myocyte apoptosis. As shown in Figure 5A, treatment with Ad-Fstl1 reduced the frequency of TUNEL-positive cells under conditions of hypoxia/reoxygenation stress. Quantification of TUNEL-positive myocytes revealed that Ad-Fstl1 significantly reduced TUNEL-positive cell number after hypoxia/reoxygenation compared with Ad-βgal-transfected cells (P=0.008). To corroborate the effects of Fstl1 on apoptosis, nucleosome fragmentation was assessed by enzyme-linked immunosorbent assay. As shown in Figure 5C, hypoxia/reoxygenation-induced nucleosome fragmentation was significantly suppressed by Ad-Fstl1 (P=0.004).
Figure 5.

Fstl1 inhibits hypoxia/reoxygenationinduced apoptosis. A, Serum-deprived NRVMs underwent 12 hours of hypoxia followed by 24 hours of reoxygenation. Representative photographs of cultures stained with TUNEL (green; top panels) to identify apoptotic nuclei and DAPI (blue; bottom panels) to identify total nuclei are shown. Cultures were transfected with Ad-Fstl1 or Ad-βgal before hypoxia/reoxygenation. Scale bar=100 μm. B, Quantification of TUNEL-positive cell number. *P<0.01 compared with Ad-βgal-transfected NRVMs after hypoxia/reoxygenation (H/R). C, Nucleosome fragmentation enzyme-linked immunosorbent assay shows that transduction with Ad-Fstl1 reduced apoptosis after hypoxia/reoxygenation. *P<0.01 compared with Ad-βgal-transfected NRVMs after hypoxia/reoxygenation.
To examine the functional significance of Akt and ERK in Fstl1-mediated cytoprotection, NRVMs were pretreated with specific inhibitors and subjected to hypoxia/reoxygenation. Cells were exposed to LY294002 (10 μmol/L), a PI3K inhibitor, or with U0126 (10 μmol/L), an MEK1/2 inhibitor, 3 hours before hypoxia/reoxygenation stress. Figure 6A shows representative fluorescent photographs of TUNEL staining for each experimental group. As shown in Figure 6B, transduction with Ad-Fstl1 reduced TUNEL-positive cells after exposure to hypoxia/reoxygenation compared with cells treated with Ad-βgal, but the protective action of Ad-Fstl1 was partially attenuated by preincubation with LY294002 or U0126 (P<0.05). In agreement with these findings, the inhibitory effect of Fstl1 on nucleosome fragmentation was significantly attenuated by treatment with each inhibitor (Figure 6C).
Figure 6.

Fstl1-mediated cytoprotection is mitigated by PI3K or MEK1 inhibition. A, Representative photographs of adenovirus-transduced NRVMs stained with TUNEL (green; top panels) and DAPI (blue; bottom panels). Three hours before the induction of hypoxia, NRVMs were pretreated with a PI3K inhibitor (LY294002; 10 μmol/L), a MEK1 inhibitor (U0126; 10 μmol/L), or vehicle. Apoptotic nuclei were identified by TUNEL staining. Scale bar=50 μm. B, Quantification of TUNEL-positive cells under different culture conditions. *P<0.05. C, Nucleosome fragmentation enzyme-linked immunosorbent assay showed that Fstl1-mediated protection of myocytes from apoptosis was reversed by treatment with the PI3K inhibitor or the MEK1 inhibitor. *P<0.05. H/R indicates hypoxia/reoxygenation.
Fstl1 Functions as an Endogenous Cardiac Myocyte Survival Factor
To assess the role of endogenous Fstl1, NRVMs were transfected with siRNA targeting Fstl1. Forty-eight hours after transfection, both mRNA and protein levels of Fstl1 were significantly attenuated, as shown by QRT-PCR and Western blot analysis (Figure 7A and 7B). Fstl1 protein expression level was decreased by 85% in cell lysate and by 90% in culture media by siRNA treatment (P<0.001). At this time point, nucleosome fragmentation was analyzed in serum-deprived cells. As shown in the Figure 7C, knockdown of Fstl1 resulted in increased apoptosis in normoxic cells as well as cells subjected to hypoxia/reoxygenation stress, suggesting that endogenous Fstl1 plays a role in maintaining cardiomyocyte viability. Fstl1 knockdown resulted in reduced phosphorylation of Akt at baseline but had no effect on baseline ERK signaling (Figure 7D). These Western immunoblot observations were corroborated with a second source of siRNA targeting Fstl1 (Figure 7D).
Figure 7.

Knockdown of endogenous Fstl1 increases apoptosis in response to hypoxia/reoxygenation. A, QRT-PCR analysis indicates that transduction of siRNA decreased Fstl1 mRNA by ≈70% (*P<0.05) in cultured cardiac myocytes. B, Representative immunoblot analysis of Fstl1 protein expression in cell lysates and culture media of cardiac myocyte cultures treated with siRNA directed to Fstl1 or an unrelated sequence. C, Nucleosome fragmentation assay shows that knockdown of endogenous Fstl1 expression in serum-deprived cardiac myocyte cultures increases apoptosis in cells under normoxic conditions and in cells treated with hypoxia/reoxygenation (H/R) (*P<0.05). D, Immunoblot analysis reveals that Fstl1 ablation results in decreased Akt phosphorylation, but not that of ERK, in cultured cardiac myocytes. Two commercial sources of siRNA targeting Fstl1 were employed in these assays.
Systemic Administration of Fstl1 Protects the Myocardium From Ischemia/Reperfusion Injury
To assess in vivo actions of Fstl1 on the heart, mice were injected intravenously with Ad-Fstl1 or Ad-βgal (1.0×109 plaque-forming units per mouse) before myocardial ischemia/reperfusion injury. This mode of adenovirus delivery leads to transduction of the liver but not heart,22 thereby allowing the assessment of the cardioprotective properties of the secreted form of Fstl1. Five days after the delivery of Ad-Fstl1, serum Fstl1 levels were markedly elevated (Figure 8A). At this time point, mice were exposed to 30 minutes of myocardial ischemia followed by 24 hours of reperfusion. Figure 8B shows representative heart sections after staining for 2-3-5-triphenyl tetrazolium chloride. Prior treatment with Ad-Fstl1 resulted in a 66.0% decrease in myocardial infarct size compared with mice treated with Ad-βgal (Figure 8C). To investigate the extent of apoptosis in the area at risk, TUNEL staining on the different experimental groups were performed. Representative fluorescent photographs of each group are shown in Figure 8D. Quantitative analysis indicated significantly fewer TUNEL-positive myocytes in the myocardium of mice that were treated with Ad-Fstl1 (Figure 8E; P<0.001).
Figure 8.

Fstl1 protects against myocardial ischemia/reperfusion injury in vivo. A, Western blot analysis of serum proteins from mice after the intravenous delivery of the indicated adenoviral vector. Sera were collected and analyzed 5 days after adenovirus delivery. Serum Fstl1 level was markedly increased by administration of Ad-Fstl1. B, Representative photographs of heart sections stained with Evans blue and 2-3-5-triphenyl tetrazolium chloride to detect the infarction zone resulting from 30 minutes of ischemia and 24 hours of reperfusion. Ischemia/reperfusion injury was performed 5 days after adenoviral injection. C, Quantification of infarction size of each experimental group (n=10 for Ad-βgal group; n=7 for Ad-Fstl1 group). *P<0.05 compared with Ad-βgal-injected group. D, Representative fluorescent images of heart sections stained with TUNEL (green), sarcomeric actin (red), and DAPI (blue). Scale bar=100 μm. E, Quantification of TUNEL-positive myocyte number showed that administration of Ad-Fstl1 decreased apoptosis (n=3 for each group). *P<0.01 compared with Ad-βgal-injected group.
Discussion
In this study, we have found that Fstl1 transcript and protein are upregulated in the myocardium after Akt1 transgene activation. Our studies have also revealed that Fstl1 is a secreted protein and that its overexpression will promote Akt and ERK1/2 signaling in cardiac myocytes and protect cardiac myocytes from hypoxia/reoxygenation-induced apoptosis through activation of these pathways. Conversely, siRNA-mediated knockdown of Fstl1 led to an increase in the frequency of stress-induced apoptosis, and this was associated with a reduction in basal Akt phosphorylation. Fstl1 expression was also upregulated in the heart after pathological stimuli including pressure overload, ischemia/reperfusion, and permanent LAD ligation. Finally, the systemic delivery of Fstl1 protected myocardium from ischemia/reperfusion injury, which was accompanied with a reduction in apoptosis.
A number of lines of evidence suggest that Fstl1 is secreted from cardiac myocytes. Cardiac myocyte cultures display immunodetectable Fstl1 protein in the cell media, and this signal is increased when cultures are transduced with the Fstl1 gene. The secreted form of Fstl1 has a larger molecular weight, presumably the result of protein glycosylation.23 Furthermore, an appreciable increase in the high-molecular-weight form of Fstl1 could be detected in mouse serum after myocardial infarction. Fstl1 has also been detected in human serum,23 indicating that Fstl1 could have diagnostic utility. The heart secretes factors that maintain its performance and produce systemic actions. For example, atrial natriuretic peptide and brain natriuretic peptide are well-known cardiac hormones produced in the heart that serve as therapeutic or diagnostic agents.24,25 Therefore, newly identified factors secreted by the myocardium, such as Fstl1, could also be considered candidates for clinical applications.
Recent reports have proposed that Akt-activated myocardium produces factors that are important in maintenance of cardiac homeostasis. Our previous study revealed that vascular endothelial growth factor and angiopoietin-2 are upregulated in Akt-activated myocardium,8 and vascular endothelial growth factor secretion was found to be necessary for compensatory hypertrophy.8,26 Others have demonstrated that mesenchymal stem cells expressing Akt improve cardiac performance when implanted into myocardium after myocardial infarction and that factors secreted from the Akt-activated mesenchymal stem cells confer the cardioprotective benefits.17 The data presented here provide further support of the hypothesis that Akt-activated cells are a source of secreted factors that are important in cardiac repair and regeneration.
The follistatin family of proteins is generally believed to function by binding to and modifying the function of members of the TGF-β superfamily.27 Follistatin binds to growth and differentiation factor 8 (GDF8), also referred to as myostatin, as well as activin A and B, and various bone morphogenic proteins (BMP) including BMP-2, -4, and -7. Similarly, Fstl3 binds to a subset of these TGF-β superfamily members including myostatin. Myostatin (GDF8) is of interest because it is expressed by the heart and is reported to have antihypertrophic and antiproliferative actions on cultured cardiac myocytes.28,29 However, it is controversial whether myostatin controls heart size in vivo.30,31 It is also of interest that GDF15 is upregulated in the heart by pressure overload or ischemic injury, and it exhibits cardioprotective functions.32,33 It is also reported that serum levels of GDF15 serve as an independent predictor of adverse events in patients with chronic heart failure and acute coronary syndrome.34,35 Finally, BMP-2 has been reported to act on cardiac myocytes to inhibit apoptosis via the SMAD pathway.36
In contrast to the aforementioned considerations, it is not clear whether Fstl1 functions by binding to TGF-β superfamily proteins in a manner similar to follistatin or Fstl3.37 In this regard, follistatin and Fstl3 display a 29% amino acid sequence identity in mice, but Fstl1 shares only 7% homology with follistatin and 6% homology with Fstl3. Currently, we do not favor the hypothesis that Fstl1 acts indirectly on cardiac myocytes through its ability to bind to TGF-β superfamily protein members. In cell culture experiments, we found that Fstl1 expression activated signaling pathways and promoted cell viability in response to hypoxia/reoxygenation. Because these cell culture experiments were performed in serum-free media, it is unlikely that Fstl1 alters cellular responses in cardiac myocytes solely through its ability to modulate the function of the binding partner. Another protein with higher homology to Fstl1 than follistatin or Fstl3 is SPARC, which exhibits a 14% sequence identity. Thus, Fstl1 may be functionally more similar to SPARC than to follistatin and Fstl3, and the structural similarity with SPARC suggests that Fstl1 may function to modulate intracellular signaling by controlling cell interactions with extracellular matrix. Additional experiments will be required to elucidate the receptor-mediated signaling events downstream of Fstl1 in the heart.
Adenovirus-mediated Fstl1 overexpression led to the up-regulation of Akt and ERK signaling in cardiac myocytes and improved cardiac myocyte survival. Furthermore, conditioned media from myocytes transduced with Ad-Fstl1 activated Akt signaling and protected against hypoxia/reoxygenation stress-induced apoptosis in cultures of NRVMs (Y. Oshima, MD, PhD, unpublished data, 2008). Because Akt and ERK are involved in cellular survival,2,21,38 it is likely that the cardioprotective actions of exogenous Fstl1 result from the upregulation of these signaling systems. Consistent with this hypothesis, treatment with inhibitors of PI3K or ERK diminished the antiapoptotic activity of exogenous Fstl1. Furthermore, knockdown of endogenous Fstl1 led to a reduction in Akt phosphorylation and an increase in the frequency of myocyte apoptosis. When it is considered that Fstl1 is upregulated in the Akt-activated heart, it appears that Fstl1 could act as a positive feedback loop to promote myocyte survival, or it could act to promote the survival of neighboring myocytes. In this regard, it has been reported that myocyte survival is also modulated by Pim-1, heme oxygenase-1, and hypoxia-inducible factor-1α, which function within the Akt signaling network.39-41 Further studies are needed to assess whether the prosurvival function of secreted Fstl1 is mediated by these other signaling proteins.
In conclusion, these data show for the first time that Fstl1 is secreted by cardiac myocytes and that this factor is upregulated in the heart by an activation of Akt signaling or by injury. Overexpression of Fstl1 is cardioprotective, whereas ablation of Fstl1 leads to an increase in cardiac myocyte apoptosis. We propose that Fstl1 is a cardioprotective factor that is secreted by the heart.
CLINICAL PERSPECTIVE.
On injury, the heart secretes factors that influence its function. It is widely recognized that the protein kinase Akt plays an important role in regulating intracellular signaling pathways that control cellular survival and physiological growth. To identify novel factors involved in regulating cardiac function, we sought to isolate and characterize proteins that are secreted from murine heart in response to myocardial Akt activation. The secreted protein follistatin-like 1 (Fstl1) is upregulated in heart in response to transgenic Akt activation as well as ischemia/reperfusion injury, pressure overload, and myocardial infarction. Delivery of the Fstl1 gene to mice protected the heart from ischemia/reperfusion injury and reduced cardiac myocyte death, whereas ablation of Fstl1 expression in myocytes led to an increase in apoptotic cell death in vitro. Thus, Fstl1 functions as an endogenous cardiac myocyte survival factor that is secreted by the heart in response to stress. As such, Fstl1 may serve a useful diagnostic or therapeutic function.
Supplementary Material
Acknowledgments
Sources of Funding This study was funded by National Institutes of Health grants HL77774, HL86785, AG15052, and HL81587 to Dr Walsh. Dr Ouchi was supported by American Heart Association grant 0635593T. Dr Pimentel was supported by National Institutes of Health grant HL71563. Dr Oshima was supported by American Heart Association grant 0625867T and a grant from the Uehara Memorial Foundation.
Footnotes
Disclosures Nara Bioscience, Inc, has licensed intellectual property related to Fstl1 from Boston University. Dr Walsh is a founder and stockholder of Nara Bioscience. No research funding was provided by Nara Bioscience. The other authors report no conflicts.
References
- 1.Shiojima I, Walsh K. Regulation of cardiac growth and coronary angiogenesis by the Akt/PKB signaling pathway. Genes Dev. 2006;20:3347–3365. doi: 10.1101/gad.1492806. [DOI] [PubMed] [Google Scholar]
- 2.Fujio Y, Nguyen T, Wencker D, Kitsis RN, Walsh K. Akt promotes survival of cardiomyocytes in vitro and protects against ischemiareperfusion injury in mouse heart. Circulation. 2000;101:660–667. doi: 10.1161/01.cir.101.6.660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Miao W, Luo Z, Kitsis RN, Walsh K. Intracoronary, adenovirus-mediated Akt gene transfer in heart limits infarct size following ischemiareperfusion injury in vivo. J Mol Cell Cardiol. 2000;32:2397–2402. doi: 10.1006/jmcc.2000.1283. [DOI] [PubMed] [Google Scholar]
- 4.Taniyama Y, Walsh K. Elevated myocardial Akt signaling ameliorates doxorubicin-induced congestive heart failure and promotes heart growth. J Mol Cell Cardiol. 2002;34:1241–1247. doi: 10.1006/jmcc.2002.2068. [DOI] [PubMed] [Google Scholar]
- 5.Condorelli G, Drusco A, Stassi G, Bellacosa A, Roncarati R, Iaccarino G, Russo MA, Gu Y, Dalton N, Chung C, Latronico MV, Napoli C, Sadoshima J, Croce CM, Ross J., Jr Akt induces enhanced myocardial contractility and cell size in vivo in transgenic mice. Proc Natl Acad Sci U S A. 2002;99:12333–12338. doi: 10.1073/pnas.172376399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Matsui T, Li L, Wu JC, Cook SA, Nagoshi T, Picard MH, Liao R, Rosenzweig A. Phenotypic spectrum caused by transgenic overexpression of activated Akt in the heart. J Biol Chem. 2002;277:22896–22901. doi: 10.1074/jbc.M200347200. [DOI] [PubMed] [Google Scholar]
- 7.Taniyama Y, Ito M, Sato K, Kuester C, Veit K, Tremp G, Liao R, Colucci WS, Ivashchenko Y, Walsh K, Shiojima I. Akt3 overexpression in the heart results in progression from adaptive to maladaptive hypertrophy. J Mol Cell Cardiol. 2005;38:375–385. doi: 10.1016/j.yjmcc.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 8.Shiojima I, Sato K, Izumiya Y, Schiekofer S, Ito M, Liao R, Colucci WS, Walsh K. Disruption of coordinated cardiac hypertrophy and angiogenesis contributes to the transition to heart failure. J Clin Invest. 2005;115:2108–2118. doi: 10.1172/JCI24682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shiraishi I, Melendez J, Ahn Y, Skavdahl M, Murphy E, Welch S, Schaefer E, Walsh K, Rosenzweig A, Torella D, Nurzynska D, Kajstura J, Leri A, Anversa P, Sussman MA. Nuclear targeting of Akt enhances kinase activity and survival of cardiomyocytes. Circ Res. 2004;94:884–891. doi: 10.1161/01.RES.0000124394.01180.BE. [DOI] [PubMed] [Google Scholar]
- 10.Rota M, Boni A, Urbanek K, Padin-Iruegas ME, Kajstura TJ, Fiore G, Kubo H, Sonnenblick EH, Musso E, Houser SR, Leri A, Sussman MA, Anversa P. Nuclear targeting of Akt enhances ventricular function and myocyte contractility. Circ Res. 2005;97:1332–1341. doi: 10.1161/01.RES.0000196568.11624.ae. [DOI] [PubMed] [Google Scholar]
- 11.de Bold AJ, Borenstein HB, Veress AT, Sonnenberg H. A rapid and potent natriuretic response to intravenous injection of atrial myocardial extract in rats. Life Sci. 1981;28:89–94. doi: 10.1016/0024-3205(81)90370-2. [DOI] [PubMed] [Google Scholar]
- 12.Flynn TG, de Bold ML, de Bold AJ. The amino acid sequence of an atrial peptide with potent diuretic and natriuretic properties. Biochem Biophys Res Commun. 1983;117:859–865. doi: 10.1016/0006-291x(83)91675-3. [DOI] [PubMed] [Google Scholar]
- 13.Maekawa K, Sudoh T, Furusawa M, Minamino N, Kangawa K, Ohkubo H, Nakanishi S, Matsuo H. Cloning and sequence analysis of cDNA encoding a precursor for porcine brain natriuretic peptide. Biochem Biophys Res Commun. 1988;157:410–416. doi: 10.1016/s0006-291x(88)80062-7. [DOI] [PubMed] [Google Scholar]
- 14.Funamoto M, Hishinuma S, Fujio Y, Matsuda Y, Oh H, Negoro S, Tone E, Kishimoto T, Yamauchi-Takihara K. Isolation and characterization of the murine cardiotrophin-1 gene: expression and norepinephrine-induced transcriptional activation. J Mol Cell Cardiol. 2000;32:1275–1284. doi: 10.1006/jmcc.2000.1161. [DOI] [PubMed] [Google Scholar]
- 15.Tsuruda T, Kato J, Kitamura K, Kuwasako K, Imamura T, Koiwaya Y, Tsuji T, Kangawa K, Eto T. Adrenomedullin: a possible autocrine or paracrine inhibitor of hypertrophy of cardiomyocytes. Hypertension. 1998;31:505–510. doi: 10.1161/01.hyp.31.1.505. [DOI] [PubMed] [Google Scholar]
- 16.Frost RJ, Engelhardt S. A secretion trap screen in yeast identifies protease inhibitor 16 as a novel antihypertrophic protein secreted from the heart. Circulation. 2007;116:1768–1775. doi: 10.1161/CIRCULATIONAHA.107.696468. [DOI] [PubMed] [Google Scholar]
- 17.Gnecchi M, He H, Liang OD, Melo LG, Morello F, Mu H, Noiseux N, Zhang L, Pratt RE, Ingwall JS, Dzau VJ. Paracrine action accounts for marked protection of ischemic heart by Akt-modified mesenchymal stem cells. Nat Med. 2005;11:367–368. doi: 10.1038/nm0405-367. [DOI] [PubMed] [Google Scholar]
- 18.Schiekofer S, Shiojima I, Sato K, Galasso G, Oshima Y, Walsh K. Microarray analysis of Akt1 activation in transgenic mouse hearts reveals transcript expression profiles associated with compensatory hypertrophy and failure. Physiol Genomics. 2006;27:156–170. doi: 10.1152/physiolgenomics.00234.2005. [DOI] [PubMed] [Google Scholar]
- 19.Sumitomo K, Kurisaki A, Yamakawa N, Tsuchida K, Shimizu E, Sone S, Sugino H. Expression of a TGF-beta1 inducible gene, TSC-36, causes growth inhibition in human lung cancer cell lines. Cancer Lett. 2000;155:37–46. doi: 10.1016/s0304-3835(00)00407-9. [DOI] [PubMed] [Google Scholar]
- 20.Johnston IM, Spence HJ, Winnie JN, McGarry L, Vass JK, Meagher L, Stapleton G, Ozanne BW. Regulation of a multigenic invasion programme by the transcription factor, AP-1: re-expression of a down-regulated gene, TSC-36, inhibits invasion. Oncogene. 2000;19:5348–5358. doi: 10.1038/sj.onc.1203927. [DOI] [PubMed] [Google Scholar]
- 21.Yue TL, Wang C, Gu JL, Ma XL, Kumar S, Lee JC, Feuerstein GZ, Thomas H, Maleeff B, Ohlstein EH. Inhibition of extracellular signal-regulated kinase enhances ischemia/reoxygenation-induced apoptosis in cultured cardiac myocytes and exaggerates reperfusion injury in isolated perfused heart. Circ Res. 2000;86:692–699. doi: 10.1161/01.res.86.6.692. [DOI] [PubMed] [Google Scholar]
- 22.Shibata R, Sato K, Kumada M, Izumiya Y, Sonoda M, Kihara S, Ouchi N, Walsh K. Adiponectin accumulates in myocardial tissue that has been damaged by ischemia-reperfusion injury via leakage from the vascular compartment. Cardiovasc Res. 2007;74:471–479. doi: 10.1016/j.cardiores.2007.02.010. [DOI] [PubMed] [Google Scholar]
- 23.Liu T, Qian WJ, Gritsenko MA, Camp DG, II, Monroe ME, Moore RJ, Smith RD. Human plasma N-glycoproteome analysis by immunoaffinity subtraction, hydrazide chemistry, and mass spectrometry. J Proteome Res. 2005;4:2070–2080. doi: 10.1021/pr0502065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tang WH, Francis GS, Morrow DA, Newby LK, Cannon CP, Jesse RL, Storrow AB, Christenson RH, Apple FS, Ravkilde J, Wu AH. National Academy of Clinical Biochemistry Laboratory Medicine practice guidelines: clinical utilization of cardiac biomarker testing in heart failure. Circulation. 2007;116:e99–e109. doi: 10.1161/CIRCULATIONAHA.107.185267. [DOI] [PubMed] [Google Scholar]
- 25.Colucci WS, Elkayam U, Horton DP, Abraham WT, Bourge RC, Johnson AD, Wagoner LE, Givertz MM, Liang CS, Neibaur M, Haught WH, LeJemtel TH, Nesiritide Study Group Intravenous nesiritide, a natriuretic peptide, in the treatment of decompensated congestive heart failure. N Engl J Med. 2000;343:246–253. doi: 10.1056/NEJM200007273430403. [DOI] [PubMed] [Google Scholar]
- 26.Izumiya Y, Shiojima I, Sato K, Sawyer DB, Colucci WS, Walsh K. Vascular endothelial growth factor blockade promotes the transition from compensatory cardiac hypertrophy to failure in response to pressure overload. Hypertension. 2006;47:887–893. doi: 10.1161/01.HYP.0000215207.54689.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Balemans W, Van Hul W. Extracellular regulation of BMP signaling in vertebrates: a cocktail of modulators. Dev Biol. 2002;250:231–250. [PubMed] [Google Scholar]
- 28.Morissette MR, Cook SA, Foo S, Mckoy G, Ashida N, Novikov M, Scherrer-Crosbie M, Li L, Matsui T, Brooks G, Rosenzweig A. Myostatin regulates cardiomyocyte growth through modulation of Akt signaling. Circ Res. 2006;99:15–24. doi: 10.1161/01.RES.0000231290.45676.d4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McKoy G, Bicknell KA, Patel K, Brooks G. Developmental expression of myostatin in cardiomyocytes and its effect on foetal and neonatal rat cardiomyocyte proliferation. Cardiovasc Res. 2007;74:304–312. doi: 10.1016/j.cardiores.2007.02.023. [DOI] [PubMed] [Google Scholar]
- 30.Cohn RD, Liang HY, Shetty R, Abraham T, Wagner KR. Myostatin does not regulate cardiac hypertrophy or fibrosis. Neuromuscul Disord. 2007;17:290–296. doi: 10.1016/j.nmd.2007.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Artaza JN, Reisz-Porszasz S, Dow JS, Kloner RA, Tsao J, Bhasin S, Gonzalez-Cadavid NF. Alterations in myostatin expression are associated with changes in cardiac left ventricular mass but not ejection fraction in the mouse. J Endocrinol. 2007;194:63–76. doi: 10.1677/JOE-07-0072. [DOI] [PubMed] [Google Scholar]
- 32.Xu J, Kimball TR, Lorenz JN, Brown DA, Bauskin AR, Klevitsky R, Hewett TE, Breit SN, Molkentin JD. GDF15/MIC-1 functions as a protective and antihypertrophic factor released from the myocardium in association with SMAD protein activation. Circ Res. 2006;98:342–350. doi: 10.1161/01.RES.0000202804.84885.d0. [DOI] [PubMed] [Google Scholar]
- 33.Kempf T, Eden M, Strelau J, Naguib M, Willenbockel C, Tongers J, Heineke J, Kotlarz D, Xu J, Molkentin JD, Niessen HW, Drexler H, Wollert KC. The transforming growth factor-beta superfamily member growth-differentiation factor-15 protects the heart from ischemia/reperfusion injury. Circ Res. 2006;98:351–360. doi: 10.1161/01.RES.0000202805.73038.48. [DOI] [PubMed] [Google Scholar]
- 34.Kempf T, von Haehling S, Peter T, Allhoff T, Cicoira M, Doehner W, Ponikowski P, Filippatos GS, Rozentryt P, Drexler H, Anker SD, Wollert KC. Prognostic utility of growth differentiation factor-15 in patients with chronic heart failure. J Am Coll Cardiol. 2007;50:1054–1060. doi: 10.1016/j.jacc.2007.04.091. [DOI] [PubMed] [Google Scholar]
- 35.Wollert KC, Kempf T, Peter T, Olofsson S, James S, Johnston N, Lindahl B, Horn-Wichmann R, Brabant G, Simoons ML, Armstrong PW, Califf RM, Drexler H, Wallentin L. Prognostic value of growth-differentiation factor-15 in patients with non-ST-elevation acute coronary syndrome. Circulation. 2007;115:962–971. doi: 10.1161/CIRCULATIONAHA.106.650846. [DOI] [PubMed] [Google Scholar]
- 36.Izumi M, Fujio Y, Kunisada K, Negoro S, Tone E, Funamoto M, Osugi T, Oshima Y, Nakaoka Y, Kishimoto T, Yamauchi-Takihara K, Hirota H. Bone morphogenetic protein-2 inhibits serum deprivation-induced apoptosis of neonatal cardiac myocytes through activation of the Smad1 pathway. J Biol Chem. 2001;276:31133–31141. doi: 10.1074/jbc.M101463200. [DOI] [PubMed] [Google Scholar]
- 37.Mashimo J, Maniwa R, Sugino H, Nose K. Decrease in the expression of a novel TGF beta1-inducible and ras-recision gene, TSC-36, in human cancer cells. Cancer Lett. 1997;113:213–219. doi: 10.1016/s0304-3835(97)04700-9. [DOI] [PubMed] [Google Scholar]
- 38.Bueno OF, De Windt LJ, Tymits KM, Witt SA, Kimball TR, Klevitsky R, Hewett TE, Jones SP, Lefer DJ, Peng CF, Kitsis RN, Molkentin JD. The MEK1-ERK1/2 signaling pathway promotes compensated cardiac hypertrophy in transgenic mice. EMBO J. 2000;19:6341–6350. doi: 10.1093/emboj/19.23.6341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cai Z, Zhong H, Bosch-Marce M, Fox-Talbot K, Wang L, Wei C, Trush MA, Semenza GL. Complete loss of ischaemic preconditioninginduced cardioprotection in mice with partial deficiency of HIF-1{alpha} Cardiovasc Res. 2008;77:463–470. doi: 10.1093/cvr/cvm035. [DOI] [PubMed] [Google Scholar]
- 40.Muraski JA, Rota M, Misao Y, Fransioli J, Cottage C, Gude N, Esposito G, Delucchi F, Arcarese M, Alvarez R, Siddiqi S, Emmanuel GN, Wu W, Fischer K, Martindale JJ, Glembotski CC, Leri A, Kajstura J, Magnuson N, Berns A, Beretta RM, Houser SR, Schaefer EM, Anversa P, Sussman MA. Pim-1 regulates cardiomyocyte survival downstream of Akt. Nat Med. 2007;13:1467–1475. doi: 10.1038/nm1671. [DOI] [PubMed] [Google Scholar]
- 41.Das S, Fraga CG, Das DK. Cardioprotective effect of resveratrol via HO-1 expression involves p38 map kinase and PI-3-kinase signaling, but does not involve NFkappaB. Free Radic Res. 2006;40:1066–1075. doi: 10.1080/10715760600833085. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.